www.jped.com.br
REVIEW
ARTICLE
What
is
new
in
genetics
and
osteogenesis
imperfecta
classification?
夽
Eugênia
R.
Valadares
a,∗,
Túlio
B.
Carneiro
a,
Paula
M.
Santos
b,
Ana
Cristina
Oliveira
b,
Bernhard
Zabel
caHospitaldasClínicas,FaculdadedeMedicina,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brazil bFaculdadedeOdontologia,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brazil
cPediatricClinic,FreiburgUniversity,Freiburg,Germany
Received17March2014;accepted27May2014 Availableonline18July2014
KEYWORDS
Osteogenesis imperfecta;
Osteochondrodysplasias; Collagentype1
Abstract
Objective: Literaturereviewofnewgenesrelatedtoosteogenesisimperfecta(OI)andupdate
ofitsclassification.
Sources: LiteraturereviewinthePubMedandOMIMdatabases,followedbyselectionofrelevant
references.
Summaryofthefindings: In1979,Sillenceetal.developedaclassificationofOIsubtypesbased
onclinicalfeaturesanddiseaseseverity:OItypeI,mild,common,withbluesclera;OItype II,perinatallethalform;OItypeIII,severeandprogressivelydeforming,withnormalsclera; andOItypeIV,moderateseveritywithnormalsclera.Approximately90%ofindividualswith OIareheterozygousformutationsintheCOL1A1andCOL1A2genes,withdominantpatternof inheritanceorsporadicmutations.After2006,mutationswereidentifiedintheCRTAP,FKBP10,
LEPRE1,PLOD2,PPIB,SERPINF1,SERPINH1,SP7,WNT1,BMP1,andTMEM38Bgenes,associated
withrecessiveOIandmutationintheIFITM5geneassociatedwithdominantOI.MutationsinPLS3
wererecentlyidentifiedinfamilieswithosteoporosisandfractures,withX-linkedinheritance pattern.InadditiontothegeneticcomplexityofthemolecularbasisofOI,extensivephenotypic variabilityresultingfromindividuallocihasalsobeendocumented.
Conclusions: Consideringthediscoveryofnewgenesandlimitedgenotype-phenotype
corre-lation,theuseofnext-generationsequencingtoolshasbecomeusefulinmolecularstudiesof OIcases.The recommendationoftheNosologyGroup oftheInternationalSociety of Skele-talDysplasiasistomaintaintheclassificationofSillenceastheprototypicalform,universally acceptedtoclassifythedegreeofseverityinOI,whilemaintainingitfreefromdirectmolecular reference.
©2014SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
夽 Pleasecitethisarticleas:ValadaresER,CarneiroTB,SantosPM,OliveiraAC,ZabelB.Whatisnewingeneticsandosteogenesisimperfecta classification?JPediatr(RioJ).2014;90:536---41.
∗Correspondingauthor.
E-mail:[email protected],[email protected](E.R.Valadares).
http://dx.doi.org/10.1016/j.jped.2014.05.003
PALAVRAS-CHAVE
Osteogênese imperfeita;
Osteocondrodisplasias; Colágenotipo1
Oquehádenovoemgenéticaeclassificac¸ãodeosteogêneseimperfeita?
Resumo
Objetivo: Revisãodaliteraturasobrenovosgenesrelacionadosàosteogêneseimperfeita(OI)
eatualizac¸ãodasuaclassificac¸ão.
Fontedosdados: RevisãonasbasesdedadosdoPUBMEDeOMIMcomselec¸ãodereferências
relevantes.
Síntesedosdados: Sillenceetal.,em1979,desenvolveramumaclassificac¸ãodossubtiposde
OIbaseadaemcaracterísticasclínicasegravidadedadoenc¸a:OItipoI,formaleve,comum,com esclerasazuladas;OItipoII,formaperinatalletal;OItipoIII,formagraveeprogressivamente deformantecomescleranormal;eOItipoIV,formadegravidademoderadacomescleranormal. Cercade90%dosindivíduoscomOIsãoheterozigotosparamutac¸õesemCOL1A1eCOL1A2,com padrãodeheranc¸adominanteouesporádico.Apartirde2006foramidentificadasmutac¸õesnos genesCRTAP,FKBP10,LEPRE1,PLOD2,PPIB,SERPINF1,SERPINH1,SP7,WNT1,BMP1eTMEM38B
associadasàOIrecessivaemutac¸ãoemIFITM5associadaàOIdominante.Mutac¸õesemPLS3
foram identificadas recentemente em famílias comosteoporose efraturas, compadrão de heranc¸aligadoaoX.AlémdacomplexidadegenéticadasbasesmolecularesdasOI,extensa variabilidadefenotípicaresultantedelociindividuaistambémtemsidodocumentada.
Conclusões: Faceàdescobertadenovosgeneseàcorrelac¸ãogenótipo-fenótipo limitada,o
usodeferramentasdesequenciamentodenovagerac¸ãotorna-seútilnoestudomolecularde casosdeOI.Arecomendac¸ãodoGrupodeNosologiadaSociedadeInternacionaldeDisplasias Esqueléticasémanteraclassificac¸ãodeSillencecomoaformaprototípicaeuniversalmente aceitaparaclassificarograudegravidadenaOI,elibertá-ladereferênciamoleculardireta. ©2014SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduction
Osteogenesis imperfecta (OI) is a group of clinically and genetically heterogeneous diseases characterized by sus-ceptibility to bone fractures, with variable degree of severityand presumedor provendefectsin collagen type I biosynthesis. Other manifestations include dentinogene-sis imperfecta, blue sclerae, and short stature, as well ashearingloss inadulthood.Clinicalmanifestationsrange fromseverecaseswithperinatallethalitytoasymptomatic individuals with mild predisposition to fractures, normal stature,andnormallife.1
Overall, the incidence of the different types of OI is approximately1in15,000-20,000birthsandmostcasesare due toautosomaldominant inheritancewithmutationsin
COL1A1or COL1A2genes,whichencode the␣1(I)and␣2
(I)chainsoftypeIcollagen.1
TypeIcollagen,themainstructuralproteinofthe extra-cellularmatrixofbone, skin,andtendons, consistsoftwo pro-␣-1chainsandonepro-␣-2chainthatinterweave,
form-ingarigidtriplehelix.Each␣chaincontainsN-(amino)and
C-(carboxy)terminalpropeptidesandacentraldomain con-sisting of 338 repeats of Gly-XY, where X and Y exclude cysteineandtryptophan,andwhichoftenare,respectively, prolineandhydroxyproline.Glycine,asthesmallestamino acid,istheonlyresiduethatcanoccupytheaxialposition ofthetriplehelix,sothatanychangeinaglycineresidue willresultinthedisruptionofthehelicalstructure.2,3
Mutations inCOL1A1andCOL1A2genesalterthe struc-tureortheamountoftypeIcollagen,resultinginaskeletal phenotypethatrangesfromsubclinicaltolethal.1
These patients exhibit qualitative and quantitative abnormalitiesintypeIcollagen duetothedominant neg-ative effectof the mutation, asthe mutantpro-␣ chains
areincorporatedintothetypeIprocollagenmoleculesthat also contain normal pro-␣ chains. As a rule, when there
is substitution of glycine in the ␣1 chain, the phenotype
willdependonthepositionofthesubstitution:C-terminal substitutions result in severe disease phenotype, and N-terminalsubstitutionsyieldmilderphenotypes.4,5Residues withlargelateralchainsorchargedresiduesarehighly dis-ruptive of the triple structure, regardless of where they arelocated.Differentphenotypeshavebeenfoundwiththe samemutation.6
Inaconsortiumcreatedin2007tostudyOI-causing muta-tionsintypeIcollagengenes,1,832independentmutations wereidentified;682resultedinthesubstitutionofglycine residuesinthetriplehelixdomainoftheencodedprotein, and150insplicesites.6
Based on clinical, radiographic, and skeletal findings, modeofinheritance,andmoleculargeneticanalyses,new OI types have been identified since 2006 through exome sequencing.Thepresentstudyaimedtoreviewthe classifi-cationofOIandtoupdatenewrelatedgenes.ThePubMed andOnlineMendelianInheritanceinMan(OMIM)databases wereused.7
SillenceClassification
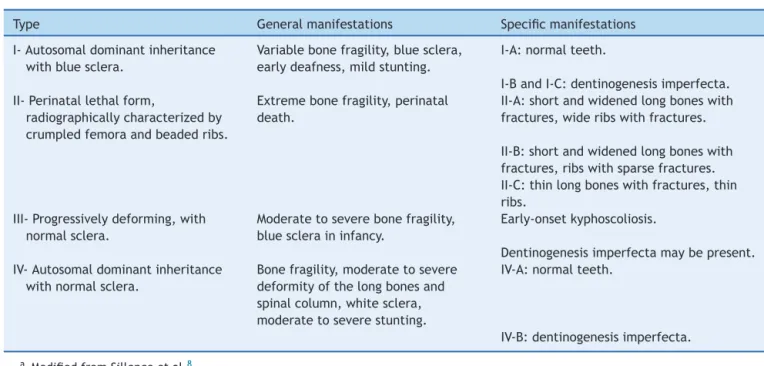
Table1 ClassificationofOI.
Type Generalmanifestations Specificmanifestations
I-Autosomaldominantinheritance withbluesclera.
Variablebonefragility,bluesclera, earlydeafness,mildstunting.
I-A:normalteeth.
I-BandI-C:dentinogenesisimperfecta. II-Perinatallethalform,
radiographicallycharacterizedby crumpledfemoraandbeadedribs.
Extremebonefragility,perinatal death.
II-A:shortandwidenedlongboneswith fractures,wideribswithfractures.
II-B:shortandwidenedlongboneswith fractures,ribswithsparsefractures. II-C:thinlongboneswithfractures,thin ribs.
III-Progressivelydeforming,with normalsclera.
Moderatetoseverebonefragility, bluescleraininfancy.
Early-onsetkyphoscoliosis.
Dentinogenesisimperfectamaybepresent. IV-Autosomaldominantinheritance
withnormalsclera.
Bonefragility,moderatetosevere deformityofthelongbonesand spinalcolumn,whitesclera, moderatetoseverestunting.
IV-A:normalteeth.
IV-B:dentinogenesisimperfecta.
aModifiedfromSillenceetal.8
common,withbluesclera;OItypeII,perinatallethalform; OItypeIII,severeandprogressivelydeforming,withnormal sclera;andOItypeIV,moderateseveritywithnormalsclera. The classification of Sillence hasbeen repeatedly revised whennewcausativegenesforOIareidentified.
ExpandedClassification
ThemoleculargeneticclassificationofOIhasshowntobe veryheterogeneous,withdifferentpatternsofinheritance andwidevariabilityofclinicalseverity.10
Glorieuxetal.11 describedanautosomaldominantform ofOI,similartoOISillencetypeIV,butwithdistinctclinical, histological, and molecular characteristics. No mutations werefound inCOL1A1 andCOL1A2and, therefore,it was called OI typeV (OMIM #610967) by theauthors. Approx-imately 65% of affected individuals develop hyperplastic callusafterfracturesor surgicalinterventions, considered apathognomoniccharacteristic.12Onlyin2012wereIFITM5 mutations identifiedin patients with typeV OI, the gene encodinginterferon-inducedtransmembraneprotein5,by sequencingoftheentire exome.12---14 Theencodedprotein hasaroleinearlymineralization,butitsmechanismremains unknown.10
In 2006, a CRTAP gene mutation was identified asthe firstgeneticcauseoflethalrecessiveOI.15Sincethen,new mutationsingenesthatcauserecessiveOIhavebeen identi-fiedbyexomesequencing,suchasFKBP10,LEPRE1,PLOD2, PPIB,SERPINF1, SERPINH1, SP, BMP1,and TMEM38B. Each of these genes received an OI type number in the OMIM database,following thesequence numbersof theSillence classification.
OItypeVI(OMIM#613982)isanautosomalrecessiveform ofthediseasethatcanbecausedby ahomozygous muta-tioninthegeneSERPINF1inchromosome17p13.3,causing amineralizationdefect.14Accordingtotheclassificationof Sillenceetal.,thephenotypeiscompatiblewithtypeIVor typeIII.16,17
OItypeVII(MIM#610682)isalethalautosomalrecessive formofOI,causedbyamutationinCRTAPgenein homozy-gosityorcompoundheterozygosityinchromosome3p22.It accountsfor2%to3%ofcasesoflethalOI.15
Cabraletal.18 describedaformofautosomalrecessive OI, called OI type VIII (OMIM #610915), which is charac-terized by white sclera, severe growth impairment, very poorskeletalmineralization,andbulbousmetaphyses.This formiscausedbymutationsinthegeneencodingleprecan (LEPRE1)inchromosome1p34.2,associatedwithsevereor lethalOI.
OItypeIX(OMIM#259440)isanautosomalrecessiveform ofOIcorrespondingtoclinicallyseveretypesII/IIIofthe Sil-lenceclassification.19Therearenoreportsofdentinogenesis imperfecta.Itcanbecausedbyahomozygousmutationin thePPIBgeneinchromosome15q22.31.
OI type X (OMIM #613848) is an autosomal recessive form ofthe disease that can becaused by a homozygous mutation in the gene SERPINH1 in chromosome 11q13.5. It ischaracterized bybone deformities andmultiple frac-tures,generalized osteopenia,dentinogenesisimperfecta, andbluesclera.SERPINH1encodesacollagen-binding pro-teinthatactsasachaperoneintheendoplasmicreticulum andthus,individualswithmutationsinthisgenehavecells thatdonotproduceovermodifiedtypeIcollagen.20
OItypeXI(OMIM#610968)isanautosomalrecessiveform of the disease caused by a homozygous mutation in the
FKBP10geneinchromosome17q21,alsorelatedtoa chap-erone defect.1 Patients withtype OI type XI have severe progressivedeformation andmayhave jointcontractures. Patientsdonothavedentinogenesisimperfecta.21---23
OI type XII (OMIM #613849) is an autosomal reces-sive form, which can be caused by mutation in the SP7
Table2 ExpandedclassificationofOI.a
TypeofOI Inheritance Phenotype Geneticdefect
ClassicalSillenceTypes
I AD Mild COL1A1
X-linked Mild PLS3
II AD Letal COL1A1orCOL1A2
III AD Progressivedeformity COL1A1orCOL1A2
IV AD Moderate COL1A1orCOL1A2
V AD Moderate,hypertrophiccallusand ossificationoftheinterosseousmembrane
IFITM5
VI AR Moderatetosevere SERPINF1
VII AR Severetoletal CRTAP
VIII AR Severetoletal LEPRE1
IX AR Severetoletal PPIB
X AR Severe SERPINH1
XI AR Progressivedeformity,contractures FKBP10
XII AR Moderate SP7
XIII AR Severe BMP1
XIV AR Variableseverity TMEM38B
XV AR
AD
Variableseverity Early-onsetosteoporosis
WNT1
AD,autosomaldominant;AR,autosomalrecessive.
a AdaptedfromForlinoetal.1
OItypeXIII(OMIM#614856)wasdescribedascausedby a homozygousmutationin the geneBMP1 in chromosome 8p21.25,26
Shaheenetal.27describedOItypeXIV(OMIM#615066), an autosomal recessive form characterized by varying degreesofseveritywithmultiplefracturesandosteopenia, withnormaldentition,sclera,andhearing.Fracturesoccur prenatallyoratapproximately6yearsofage.Itiscausedby ahomozygousmutationinthegeneTMEM38Binchromosome 9q31.
OI typeXV (OMIM#615220) has been designated based on the identification of mutations in WNT1.28---30 Keupp et al.30 reported that WNT1 hypofunctional alleles result in phenotypes withlow bone mass in humans. They veri-fiedthatmutationsintherecessiveinheritedgeneleadto phenotypesofvaryingseverity,rangingfrommildto progres-sivelydeforming,whichcanoccasionallyleadtoearlyinfant death.Theyalsodetectedfamiliesthathadearly osteoporo-siswiththeautosomaldominantpatternofinheritance,with aheterozygousmutationinWNT1.
TherecessiveformsofOIwithmoderatetolethal phen-otypes are caused by defects in genes whose products interactwithcollagentypeI.Mostrecessivecaseshavenull mutationsin genesencodingproteins involvedin prolyl 3-hydroxylationofcollagen (CRTAP, LEPRE1,andPPIB),orin those responsible for the correct helical folding (FKBP10
andSERPINH1).TypesVII,VIII,andIXarecausedbydefects in3-hydroxylation.1Thegenotype-phenotypecorrelationin recessiveformshasbeensuggested.31
In2013,PLS3mutationswereidentifiedinfamilieswith osteoporosisandfracturesmanifestinginchildhood,withan X-linkedpatternofinheritance.32
Table 2 summarizes the classification based on the involvedgenes.
In2010,vanDijketal.33proposedarevisedclassification ofOI,mentioning thecausativegeneandthe correspond-ingclinicalpictureonlyfortypesItoVI.TypesVIIandVIII wereexcluded,asthosetypeswereaddedbygenetic crite-ria, although their clinical and radiological findings were indistinguishablefromthoseintypesIItoIV.Theproposed classificationleavesroomfor newgenesdiscoveredasthe causeofOIuntilthefullextentofheterogeneityisknown.34
OIClassificationbytheInternationalSocietyof
SkeletalDysplasias
Table3 OIclassificationaccordingtotheInternationalSocietyofSkeletalDysplasias withadditionofnewlydiscoveredgenes.
OsteogenesisImperfecta Inheritance Genes
Nondeformingosteogenesisimperfecta(typeI) AD X-linked
COL1A1,COL1A2
PLS3
Perinatallethal(typeII) AD,AR COL1A1,COL1A2,CRTAP,LEPRE1,PPIB,BMP1
Progressivelydeforming(typeIII) AD,AR COL1A1,COL1A2,CRTAP,LEPRE1,PPIB,
FKBP10,SERPINH1,SERPINF1,WNT1
Moderate(typeIV) AD,AR COL1A1,COL1A2,CRTAP,FKBP10,SP7,
SERPINF1,WNT1,TMEM38B
Withcalcificationoftheinterosseousmembrane and/orhypertrophiccallus(typeV)
AD IFITM5
AD,autosomaldominant;AR,autosomalrecessive.
aWarmanetal.35
Conclusion
In practice, in spite of the complex genotypicvariability of OI demonstrated in recent years, its phenotypes are still classified according to Sillence. Genotypic investiga-tion should be indicated, especially in cases suggesting autosomalrecessiveinheritance,aimedatgenetic counsel-ing.ThemolecularstudyshouldbeperformedusingSanger sequencingoftheseveralnewgenes,orbynext-generation sequencing.Exome sequencingis useful whenthere is no panelof available genes, or when the involved genes are notknown.
Funding
CNPq(Conselho Nacionalde Desenvolvimento Científico e Tecnológico).
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgements
Tothe ConselhoNacionaldeDesenvolvimento Científico e Tecnológico (CNPq) for the post-doctoral grant given to Eugênia Ribeiro Valadares in 2013 in the Genetics Sector ofthe PediatricClinicofFreiburg University,Germany, to developthe project‘‘Investigationof osteogenesis imper-fectathroughtheanalysisofknowngenesandnew candi-date genesin Brazilian and German patients’’, underthe supervisionofProf.Dr.BernhardZabel,Dr.Pablo Villavicen-cioLorini,andDr.EkkehartLausch,remarkableindividuals.
References
1.ForlinoA,CabralWA,BarnesAM,MariniJC.Newperspectiveson osteogenesisimperfecta.NatRevEndocrinol.2011;7:540---57. 2.ColeWG.Themolecularpathologyofosteogenesisimperfecta.
ClinOrthopRelatRes.1997;343:235---48.
3.ByersPH,WallisGA,WillingMC.Osteogenesisimperfecta: trans-lationofmutationtophenotype.JMedGenet.1991;28:433---42. 4.StaceyA,BatemanJF,ChoiT.Perinatallethalintransgenicmice bearing anengineeredmutantpro-[alpha]1(I)collagengene. Nature.1988;332:131---6.
5.ColeWG,DalgleisR.Perinatallethalosteogenesis.JMedGenet. 1995;32:284---9.
6.MariniJC,ForlinoA, CabralWA,BarnesAM,SanAntonioJD, MilgromS,etal.Consortiumforosteogenesisimperfecta muta-tionsinthehelicaldomainoftypeI collagen:regions richin lethalmutationsalignwithcollagenbindingsitesforintegrins andproteoglycans.HumMutat.2007;28:209---21.
7.Online Mendelian Inheritance in Man, OMIM®.
McKusick-NathansInstituteofGeneticMedicine,JohnsHopkinsUniversity (Baltimore, MD), [cited 14 Mar 2014]. Available from: http://omim.org/
8.SillenceDO,SennA,DanksDM.Geneticheterogeneityin osteo-genesisimperfecta.JMedGenet.1979;16:101---16.
9.SillenceDO.Osteogenesisimperfect:anexpandingpanorama ofvariants.ClinOrtop.1981;159:11---25.
10.MariniJC,BlissetAR.Newgenesinbonedevelopment:what’s new in osteogenesis imperfecta. J Clin Endocrinol Metab. 2013;98:3095---103.
11.GlorieuxFH,RauchF,PlotkinH,WardL, TraversR,Roughley P,etal.TypeVosteogenesisimperfecta:anewformofbrittle bonedisease.JBoneMinerRes.2000;15:1650---8.
12.SemlerO,GarbesL,KeuppK,SwanD,ZimmermannK,Becker J, et al. A mutationin the5′-UTR ofIFITM5 creates an in-framestartcodonandcausesautosomal-dominantosteogenesis imperfectatypeVwithhyperplasticcallus.AmJHumGenet. 2012;91:349---57.
13.ChoTJ,LeeKE,LeeSK,SongSJ,KimKJ,JeonD,etal.Asingle recurrentmutationinthe5’-UTRofIFITM5causesosteogenesis imperfectatypeV.AmJHumGenet.2012;91:343---8.
14.BalasubramanianM,ParkerMJ,DaltonA,GiuntaC,LindertU, PeresLC,etal.Genotype-phenotypestudyintypeV osteogen-esisimperfecta.ClinDysmorphol.2013;22:93---101.
15.Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR,etal.Deficiencyofcartilage-associatedproteinin reces-sivelethalosteogenesisimperfecta.NEnglJMed.2006;355: 2757---64.
16.BeckerJ,SemlerO,GilissenC,LiY,BolzHJ,GiuntaC,etal. AmJHumGenet.2011;88:362---71.
17.GlorieuxFH,WardLM,RauchF,LalicL,RoughleyPJ,TraversR. OsteogenesisimperfectatypeVI:aformofbrittlebonedisease withamineralizationdefect.JBoneMinerRes.2002;17:30---8. 18.Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S,etal. Prolyl3-hydroxylase1 deficiencycausesa recessive metabolicbonedisorderresemblinglethal/severeosteogenesis imperfecta.NatureGenet.2007;39:359---65.
19.vanDijkFS,NesbittIM,ZwikstraEH,NikkelsPG,PiersmaSR, FratantoniSA,etal.PPIBmutationscausesevereosteogenesis imperfecta.AmJHumGenet.2009;85:521---7.
resultsinsevererecessiveosteogenesisimperfecta.AmJHum Genet.2010;86:389---98.
21.AlanayY,AvayganH,CamachoN,UtineGE,BodurogluK,Aktas D,etal.MutationsinthegeneencodingtheRERproteinFKBP65 causeautosomal-recessiveosteogenesisimperfecta.AmJHum Genet.2010;86:551---9.
22.KelleyBP,MalfaitF,BonafeL,BaldridgeD,HomanE,SymoensS, etal.MutationsinFKBP10causerecessiveosteogenesis imper-fectaandBrucksyndrome.JBoneMinerRes.2011;26:666---72. 23.ShaheenR, Al-OwainM,FaqeihE,Al-HashmiN,Awaji A,
Al-ZayedZ,etal.MutationsinFKPB10causebothBrucksyndrome and isolated osteogenesisimperfecta in humans. Am J Med Genet.2011;155A:1448---52.
24.LapunzinaP,AglanM,TemtamyS,Caparros-MartinJA,Valencia M,Leton R,et al. Identificationof aframeshift mutationin Osterixinapatientwithrecessiveosteogenesisimperfecta.Am JHumGenet.2010;87:110---4.
25.AsharaniPV,KeuppK,SemlerO,WangW,LiY,ThieleH,etal. AttenuatedBMP1function compromisesosteogenesis,leading tobonefragilityin humansand zebrafish. AmJHum Genet. 2012;90:661---74.
26.Martinez-GlezV,ValenciaM,Caparros-MartinJA,AglanM, Tem-tamyS,TenorioJ,et al.Identificationofamutationcausing deficientBMP1/mTLDproteolyticactivityinautosomal reces-siveosteogenesisimperfecta.HumMutat.2012;33:343---50. 27.ShaheenR,Alazami AM,Alshammari MJ,FaqeihE, Alhashmi
N,MousaN,etal.Studyofautosomalrecessiveosteogenesis imperfectainArabiarevealsanovellocusdefinedbyTMEM38B mutation.JMedGenet.2012;49:630---5.
28.FahiminiyaS,MajewskiJ,MortJ,MoffattP,GlorieuxFH,Rauch F.MutationsinWNT1areacauseofosteogenesisimperfecta.J MedGenet.2013;50:345---8.
29.Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, TemmeRT,etal.WNT1Mutationsinfamiliesaffectedby moder-atelysevereandprogressiverecessiveOsteogenesisImperfecta. AmJHumGenet.2013;92:590---7.
30.KeuppK,BeleggiaF,KayseriliH,BarnesAM,SteinerM,SemlerO, etal.MutationsinWNT1causedifferentformsofbonefragility. AmJHumGenet.2013;92:565---74.
31.Caparrós-Martin JA, Valencia M, Pulido V, Martínez-Glez V, Rueda-ArenasI,AmrK,etal.Clinicalandmolecularanalysisin familieswithautosomalrecessiveosteogenesisimperfect iden-tifiesmutationsinfivegenesandsuggestsgenotype---phenotype correlations.AmJMedGenetPartA.2013;161A:1354---69. 32.vanDijkFS,ZillikensMC,MichaD,RiesslandM,MarcelisCLM,
Die-SmuldersCE,etal.PLS3mutationsinX-linkedosteoporosis withfractures.NEnglJMed.2013;369:1529---36.
33.vanDijkFS,PalsG,VanRijnRR,NikkelsPG,CobbenJM. Classi-ficationofosteogenesisimperfectarevisited.EurJMedGenet. 2010;53:1---5.
34.van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, RohrbachM,etal.EMQNbestpracticeguidelinesforthe lab-oratorydiagnosisofosteogenesisimperfect.EurJHumGenet. 2012;20:11---9.