www.jped.com.br
ARTIGO
DE
REVISÃO
What
is
new
in
genetics
and
osteogenesis
imperfecta
classification?
夽
Eugênia
R.
Valadares
a,∗,
Túlio
B.
Carneiro
a,
Paula
M.
Santos
b,
Ana
Cristina
Oliveira
be
Bernhard
Zabel
caHospitaldasClínicas,FaculdadedeMedicina,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brasil bFaculdadedeOdontologia,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brasil
cClínicaPediátricadaUniversidadedeFreiburg,Freiburg,Alemanha
Recebidoem17demarçode2014;aceitoem27demaiode2014
KEYWORDS
Osteogenesis imperfecta;
Osteochondrodysplasias; Collagentype1
Abstract
Objective: Literaturereviewofnewgenesrelatedtoosteogenesisimperfecta(OI)andupdate
ofitsclassification.
Sources: LiteraturereviewinthePubMedandOMIMdatabases,followedbyselectionofrelevant
references.
Summaryofthefindings: In1979,Sillenceetal.developedaclassificationofOIsubtypesbased
onclinicalfeaturesanddiseaseseverity:OItypeI,mild,common,withbluesclera;OItype II,perinatallethalform;OItypeIII,severeandprogressivelydeforming,withnormalsclera; andOItypeIV,moderateseveritywithnormalsclera.Approximately90%ofindividualswith OIareheterozygousformutationsintheCOL1A1andCOL1A2genes,withdominantpatternof inheritanceorsporadicmutations.After2006,mutationswereidentifiedintheCRTAP,FKBP10,
LEPRE1,PLOD2,PPIB,SERPINF1,SERPINH1,SP7,WNT1,BMP1,andTMEM38Bgenes,associated
withrecessiveOIandmutationintheIFITM5geneassociatedwithdominantOI.Mutationsin PLS3wererecentlyidentifiedinfamilieswithosteoporosisandfractures,withX-linked inhe-ritancepattern.InadditiontothegeneticcomplexityofthemolecularbasisofOI,extensive phenotypicvariabilityresultingfromindividuallocihasalsobeendocumented.
Conclusions: Consideringthediscoveryofnewgenesandlimitedgenotype-phenotype
corre-lation,theuseofnext-generationsequencingtoolshasbecomeusefulinmolecularstudiesof OIcases.The recommendationoftheNosologyGroup oftheInternationalSociety of Skele-talDysplasiasistomaintaintheclassificationofSillenceastheprototypicalform,universally acceptedtoclassifythedegreeofseverityinOI,whilemaintainingitfreefromdirectmolecular reference.
©2014SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
DOIserefereaoartigo:http://dx.doi.org/10.1016/j.jped.2014.05.003
夽 Comocitaresteartigo:ValadaresER,CarneiroTB,SantosPM,OliveiraAC,ZabelB.Whatisnewingeneticsandosteogenesisimperfecta
classification?JPediatr(RioJ).2014;90:536---41. ∗Autorparacorrespondência.
E-mail:[email protected](E.R.Valadares).
PALAVRAS-CHAVE
Osteogênese imperfeita;
Osteocondrodisplasias; Colágenotipo1
Oquehádenovoemgenéticaeclassificac¸ãodeosteogêneseimperfeita?
Resumo
Objetivo: Revisãodaliteraturasobrenovosgenesrelacionadosàosteogêneseimperfeita(OI)
eatualizac¸ãodasuaclassificac¸ão.
Fontedosdados: RevisãonasbasesdedadosdoPUBMEDeOMIMcomselec¸ãodereferências
relevantes.
Síntesedosdados: Sillenceetal.,em1979,desenvolveramumaclassificac¸ãodossubtiposdeOI
baseadaemcaracterísticasclínicasegravidadedadoenc¸a:OItipoI,formaleve,comum,com esclerasazuladas;OItipoII,formaperinatalletal;OItipoIII,formagraveeprogressivamente deformantecomescleranormal;eOItipoIV,formadegravidademoderadacomescleranormal. Cercade90%dosindivíduoscomOIsãoheterozigotosparamutac¸õesemCOL1A1eCOL1A2,com padrãodeheranc¸adominanteouesporádico.Apartirde2006foramidentificadasmutac¸õesnos genesCRTAP,FKBP10,LEPRE1,PLOD2,PPIB,SERPINF1,SERPINH1,SP7,WNT1,BMP1eTMEM38B
associadasàOIrecessivaemutac¸ãoemIFITM5associadaàOIdominante.Mutac¸õesemPLS3
foram identificadas recentemente em famílias comosteoporose efraturas, compadrão de heranc¸aligadoaoX.AlémdacomplexidadegenéticadasbasesmolecularesdasOI,extensa variabilidadefenotípicaresultantedelociindividuaistambémtemsidodocumentada.
Conclusões: Faceàdescobertadenovosgeneseàcorrelac¸ãogenótipo-fenótipo limitada,o
usodeferramentasdesequenciamentodenovagerac¸ãotorna-seútilnoestudomolecularde casosdeOI.Arecomendac¸ãodoGrupodeNosologiadaSociedadeInternacionaldeDisplasias Esqueléticasémanteraclassificac¸ãodeSillencecomoaformaprototípicaeuniversalmente aceitaparaclassificarograudegravidadenaOI,elibertá-ladereferênciamoleculardireta. ©2014SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduc
¸ão
Osteogêneseimperfeita(OI)éumgrupodedoenc¸asclinica egeneticamenteheterogêneo,caracterizadopor suscetibi-lidadeafraturasósseascomgravidadevariávele defeitos presumidos ou comprovados na biossíntese de colágeno tipoI.Outrasmanifestac¸õessãodentinogêneseimperfeita, escleras azuis, baixa estatura, e perda auditiva na idade adulta.Asmanifestac¸õesclínicasvariamemumcontinuum que vaidesde casosgraves, com letalidadeperinatal,até indivíduos assintomáticos,compredisposic¸ãoleve a fratu-ras,estaturaevidanormais.1
Em, a incidência dos vários tipos de OI é de cerca de
1 em 15.000-20.000 nascimentos, a maioria de heranc¸a
autossômicadominantepormutac¸ãoemCOL1A1ouCOL1A2,
quecodificamascadeias␣1(I)e␣2(I)decolágenotipoI.1
OcolágenotipoI,principalproteínaestruturaldamatrix
extracelulardosossos,peleetendões,écompostodeduas
pró-cadeias␣-1 eumapró-cadeia ␣-2,que seentrelac¸am
formandotripla hélice rígida. Cadacadeia ␣ contém
pró--peptídeosterminaisnasextremidadesC-terminal(carboxi)
e N-terminal (amino) e um domínio central composto de
338repetic¸õesdeGly-X-Y,ondeoXeoYexcluemcisteína
etriptofano,efrequentementesão,respectivamente,
pro-linaehidroxiprolina.Aglicina,porseromenoraminoácido,
éoúnicoresíduocapazdeocuparaposic¸ãoaxialdatripla
hélice,demodoquequalqueralterac¸ãoemumresíduode
glicinaacarretarádesorganizac¸ãodaestruturahelicoidal.2,3
As mutac¸õesemCOL1A1 eCOL1A2alteram aestrutura
ouaquantidadedecolágenotipoIecausamumfenótipo
esqueléticoquevariadesubclínicoaletal.1Estespacientes
apresentamanomaliasqualitativasequantitativasno
colá-genotipoIdevidoaoefeitodominantenegativodamutac¸ão,
já que as pró-cadeias ␣ mutantes são incorporadas nas
moléculasdepró-colágenotipoI,quecontêmtambém
pró--cadeias␣normais.Comoregra,quandohásubstituic¸ãoda
glicinanacadeia␣1,eofenótipovaidependerdaposic¸ãoda
substituic¸ão:substituic¸õesC-terminaiscausamumfenótipo
gravedadoenc¸a e assubstituic¸õesN-terminais, fenótipos
maisleves.4,5Resíduoscomcadeiaslateraisgrandesou
car-regadossãoaltamentedesorganizadoresdaestruturatripla,
nãoimportandoondeestejamlocalizados.Diferentes
fenó-tipostêmsidoencontradoscomamesmamutac¸ão.6
Emconsórciorealizadoem2007paraestudodemutac¸ões
causadorasdeOInosgenesdecolágeno1,foram
identifi-cadas1.832mutac¸õesindependentes,sendo682resultado
de substituic¸ão de resíduos de glicina no domínio da
tri-plahélice da proteínacodificada e 150 de locais de sítio
desplice.6
Combaseemachadosclínicos,achadosradiográficosdo
esqueleto,mododeheranc¸aeanálisesgenéticas
molecula-res,novasOIvêmsendoidentificadasapartirde2006,por
meiode sequenciamento do exoma. O presente trabalho
tevepor objetivo rever a classificac¸ão das OIe atualizar
osnovosgenes relacionados.Foramutilizadasasbasesde
dadosdoPUBMEDedoMendelianInheritanceinMan(MIM).7
Classificac¸ãodeSillence
Devido à variabilidade fenotípica considerável, Sillence
Tabela1 Classificac¸ãodeOIa
Tipo Manifestac¸õesgerais Manifestac¸õesespecíficas
I-OIdeheranc¸aautossômica dominantecomescleraazulada.
Fragilidadeósseavariável,escleraazulada, surdezprecoce,baixaestaturaleve.
IA:dentesnormais.IBeIC:dentinogênese imperfecta.
II-OIperinatalletal
radiograficamentecomfêmures sanfonadosecostelasem rosário.
Fragilidadeósseaextrema,morteperinatal. IIA:ossoslongoscurtosealargadoscom fraturas,costelaslargascomfraturas.IIB: ossoslongoscurtosealargadoscom fraturas,costelascomfraturasesparsas. IIC:ossoslongosfinoscomfraturas, costelasfinas.
III-OIprogressivamente deformantecomesclera normal.
Fragilidadeósseamoderadaagrave, esclerasazuladasnainfância.
Cifoescolioseprecoce.Dentinogênese imperfectapodeestarpresente.
IV-OIdeheranc¸aautossômica dominantecomescleranormal
Fragilidadeóssea,deformidadedosossos longosecolunadegraumoderadoagrave, esclerabranca,baixaestaturamoderadaa grave.
IVA:dentesnormais.IVB:dentinogênese imperfecta.
aModificadadeSillence.8
baseadaem característicasclínicasegravidadedadoenc¸a
(tabela1):OItipoI,formaleve,comum,comescleras
azula-das;OItipoII,formaperinatalletal;OItipoIII,formagrave
eprogressivamentedeformante,comescleranormal;eOI
tipoIV,formadegravidademoderada,comescleranormal.
Aclassificac¸ãodeSillencevemsendorepetidamenterevista
emmomentos deidentificac¸ão denovosgenescausadores
daOI.
Classificac¸ãoexpandida
A classificac¸ão genética molecular de OItem se revelado muito heterogênea, com diversos padrões de heranc¸a e amplavariabilidadedegravidadeclínica.10
Glorieux et al.11 descreveram uma forma
autossô-mica dominante de OI, similar à OI tipo IV de Sillence,
mas com características clínica, histológica e
molecu-lar distintas. Não foi encontrada mutac¸ão em COL1A1
e COL1A2, sendo então nomeada pelos autores de OI
tipo V (MIM #610967). Cerca de 65% dos indivíduos
afe-tados desenvolvem calos hiperplásicos após fraturas ou
intervenc¸ões cirúrgicas, considerada uma característica
patognomônica.12 Somente em 2012 foram identificadas
mutac¸õesnoIFITM5em pacientescomOItipoV,geneque
codificaa proteína 5 transmembrana interferon-induzida,
por sequenciamento de todo o exoma.12---14 A proteína
codificada tem papel na mineralizac¸ão precoce, mas seu
mecanismoédesconhecido.10
Em2006,mutac¸ãonogeneCRTAPfoiidentificadacomo
primeiracausagenéticadeOIrecessivaletal.15Apartirde,
mutac¸õesemnovosgenesquecausamOIrecessivatêmsido
identificadasporsequenciamentodoexoma,comoFKBP10,
LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP, BMP1 e
TMEM38B.Cadaumdestesgenesrecebeuumnúmerodetipo
deOInabasededadosMIM,dandosequênciaaosnúmeros
declassificac¸ãodeSillence.
AOIchamadatipoVI(MIM#613982)éumaforma
autos-sômica recessiva da doenc¸a, que pode ser causada por
mutac¸ãohomozigóticanogeneSERPINF1em17p13.3,com
defeitodemineralizac¸ão.14Pelaclassificac¸ãodeSillence,o
fenótipoécompatívelcomotipoIVoutipoIII.16,17
OItipoVII(MIM#610682)éumaformaautossômica
reces-siva letal de OI causada por mutac¸ão no gene CRTAP em
homozigoseouheterozigosecompostanocromossomo3p22.
Éresponsávelpor2a3%doscasosdeOIletal.15
Cabral et al.18 descreveram uma forma de OI
autos-sômica recessiva, denominada OI tipoVIII (MIM #610915),
quesecaracterizaporesclerabranca,gravedeficiênciade
crescimento,mineralizac¸ãoesqueléticamuitodeficientee
metáfisesbulbosas. Esta formaécausada pormutac¸ãono
genequecodificaleprecan(LEPRE1),em1p34.2,associada
àOIgraveouletal.
OItipoIX(MIM#259440)éumaformaautossômica
reces-sivadeOIcorrespondenteaostipos clinicamentegravesII
/ III da classificac¸ão de Sillence.19 Não há relato de
den-tinogênese imperfeita.Ela podeser causadapor mutac¸ão
homozigóticadogenePPIBem15q22.31.
OItipoX(MIM#613848)éumaformaautossômica
reces-siva,quepodesercausadaporumamutac¸ãohomozigótica
dogeneSERPINH1nocromossomo11q13.5.Écaracterizada
por deformidades ósseas e fraturas múltiplas, osteopenia
generalizada,dentinogêneseimperfeitae escleraazulada.
SERPINH1 codifica uma proteína de colágeno de ligac¸ão
que funciona como uma chaperona no retículo
endoplas-mático,motivopeloqualosindivíduoscommutac¸ãoneste
geneapresentamcélulasquenãoproduzemcolágenotipoI
supermodificado.20
OItipoXI(MIM#610968)éumaformaautossômica
reces-siva causada por uma mutac¸ão homozigótica do gene
FKBP10 em 17q21, também relacionada a defeito
de chaperona.1 Pacientes com OI tipo XI apresentam
deformac¸ão progressiva grave e podem ter contraturas
articulares. Os pacientes não apresentam dentinogênese
imperfeita.21---23
OI tipo XII (MIM #613849) é uma forma autossômica
recessiva, que pode ser causada por mutac¸ão no gene
SP7 em 12q13.13. Clinicamente é caracterizada por
fra-turas recorrentes,deformac¸ões ósseas leves,osteoporose
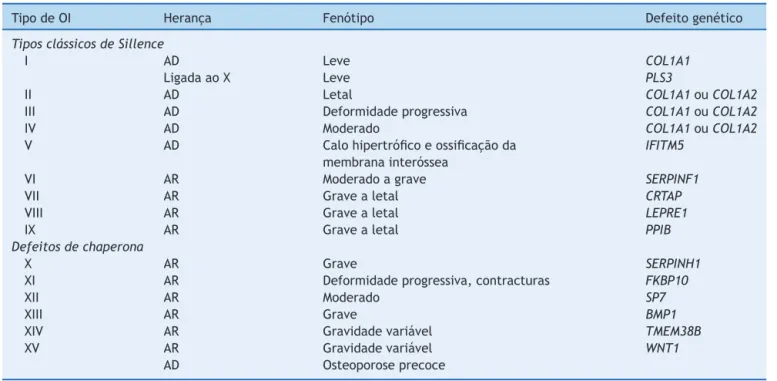
Tabela2 NosologiadaOIa
TipodeOI Heranc¸a Fenótipo Defeitogenético
TiposclássicosdeSillence
I AD Leve COL1A1
LigadaaoX Leve PLS3
II AD Letal COL1A1ouCOL1A2
III AD Deformidadeprogressiva COL1A1ouCOL1A2
IV AD Moderado COL1A1ouCOL1A2
V AD Calohipertróficoeossificac¸ãoda membranainteróssea
IFITM5
VI AR Moderadoagrave SERPINF1
VII AR Gravealetal CRTAP
VIII AR Gravealetal LEPRE1
IX AR Gravealetal PPIB
Defeitosdechaperona
X AR Grave SERPINH1
XI AR Deformidadeprogressiva,contracturas FKBP10
XII AR Moderado SP7
XIII AR Grave BMP1
XIV AR Gravidadevariável TMEM38B
XV AR Gravidadevariável WNT1
AD Osteoporoseprecoce
AD,autossômicadominante;AR,autossômicarecessiva.
a AdaptadadeForlinoetal.1
dentinogênese imperfeita, audic¸ão normal e esclerótica
branca.24
OItipoXIII(MIM#614856)foidescritopormutac¸ão
homo-zigóticanogeneBMP1nocromossomo8p21.25,26
Shaheenetal.27descreveramaOItipoXIV(MIM#615066),
umaforma autossômicarecessiva caracterizada por graus
variáveisdegravidadedemúltiplasfraturaseosteopenia,
comdentes,escleraeaudic¸ãonormais.Fraturasocorremno
pré-natalouporvoltadosseisanosdeidade.Écausadapor
mutac¸ão homozigótica no gene TMEM38B no cromossomo
9q31.
OI tipo XV (MIM #615220) foi nomeada a partir da
identificac¸ão de mutac¸ões em WNT1.28---30 Keupp et al.30
reportaram que osaleloshipofuncionais deWNT1 causam
fenótipos combaixa massa ósseaem humanos.
Identifica-ram que mutac¸ões no gene herdadas de forma recessiva
levamafenótiposdegravidadesvariáveis,variandode
for-masmoderadasaprogressivamentedeformantes,podendo,
ocasionalmente, levar à morte infantil precoce.
Detecta-ramtambémfamíliascompadrãoautossômicodominantede
osteoporoseprecoceapresentandomutac¸ãoheterozigótica
emWNT1.
AsformasrecessivasdeOIcomfenótiposletaisa
mode-rados são causadaspor defeitos em genes cujos produtos
interagem com o colágeno tipo I. A maioria dos casos
recessivos tem mutac¸ões nulas em genes que codificam
proteínas envolvidas na prolil 3-hidroxilac¸ão do colágeno
(CRTAP, LEPRE1 e PPIB) ou as responsáveis pela
cor-reta dobragemhelical (FKBP10 e SERPINH1). Ostipos VII,
VIII e IX são causados por defeitos de 3-hidroxilac¸ão.1 A
correlac¸ãogenótipo-fenótiponasformasrecessivastemsido
sugerida.31
Em2013mutac¸õesemPLS3foramidentificadasem
famí-liascomosteoporoseefraturassemanifestandonainfância,
deheranc¸aligadaaoX.32
A tabela 2 resume a classificac¸ão baseada em genes envolvidos.
Em2010,vanDijketal.33propuseramumaclassificac¸ão
revisadadasOI,mencionandoo genecausadoreoquadro
clínico indicado apenas para os tipos de I a VI. Os tipos
VIIe VIII foramexcluídos,umavez queestes haviamsido
adicionadospor critérios genéticos, embora seus achados
clínicose radiológicosfossemindistinguíveisdos tiposII a
IV.Aclassificac¸ãoproposta deixaespac¸oparanovosgenes
descobertoscomocausadeOIatéqueaextensãoda
hete-rogeneidadesejaconhecida.34
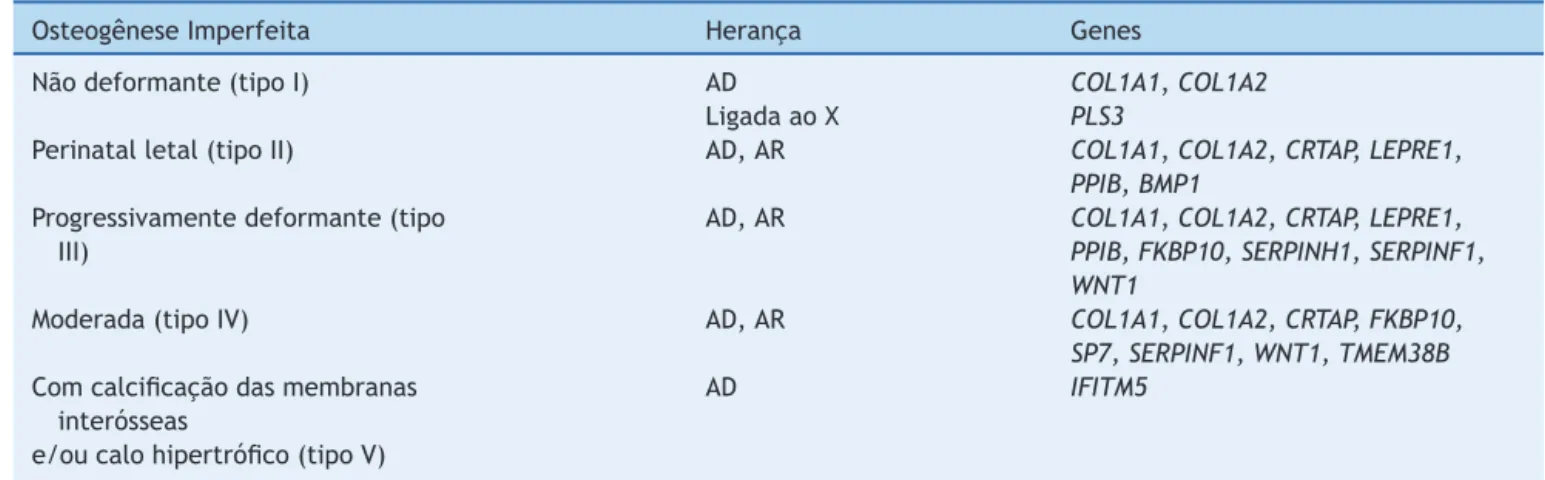
Classificac¸ãodasOIpelaSociedadeInternacional deDisplasiasEsqueléticas
Pela alta complexidade genética das bases moleculares das OI e extensa variabilidade fenotípica resultante de loci individuais descrita nos últimos anos, parecia insus-tentável manter correlac¸ões entre os tipos de Sillence e sua base molecular. Porém, a proliferac¸ão dos tipos de OIpara refletir cada geneseparadamente, defendida por alguns, se tornou mais confusa do que útil na prática clínica. Por esses motivos,o Grupo deNosologia da Soci-edadeInternacionaldeDisplasiasEsqueléticas,reunidoem 2009,recomendoumanteraclassificac¸ãodeSillencecomo aforma prototípicae universalmenteaceita para classifi-car o grau de gravidade na OI e libertá-la de referência moleculardireta.35 Assim,comolistadasnatabela3,asOI
foramagrupadasem cinco categoriasclínicas, e osvários
genesquepodemcausarOIforamlistadosseparadamente.
AcrescentamosàtabelaoriginalosgenesIFITM5,SERPINF1,
BMP1,WNT1,TMEM38Be PLS3,descobertosdepoisdasua
Tabela3 Classificac¸ãodasOIpelaSociedadeInternacionaldeDisplasiasEsqueléticasacomacréscimodegenesrecentemente
descobertos
OsteogêneseImperfeita Heranc¸a Genes
Nãodeformante(tipoI) AD COL1A1,COL1A2
LigadaaoX PLS3
Perinatalletal(tipoII) AD,AR COL1A1,COL1A2,CRTAP,LEPRE1,
PPIB,BMP1
Progressivamentedeformante(tipo III)
AD,AR COL1A1,COL1A2,CRTAP,LEPRE1,
PPIB,FKBP10,SERPINH1,SERPINF1,
WNT1
Moderada(tipoIV) AD,AR COL1A1,COL1A2,CRTAP,FKBP10,
SP7,SERPINF1,WNT1,TMEM38B
Comcalcificac¸ãodasmembranas interósseas
AD IFITM5
e/oucalohipertrófico(tipoV)
AD,autossômicadominante;AR,autossômicarecessiva.
aWarmanetal.35
Conclusão
Naprática,apesardacomplexavariabilidadegenotípicada OI evidenciada nos últimos anos, seus fenótipos ainda se enquadramnaclassificac¸ãodeSillence.Ainvestigac¸ão geno-típica deveser indicada especialmentenos casos em que sugiram heranc¸a autossômica recessiva, a fim de aconse-lhamentogenético.Oestudo moleculardeveser feitopor meiodesequenciamentodeSangerdosdiversosnovosgenes ouporsequenciamentodenovagerac¸ão.Osequenciamento doexomatemutilidadequandonãoháumpaineldegenes disponível,ouquandonãoseconhecemosgenesenvolvidos.
Financiamento
CNPq(Conselho Nacionalde Desenvolvimento Científico e Tecnológico).
Conflitos
de
interesse
Osautoresdeclaramnãohaverconflitosdeinteresse.
Agradecimentos
AoConselhoNacionaldeDesenvolvimentoCientíficoe Tec-nológico (CNPq) por ter proporcionado, em 2013, a bolsa de pós-doutorado de Eugênia Ribeiro Valadares no setor degenéticadaClínicaPediátricadaUniversidadede Frei-burg,Alemanha,paradesenvolveroprojeto‘‘Investigac¸ão de osteogênese imperfeita pela análise dos genes conhe-cidose novosgenes candidatosem pacientesbrasileirose alemães’’,sobsupervisãodoProf.Dr.BernhardZabel,doDr. PabloVillavicencioLoriniedoDr.EkkehartLausch,pessoas donossomaioraprec¸o.
Referências
1.ForlinoA,CabralWA,BarnesAM,MariniJC.Newperspectiveson osteogenesisimperfecta.NatRevEndocrinol.2011;7:540---57.
2.ColeWG.Themolecularpathologyofosteogenesisimperfecta. ClinOrthopRelatRes.1997;343:235---48.
3.ByersPH,WallisGA,WillingMC.Osteogenesisimperfecta: trans-lationofmutationtophenotype.JMedGenet.1991;28:433---42. 4.StaceyA,BatemanJF,ChoiT.Perinatallethalintransgenicmice bearinganengineered mutantpro-[alpha]1(I)collagengene. Nature.1988;332:131---6.
5.ColeWG,DalgleisR.Perinatallethalosteogenesis.JMedGenet. 1995;32:284---9.
6.MariniJC,ForlinoA, CabralWA,BarnesAM,SanAntonioJD, MilgromS,etal.Consortiumforosteogenesisimperfecta muta-tionsinthehelicaldomainoftypeI collagen:regions richin lethalmutationsalignwithcollagenbindingsitesforintegrins andproteoglycans.HumMutat.2007;28:209---21.
7.OnlineMendelianInheritanceinMan,OMIM®.McKusick-Nathans
Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), [cited 14 Mar 2014]. Available from: http://omim.org/
8.SillenceDO,SennA,DanksDM.Geneticheterogeneityin oste-ogenesisimperfecta.JMedGenet.1979;16:101---16.
9.SillenceDO.Osteogenesisimperfect:anexpandingpanorama ofvariants.ClinOrtop.1981;159:11---25.
10.MariniJC,BlissetAR.Newgenesinbonedevelopment:what’s new in osteogenesis imperfecta. J Clin Endocrinol Metab. 2013;98:3095---103.
11.GlorieuxFH,RauchF,PlotkinH,WardL, TraversR,Roughley P,etal.TypeVosteogenesisimperfecta:anewformofbrittle bonedisease.JBoneMinerRes.2000;15:1650---8.
12.SemlerO,Garbes L,KeuppK, SwanD,Zimmermann K, Bec-ker J,et al. A mutationinthe 5′-UTR ofIFITM5 creates an
in-framestartcodonandcausesautosomal-dominant osteoge-nesisimperfecta typeV withhyperplasticcallus. AmJ Hum Genet.2012;91:349---57.
13.ChoTJ,LeeKE,LeeSK,SongSJ,KimKJ,JeonD,etal.Asingle recurrentmutationinthe5’-UTRofIFITM5causesosteogenesis imperfectatypeV.AmJHumGenet.2012;91:343---8.
14.BalasubramanianM,ParkerMJ,DaltonA,GiuntaC,LindertU, PeresLC,etal.Genotype-phenotypestudyintypeV osteoge-nesisimperfecta.ClinDysmorphol.2013;22:93---101.
15.Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355:2757---64.
17.GlorieuxFH,WardLM,RauchF,LalicL,RoughleyPJ,TraversR. OsteogenesisimperfectatypeVI:aformofbrittlebone dise-asewithamineralizationdefect.JBoneMinerRes.2002;17: 30---8.
18.CabralWA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S,etal. Prolyl3-hydroxylase1deficiency causesa recessive metabolicbonedisorderresemblinglethal/severeosteogenesis imperfecta.NatureGenet.2007;39:359---65.
19.vanDijkFS,NesbittIM,ZwikstraEH,NikkelsPG,PiersmaSR, FratantoniSA,etal.PPIBmutationscausesevereosteogenesis imperfecta.AmJHumGenet.2009;85:521---7.
20.ChristiansenHE,SchwarzeU,PyottSM,AlSwaidA,AlBalwiM, AlrasheedS,etal.Homozygosityforamissensemutationin SER-PINH1,whichencodesthecollagenchaperoneproteinHSP47, resultsinsevererecessiveosteogenesisimperfecta.AmJHum Genet.2010;86:389---98.
21.AlanayY,AvayganH,CamachoN,UtineGE,BodurogluK,Aktas D,etal.MutationsinthegeneencodingtheRERproteinFKBP65 causeautosomal-recessiveosteogenesisimperfecta.AmJHum Genet.2010;86:551---9.
22.KelleyBP, Malfait F,BonafeL, Baldridge D,HomanE, Symo-ensS,etal.MutationsinFKBP10causerecessiveosteogenesis imperfectaand Brucksyndrome.JBone MinerRes.2011;26: 666---72.
23.ShaheenR, Al-OwainM,FaqeihE,Al-HashmiN,Awaji A, Al-ZayedZ,etal.MutationsinFKPB10causebothBrucksyndrome and isolated osteogenesisimperfecta in humans. Am J Med Genet.2011;155A:1448---52.
24.LapunzinaP,AglanM,TemtamyS,Caparros-MartinJA,Valencia M,Leton R,et al. Identificationof aframeshift mutationin Osterixinapatientwithrecessiveosteogenesisimperfecta.Am JHumGenet.2010;87:110---4.
25.AsharaniPV,KeuppK,SemlerO,WangW,LiY,ThieleH,etal. AttenuatedBMP1function compromisesosteogenesis,leading tobonefragilityin humansand zebrafish. AmJHum Genet. 2012;90:661---74.
26.Martinez-GlezV,ValenciaM,Caparros-MartinJA,AglanM, Tem-tamyS,TenorioJ,etal.Identificationofamutationcausing deficientBMP1/mTLDproteolyticactivityinautosomal reces-siveosteogenesisimperfecta.HumMutat.2012;33:343---50. 27.ShaheenR, Alazami AM,Alshammari MJ,FaqeihE,Alhashmi
N,MousaN,etal.Studyofautosomalrecessiveosteogenesis imperfectainArabiarevealsanovellocusdefinedbyTMEM38B mutation.JMedGenet.2012;49:630---5.
28.FahiminiyaS,MajewskiJ,MortJ,MoffattP,GlorieuxFH,Rauch F.MutationsinWNT1areacauseofosteogenesisimperfecta. JMedGenet.2013;50:345---8.
29.KeuppK,BeleggiaF,KayseriliH,BarnesAM,SteinerM,SemlerO, etal.MutationsinWNT1causedifferentformsofbonefragility. AmJHumGenet.2013;92:565---74.
30.Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, et al. WNT1 Mutations in families affected by moderately severe and progressive recessive Osteogenesis Imperfecta.AmJHumGenet.2013;92:590---7.
31.Caparrós-Martin JA, Valencia M, Pulido V, Martínez-Glez V, Rueda-ArenasI,AmrK,etal.Clinicalandmolecularanalysisin familieswithautosomalrecessiveosteogenesisimperfect iden-tifiesmutationsinfivegenesandsuggestsgenotype---phenotype correlations.AmJMedGenetPartA.2013;161A:1354---69. 32.vanDijkFS,ZillikensMC,MichaD,RiesslandM,MarcelisCLM,
Die-SmuldersCE,etal.PLS3mutationsinX-linkedosteoporosis withfractures.NEnglJMed.2013;369:1529---36.
33.vanDijkFS,PalsG,VanRijnRR,NikkelsPG,CobbenJM. Classi-ficationofosteogenesisimperfectarevisited.EurJMedGenet. 2010;53:1---5.
34.van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, RohrbachM,etal.EMQNbestpracticeguidelinesforthe labo-ratorydiagnosisofosteogenesisimperfect.EurJHumGenet. 2012;20:11---9.