Aspectos eletrônicos e estruturais do grafeno e derivados:: um estudo teórico-experimental
Texto
Imagem
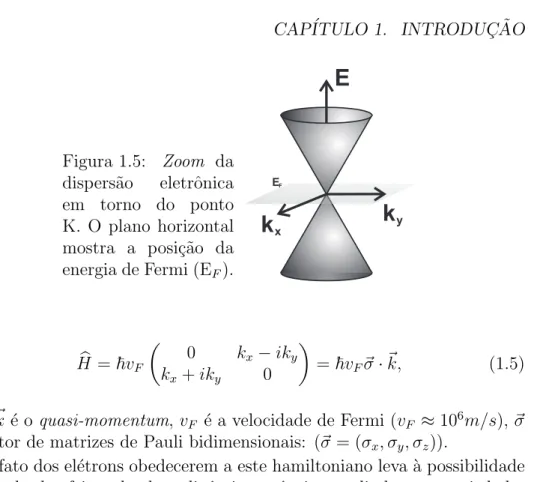
![Figura 1.4: Dispers˜ao eletrˆonica do grafeno de uma ´ unica camada calculada por (a) tight binding, figura adaptada da referˆencia [8]; e (b) primeiros princ´ıpios, utilizando a Teoria do Funcional da Densidade](https://thumb-eu.123doks.com/thumbv2/123dok_br/15709329.121113/14.892.135.701.394.576/eletrˆonica-calculada-adaptada-referˆencia-primeiros-utilizando-funcional-densidade.webp)
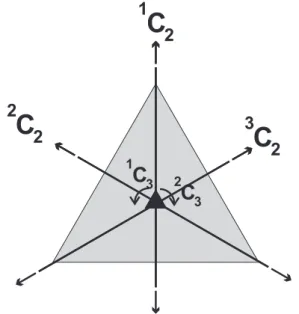
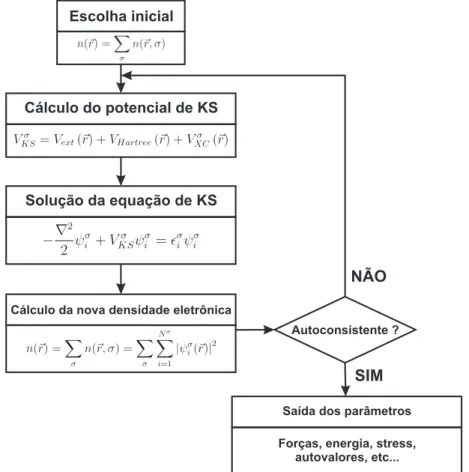
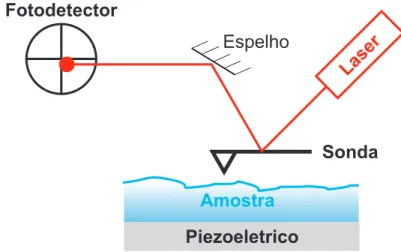
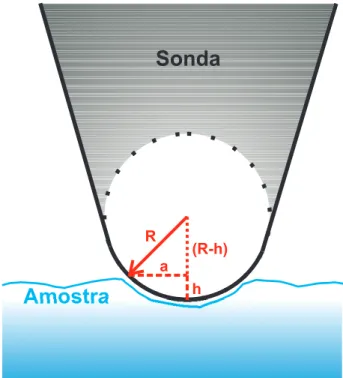
Documentos relacionados
◦ Os filtros FIR implementados através de estruturas não recursivas têm menor propagação de erros. ◦ Ruído de quantificação inerente a
Os candidatos reclassificados deverão cumprir os mesmos procedimentos estabelecidos nos subitens 5.1.1, 5.1.1.1, e 5.1.2 deste Edital, no período de 15 e 16 de junho de 2021,
Todavia, há poucos trabalhos sobre interferência de herbicidas no crescimento da cultura; portanto, visando avaliar os efeitos de amicarbazone e diuron + paraquat, estes foram
Para Souza (2004, p 65), os micros e pequenos empresários negligenciam as atividades de planejamento e controle dos seus negócios, considerando-as como uma
A direção dos Serviços Farmacêuticos Hospitalares (SFH) fica sempre a cargo de um farmacêutico. São várias as funções desempenhadas pelo FH no decorrer da sua atividade
Contudo, não é possível imaginar que essas formas de pensar e agir, tanto a orientada à Sustentabilidade quanto a tradicional cartesiana, se fomentariam nos indivíduos
A variável em causa pretende representar o elemento do modelo que se associa às competências TIC. Ainda que o Plano Tecnológico da Educação preveja a conclusão da
Entre as atividades, parte dos alunos é também conduzida a concertos entoados pela Orquestra Sinfônica de Santo André e OSESP (Orquestra Sinfônica do Estado de São
