Dissertação de candidatura ao grau de Mestre - Versão Definitiva -
Avaliação da Expressão Génica num Modelo de Neoplasia
Experimental da Mama
Mestrado em Biologia Clínica Laboratorial
Lúcia Maria Castanheira Gonçalves
Os orientadores:
Fernanda de Seixas Travassos Laboratório de Histologia e Anatomia Patológica-ECAV-UTAD António Silvério Cabrita Serviço de Patologia Experimental-FMUC
As doutrinas expostas neste trabalho são da exclusiva responsabilidade do autor
Ao finalizar este trabalho, gostaria de agradecer e reconhecer o apoio solicitado por todos aqueles que de forma directa ou indirecta contribuíram para a realização do mesmo.
Começo por exprimir o meu agradecimento à Universidade de Trás-os-Montes e Alto Douro, na pessoa do seu Magnifico Reitor, Senhor Professor Doutor Carlos Alberto Sequeira, pelas condições de trabalho que me tem proporcionado desde o inicio da minha admissão nesta Universidade.
À coordenação do Mestrado em Biologia Clínica Laboratorial, pelas suas orientações e pela aceitação do meu plano de dissertação de mestrado.
Ao Professor Doutor António Silvério Cabrita, expresso a minha gratidão por ter aceitado ser meu orientador e por ter proporcionado a realização deste trabalho experimental no Serviço de Patologia Experimental da Faculdade de Medicina da Universidade de Coimbra. Gostaria de agradecer igualmente o seu apoio, disponibilidade, transmissão de conhecimentos aquando da realização deste trabalho, bem como a sua capacidade científica demonstrada desde que o conheço. Foi um grande privilégio para mim ter realizado este estudo com o Senhor Professor. Agradeço ainda a sua disponibilidade na correcção desta dissertação.
Á Professora Doutora Fernanda Seixas Travassos, manifesto o meu agradecimento por ter aceite ser minha co-orientadora, pelo incentivo, pela colaboração e disponibilidade demonstradas. Agradeço ainda a disponibilidade de bibliografia e ainda as correcções da dissertação por si efectuadas.
Ao Laboratório de Bioquímica Genética, CNC, na pessoa da Professora Doutora Maria Manuela Grazina, expresso a minha gratidão por toda a colaboração prestada durante a elaboração deste trabalho. A sua disponibilidade e os seus conselhos foram essenciais para a obtenção deste trabalho. Agradeço também por me terem recebido na suas instalações.
A todo o pessoal do Laboratório de Bioquímica Genética, CNC, pelo carinho e atenção que me demonstraram durante a realização do meu trabalho. Um Obrigada
Agradeço a todo o pessoal do Serviço de Patologia Experimental da FMUC, que contribuíram para a realização deste trabalho experimental.
A todos os docentes do Mestrado em Biologia Clínica Laboratorial, agradeço todos os ensinamentos transmitidos que tanto contribuíram para a minha formação académica, particularmente, à professora Maria dos Anjos Pires, queria dar uma palavra especial, pela amabilidade que mostrou ao disponibilizar-se para me auxiliar na concretização desta dissertação, muito obrigada!
A todos os meus colegas do Mestrado em Biologia Clínica Laboratorial agradeço os agradáveis momentos que passámos juntos.
Aos meus fiéis amigos, agradeço tudo o que fizeram por mim desde que entrei nesta Universidade, ajudando-me incansavelmente nos bons e maus momentos da minha vida. Dou graças a todos vós por ser o que sou hoje, e disso nunca me esquecerei!
Á minha família, em especial a minha mãe, um muito obrigado pelo constante apoio e carinho revelado ao longo do tempo. Sei que sem vós, nunca seria capaz de atingir os meus objectivos, a todos os níveis, ficando em divida para convosco durante toda a minha vida.
Trabalhos Apresentados
Apresentação Oral:
“Transcriptómica num Modelo de Neoplasia Experimental da Mama”. II Congresso Luso-Brasileiro de Experimentação Animal. Faculdade de Medicina da Universidade de Coimbra. 11 de Dezembro de 2012. Coimbra
A proliferação incontrolada das células, que representa a essência da doença neoplásica, envolve não só a desregulação da proliferação mas também o ajustamento metabólico de modo a sustentar o crescimento e divisão celular. O ajustamento do metabolismo em células neoplásicas foi observado pela primeira vez por Warburg e desde então vários têm sido os estudos que envolvem a avaliação da expressão genes envolvidos no metabolismo.
Este estudo experimental foi executado para estudar a variação da expressão génica de três genes: Citrato sintase (ciclo de Krebs), Acetil-CoA Carboxilase α (sintese de ácidos gordos) e Glucose-6-fosfatase (gliconeogénese e Glicogenólise) em quatro grupos histológicos de lesões em relação a tecido mamário normal.
Foram recolhidas amostras de glândulas mamárias de 10 fêmeas de ratos Sprague-Dawley previamente induzidas com 7,12 - Dimetilbenzantraceno (DMBA) numa concentração de 65 mg/Kg de animal. Foi retirada uma secção das amostra para histologia clássica. Os níveis de expressão foram avaliados comparando os níveis de expressão dos genes-alvo com o gene actina β (controlo endógeno). Os dados obtidos foram tratados estatisticamente pelo teste ANOVA.
A expressão do gene Citrato síntase é diminuída ao longo da progressão da doença neoplásica, permitindo um distinção significativa entre as neoplasias benignas e malignas uma vez que a expressão é bastante inferior nestas últimas. A expressão do gene Acetil coA carboxilase α é ligeiramente aumentada nas neoplasias benignas e significativamente inferior nas neoplasias malignas. A expressão do gene Glucose-6-fosfatase apresenta amplos valores de expressão desde valores próximos de zero até valores várias vezes superiores ao normal.
Desde modo, existe uma diferença significativa entre os tumores benignos e malignos para os genes Citrato sintase e Actetil CoA carboxilase α, mas nenhum dos genes apresenta diferenças de expressão entre neoplasias malignas não-invasoras das não-invasoras. A expressão do gene Glucose-6-fosfatase não permite qualquer distinção entre os grupos histológicos.
The uncontrolled proliferation of cells, which represent the essence of neoplastic disease, involves not only dysregulation of proliferative cells but also the metabolic adjustment to sustain growth and cell division. The adjustment of cellular metabolism in cancer cells was first observed by Warburg and since then has been several studies that involves the study of the expression of several genes involved in metabolism, in order to understand the fundamentals of this amendment.
This experimental study was performed to investigate the variation of gene expression of three genes: citrate synthase (Krebs cycle), acetyl-CoA carboxylase α (fatty acid synthesis), and glucose-6-phosphatase (gluconeogenesis and glycogenolysis) in four groups of histological lesions relative to normal breast tissue.
Samples were taken from mammary glands of 10 female Sprague-Dawley previously induced with 7,12-Dimethylbenzanthracene (DMBA) at a concentration of 65mg/Kg of animal. Section was taken from sample for histology. Levels of expression were assessed by comparing expression levels of target genes with β actin gene (endogenous control). The data were processed by ANOVA statistic program.
Citrate synthase gene expression is decreased along the progression of the neoplastic disease, allowing a significant distinction between benign and malignant neoplasms since the expression is far below the latter. The gene expression of acetyl CoA carboxylase α is slightly increased in benign and significantly lower in malignant neoplasms. The gene expression Glucose-6-phosphatase has wide expression values from values approaching zero values to several times the normal.
In this way, there is a significant difference between benign and malignant tumors for the genes citrate synthase and acetyl CoA carboxylase α , but none of the genes shows differences in expression between malignant non-invasive tumors. The gene expression of glucose-6-phosphatase allows no distinction between the histologic groups.
Acaca – acetil-coA carboxilase alfa ATP – adenosina trifosfato
Bcl 2 – B-cell lynphoma 2 – linfoma das células B ºC – graus Celcius
CA IX – anidrase carbónica IX
cDNA - complementary DesoxiriboNucleic Acid – ácido desoxiribonucleico complementar
cm3 – centímetro cúbico
CNC – Centro de Neurociências e Biologia Molecular de Coimbra Cs – Citrato sintase
CT – threshold cycle
DEPC – dietil-pirocarbonato
DMBA – 7, 12 - dimetilbenzantraceno
DNA – Desoxirribonucleic Acid – ácido desoxirribonucleico EU – European Union – União Europeia
18F-FDG – Fluorodeoxyglucose 18F – Fluorodesoxiglucose 18F FGF – fibroblastic growth factor – fator de crescimento fibroblástico FMUC – Faculdade de Medicina de Coimbra
g – grama
G6pc – Glucose-6-fosfatase sub-unidade catalítica Glut 1- transportador da glucose 1
H&E – hematoxilina e eosina
HER2 - Human Epidermal Receptor 2 – receptor do factor de crescimento epidérmico humano 2
HIF – Hipoxia Inductible Factor – Factor induzido por hipóxia
Kg – quilograma
LBG – Laboratório de Bioquímica Genética µ L – microlitro
MCT – transportadores de monocarboxilatos
MDSC – myeloid-derived suppressor cells – células supressoras derivadas da linha mieloide
µg – micrograma mg – miligrama mL – mililitro min – minuto
NAD - Nicotinamide adenine dinucleotide – dinucleótido adenina nicotinamida NCBI – National center for Biotechnology Information
NK – “natural killer” NMU – N-nitrometilureia
PCR – polymerase chain reaction - reação em cadeia da polimerase RB – retinoblastoma
RE – receptor de estrogénio
RIN – RNA integrity number – número de integridade do RNA RNA – Ribonucleic Acid – ácido ribonucleico
rRNA – ácido ribonucleico ribosomal
RT-PCR – reverse transcriptase – polymerase chain reaction – reação em cadeia da polimerase reversa
s – velocidade de sedimentação
TGF-β – transforming growth factor – fator de transformação do crescimento TSP – trombospondin – trombospondina
Capitulo I – Revisão Bibliográfica 1
1. A Neoplasia da Mama 3
1.1. A visão Global 3
1.2. Da classificação histológica à molecular 3
1.3. O desenvolvimento da glândula mamária e carcinogénese 4 1.4. O microambiente durante a progressão da neoplasia 6 1.5. A carcinogénese induzida e a importância dos modelos
animais no estudo da neoplasia mamária 7
2. As capacidades biológicas adquiridas durante a carcinogénese 8
2.1. “Halmarks” das neoplasias 10
2.1.1. Sustentação da sinalização proliferativa 10 2.1.2. Evasão dos supressores de crescimento 10
2.1.3. Resistência à morte celular 11
2.1.4. Permissão da imortalidade replicativa 13
2.1.5. Indução da angiogénese 14
2.1.6. Ativação da invasão e metástases 15
2.1.7. Evasão ao sistema imune: um marcador emergente 16 2.2. A reprogramação do metabolismo: um marcador emergente 18
2.2.1. Visão global do metabolismo 18
2.2.2. O metabolismo especial das células neoplásicas 19
Capitulo II – Objectivos 22
Capitulo III – Material e Métodos 24
3.1. Indução tumoral 25
3.2. Colheita e preservação das amostras 26
3.3. Classificação histológica 26
3.4. Análise de transcritos 27
3.4.1. Tratamento do material 27
3.4.2. Extracção do RNA 27
3.4.6. Avaliação da expressão relativa dos genes-alvo 29
3.4.7. Análise Estatística 30
Capitulo IV – Resultados 31
4.1. Classificação histológica 32
4.2. Avaliação da integridade e concentração do RNA 35 4.3. Quantificação relativa da expressão relativa dos genes-alvo 37
Capitulo V – Discussão dos Resultados 40
Capitulo VI – Considerações finais 46
Índice de Figuras
Figura 1.1.1 – Possível modelo explicativo da origem dos subtipos
moleculares das neoplasias da mama 3 Figura 1.4.1 – Alterações morfológicas durante a carcinogénese 7
Figura 2.1 – Capacidades biológicas adquiridas durante o
desenvolvimento de neoplasias 9 Figura 3.4.3.1 – Imagem do chip “Agilent RNA 6000 Nanokit®” 28
Figura 4.1.1 – Ilustração dos tipos histológicos das amostras recolhidas
coradas pelo método Hematoxilina-Eosina 32 Figura 4.2.1 - Exemplo de três electroforamas obtidos com chip
“Agilent RNA 6000 Nano Kit®” 35
Figura 4.3.1- Ilustração gráfica da expressão relativa do gene Cs nos
quatro grupos histológicos em relação ao grupo controlo 37 Figura 4.3.2 - Ilustração gráfica da expressão relativa do gene Acaca nos
quatro grupos histológicos em relação ao grupo controlo 38 Figura 4.3.3 - Ilustração gráfica da expressão relativa do gene G6pc nos
Tabela 4.1.1 – Registo e Classificação histológica das amostras 34 Tabela 4.2.1 – Valores de concentração e integridade do RNA obtidos
1- A Neoplasia Mamária 1.1 A visão global
O cancro da mama é um dos tumores malignos mais prevalentes na mulher, com importantes implicações sociais e psicológicas, e incidência crescente sobretudo nos países desenvolvidos. Em Portugal continua a ser a primeira causa de morte por doença oncológica no sexo feminino com uma taxa de incidência de 27,7% e uma taxa de mortalidade de 15,9% (GLOBOCAN, 2008).
Durante o desenvolvimento ou regeneração de um órgão, as stem cells e as células progenitoras normais encontram-se em constante proliferação; porém, quando os órgãos se encontram formados ou regenerado, elas suspendem a sua proliferação, parecendo estar sujeitas a um controlo inibitório por mecanismos de retrocontrolo exercidos pelas células diferenciadas. Muitos autores acreditam que as lesões neoplásicas têm origem em células com propriedades estaminais (stem cells ou células progenitoras). Assim, podemos colocar a hipótese de a proliferação de certas células tumorais ser inibida pelo tecido diferenciado circundante, tal como acontece com as células normais em desenvolvimento ou em regeneração. Um estudo publicado vem corroborar a existência deste efeito inibitório das células normais sobre as tumorais através de influências parácrinas (Toillon et al, 2007). Hipoteticamente, uma mutação numa célula progenitora pode torná-la insensível a alguns dos mecanismos de inibição, começando esta célula transformada (célula estaminal cancerosa) a dividir-se de forma excessiva e autónoma simétrica ou assimétrica, dando origem a células idênticas à inicial ou mais diferenciadas, respectivamente (Martins et al, 2009).
A existência de diferentes tipos moleculares de tumores pode-se dever aos diferentes níveis na hierarquia de células progenitoras onde as mutações podem ocorrer. As alterações nas células mais primitivas e indiferenciadas poderiam estar na origem dos tumores mais indiferenciados e agressivos; do mesmo modo, as células progenitoras, oriundas das primeiras, poderiam dar origem a tumores mais diferenciados e de melhor prognóstico (Figura 1.1.1).
1.2 Da classificação histológica à molecular
A atual classificação dos carcinomas da mama pela Organização Mundial de Saúde é histológica e representa uma extensa lista, da qual os tipos histológicos mais frequentes são o carcinoma ductal invasor (50 a 75% dos casos) e o carcinoma lobular invasor (5-15%), o carcinoma ductal in situ e o carcinoma lobular in situ. Os restantes, são menos frequentes e incluem o carcinoma tubular, mucinoso, medular e metaplásico, entre outros. Os parâmetros obtidos pelo exame patológico tradicional, como tamanho, tipo e grau histológico do tumor, invasão vascular, envolvimento de gânglios linfáticos, índice de proliferação celular e expressão de recetores hormonais, são considerados fatores de prognóstico clássicos no cancro da mama. Mas, em geral, não são suficientes para prever o comportamento biológico dos carcinomas, o que motivou a procura por uma nova classificação que refletisse mais adequadamente o prognóstico dos carcinomas invasores da mama e suas possibilidades terapêuticas (Robbins e Cotran, 2005).
A utilização da técnica de microarray cDNA (complementary DesoxiriboNucleic Acid – ácido desoxiribonucleico complementar) permitiu correlacionar a diversidade fenotípica dos carcinomas da mama com a expressão génica e assim classificá-los em três grupos principais: luminal, um sub-tipo que sobre-expressa o HER2 (Human Epidermal Receptor 2 – receptor do factor de crescimento epidérmico humano 2), e um terceiro denominado de tipo basal (Perou et al, 2000;
Figura 1.1.1 - Possível modelo explicativo da origem dos subtipos moleculares de neoplasias da mama (Martins et al, 2009)
Sorlie et al, 2001; Sorlie et al, 2003). O primeiro grupo apresenta características de células luminais, ao expressar citoqueratinas 8/18 e 19 e fatores de transcrição que incluem o recetor de estrogénio. Este sub-tipo representa cerca de 70% dos cancros da mama invasores e está associado a um prognóstico mais favorável e a melhor resposta à terapêutica endócrina com tamoxifeno e inibidores da aromatase (Paredes et al, 2006). O sub-tipo que sobrexpressa o recetor tirosina-cinase HER2 (15 a 30%) apresenta pior prognóstico relativamente aos luminais, mas é elegível para o tratamento com Trastuzumab, um anticorpo monoclonal humanizado dirigido contra o domínio extracelular deste receptor. O terceiro grupo, com diferenciação basal, caracteriza-se pela ausência de receptores hormonais (receptor de estrogénio e receptor de progesterona), e de HER2, pela expressão de marcadores característicos das células basais/mioepiteliais. Os carcinomas de tipo basal, que representam cerca de 2 a 18% dos carcinomas invasivos da mama, apresentam elevada agressividade e menor taxa de resposta às terapias tradicionais, sem alvo molecular específico (Fulford et al, 2006), representando por isso uma área de interesse cada vez maior, pois a caracterização do seu perfil molecular poderá conduzir à aplicação de terapias dirigidas. Apesar desta classificação ter por base a assinatura molecular, a validação, reprodutibilidade, bem como o valor preditivo desta nova taxonomia molecular, necessita de confirmação pela análise de séries de carcinomas da mama para que possa fazer parte da prática clínica. Matos et al (2005) e Livasy et al (2006) demonstraram que é possível reconhecer os sub-tipos de cancros da mama através do fenótipo imunohistoquímico apenas com a utilização de dois anticorpos – contra RE e HER2 – que definem os sub-tipos principais: luminal (RE+/HER2-/+), o que sobreexpressa o HER2 /HER2+) e o basal (RE-/HER2-). A utilização de marcadores basais/mioepiteliais permite uma caracterização mais completa do fenótipo basal (Matos et al, 2005; Livasy et al, 2006, Schnitt, 2010).
1.3 – O desenvolvimento da glândula mamária e carcinogénese
A glândula mamária tem características anatómicas, funcionais e patológicas que a tornam num órgão único para o estudo da carcinogénese.
Primariamente, o parênquima mamário sofre grande parte do seu crescimento após a puberdade, permitindo a realização de estudos experimentais do desenvolvi-mento, realizados em idade juvenil ou adulta, e apresentando oportunidades para manipulação não disponíveis em outros órgãos. Com base nesta característica foram
elaboradas metodologias de transplantação de tecidos, tumores e células tumorais para o tecido adiposo mamário. Em segundo lugar, a acessibilidade da glândula e a organização dos ductos permite uma fácil visualização das alterações existentes (aparecimento de tumores), uma fácil colheita e manipulação cirúrgica do órgão e uma administração de moléculas específicas às células do parênquima mamário, as quais estão base da tumorigénese. Em terceiro lugar, a mama é um alvo de diferentes carcinogénios, químicos, virais ou físicos, o que permite o desenvolvimento de modelos únicos e complexos de progressão neoplásica. Em quarto lugar, a maioria dos modelos animais apresenta vários pares de glândulas mamárias o que permite manipulações nas glândulas de uma cadeia, servindo a contralateral, muitas vezes, de controlo. Finalmente, a complexidade da regulação hormonal e de factores de crescimento funciona como uma abordagem sofisticada para o estudo das acções hormonais (Medina, 1996).
Os passos iniciais da tumorigénese resultam na transformação de epitélio não neoplásico num epitélio hiperplásico que resulta numa proliferação e estratificação celular, tornando-se num crescimento em multicamadas. Qualquer que seja o mecanismo, estes passos envolvem uma desregulação das interacções célula-célula e célula-matriz que inibem o crescimento e que mantêm o epitélio em monocamada (Gateby et al, 2007). Apesar da importância destes passos, é assumido que mecanismos moleculares são de extrema importância, como o stress genotóxico, pois os tumores podem ser induzidos por produtos químicos que formam ductos com o DNA (Melendez-Colon et al, 1999). De igual forma, danos causados por radiações ou inflamação causada por espécies reactivas de oxigénio combinados com um sistema de reparação de DNA deficiente, podem também dar inicio à carcinógenese. Os eventos de iniciação da carcinogénese envolvem a activação por mutação de proto-oncogenes em oncogenes. Os Proto-oncogenes são genes constituintes das células envolvidos na regulação do ciclo celular, proliferação e diferenciação, que quando activados podem levar a um crescimento desregulado; podem constituir um marcador tumoral, podendo ser activados através de carcinógeneos químicos ou através de RNA viral, entre outros eventos genotóxicos.
A activação crónica da vias pró-proliferativas podem ser contrariadas pela acção de genes supressores tumorais, incluindo genes que intervêm nos checkpoints (Hartwell e Weinert, 1989; Vazini et al, 1999). Consequentemente, a carcinogénese pode estar
associada à perda de função dos genes supressores tumorais. Normalmente, os humanos que são heterozigóticos para os genes tumorais tem um aumento da susceptibilidade para tumores após a perda da heterozigocidade. Esta perda de heterozigocidade hereditária pode ocorrer através de mutações genéticas, de perdas cromossómicas ou através de mecanismos epigenéticos como a metilação de DNA (Pane et al, 2005).
1.4 - O microambiente durante a progressão da neoplasia
O epitélio normal é constituído por uma única camada de células epiteliais que crescem na superfície da membrana basal, cercado pelo estroma; estas células estão adjacentes aos vasos sanguíneos no outro lado da membrana basal e também são perfundidas por factores de crescimento e nutrientes. Portanto, a proliferação de células normais do epitélio no lúmem deve ser constrangida através de interacções célula-célula e célula-matriz. Se as células epiteliais morrerem, descamam para o lúmem; a perda de adesão intercelular induz a célula vizinha a dividir-se para preencher o espaço. As células proliferativas requerem a anexação da membrana basal para proliferar e sobreviver, se esta anexação for anulada a célula sofrerá apotose (Frisch e Ruoslahti, 1997). Assim a carcinogénese envolve mecanismos que suprimam este fenómeno, incluindo a aquisição da capacidade de proliferar mesmo quando não estão em contacto com a membrana basal assim como a perda da inibição por contacto célula-célula.
A transformação num tumor invasivo envolve a quebra da membrana basal, dando às células epiteliais acesso directo ao sistema vascular e ao estroma, podendo dispersar-se através do sistema vascular sanguíneo e linfático. A migração destas células através dos vasos linfáticos resulta na colonização dos gânglios linfáticos num padrão característicos. Deste modo, em tumores mamários a metastização dos gânglios linfáticos axilares é o primeiro sinal de disseminação e por isso são conhecidos como gânglios sentinela. Quer directa ou indirectamente através vias linfáticas, as células tumorais embólicas atingem o sangue. Como foi estimado, um tumor com cerca de 1cm3 liberta no sangue mais de 106 células por dia (Bockhom et al, 2007).
A hipótese da invasão mediada pelo microambiente ácido propõe que os tumores invasores, exportam H+ derivados da glicolise, o que favorece a degradação da matriz extracelular e uma invasão mais eficiente dos tecidos
1.5. A carcinogénese induzida e a importância dos modelos animais no estudo na neoplasia mamária
Vários autores (Huggins et al, 1959; Russo et al, 1982; Cheung et al, 2003) utilizaram modelos animais para estudar as neoplasias da mama da mulher, o que contribuiu largamente para o entendimento da sua oncobiologia e desenvolvimento de novas técnicas terapêuticas. Contudo são poucas as espécies que desenvolvem tumores mamários espontaneamente, destacando-se os roedores.
Para o desenvolvimento de tumores mamários em roedores, pode recorrer a várias técnicas, como a indução química, indução por vírus, transplantação de tumores humanos, modelos transgénicos e indução por radiação ionizante (Bland e Copeland, 2004; Harris et al, 2004; Russo et al, 2005a, 2005b).
O rato tem sido usado como modelo ideal nos estudos acerca dos mecanismos envolvidos na carcinogénese mamária, nomeadamente para investigar o papel protector de alguns fármacos. Primeiro, temos muita informação acerca da carcinogénese da glândula mamária do rato. Segundo, há muitas semelhanças entre as glândulas mamárias e os carcinomas da rata e da mulher nomeadamente no que se refere à histologia e carcinogénese (Russo et al, 1997).
As glândulas mamárias de várias estirpes de rato são susceptíveis à transformação química, nomeadamente, as espécies Sprague-Dawley, Búfalo/N, Lewis e Wistar-Furth (Russo et al, 2005a, 2005b; Barros et al, 2003, Imai et al, 2005). Sabe-se
Figura1.4.1- Alterações morfológicas ocorrentes durante a carcinogénese (adaptado de Robbins & Cotran, 2005)
também que, os modelos de indução química em ratos sofrem influência quanto a idade do animal, da dose do agente carcinogénico e do tipo de ração. As fêmeas devem ser virgens e submetidas a dieta com rações em “pellets” com uma percentagem de gordura de 5% (Manni et al, 1982; Assis et al, 2006).
Os dois compostos químicos mais utilizados para indução química de neoplasias mamárias são o N-nitrosometilureia (NMU) e o 7,12- Dimetilantraceno (DMBA). O NMU, induz a formação de carcinomas mamários em roedores quando administrado por via subcutânea ou intravenosa. Este composta altera em cerca de 75% a expressão do proto-oncogene ras (Russo et al, 2005a, Cha et al, 1996). O DMBA, o outro potente indutor de carcinogénese mamária em roedores; é geralmente administrado por via oral (“gavage”), misturado com óleo ou azeite. Os metabolitos do DMBA são activados no fígado e na glândula mamária (Russo et al, 2005a, Hilakivi-Clarke et al, 1997).
A carcinogénese mamária em rato, segundo Russo e Russo (1997), ocorre sobretudo nos epitélio dos ductos terminais, nos brotos alveolares ou no ducto terminal, que constituem a unidade ducto-lobular terminal na mulher. Há evidencias de que os carcinomas mamários surgem nas estruturas indiferenciadas, porém lesões benignas, como adenomas, cistos e fibroadenomas, ocorrem em estruturas mais diferenciadas quando expostas ao agente carcinogénico. Quanto mais diferenciada é a estrutura na ocasião da exposição, mais benigna é a lesão. A similaridade estrutural e funcional entre as neoplasias mamárias da fêmea de rato e as humanas, já e acima referenciadas, levou a realização de uma conferência em Hannover, na Alemanha, em 1987, que resultou na classificação de lesões não neoplásicas e de neoplasias da mama de fêmeas de rato em comparação com lesões da espécie humana, mostrando essa semelhança. Contudo existem alguns tipos de lesões em mama de rato que não são encontrados nem tem paralelo na mulher (Russo e Russo, 2000).
2 – As capacidades biológicas adquiridas durante a carcinogénese
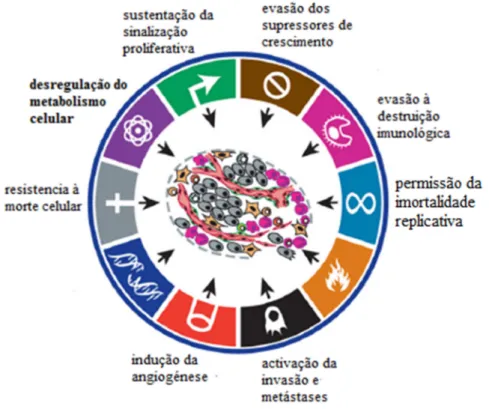
Os marcadores oncológicos compreendem seis propriedades biológicas adquiridas durante o desenvolvimento faseado das neoplasias, constituem um princípio para racionalizar a complexidade das doenças neoplásicas; estes incluem a sustentação da sinalização proliferativa, evasão aos supressores de crescimento, resistência à morte celular, imortalidade replicativa, indução da angiogénese e activação da invasão e metastização. Subjacente a estes marcadores está a instabilidade do genoma, que gera a
diversidade genética que acelera a aquisição de autonomia celular e a inflamação. O progresso conceptual da última década levou ao surgimento de mais dois possíveis marcadores: a evasão ao sistema imunológico e a reprogramação do metabolismo energético (Hanahan e Weinberg, 2011).
Fig. 2.1 – Capacidades biológicas adquiridas durante o desenvolvimento de neoplasias (adaptado de Hanahan e Weinberg, 2011).
2.1 – “Hallmarks” das neoplasias
2.1.1. - Sustentação da sinalização proliferativa
Indiscutivelmente, o traço fundamental das células neoplásicas envolve a sua capacidade de manter a proliferação. Os tecidos normais controlam cuidadosamente a produção e a libertação de sinais promotores de crescimento que induzem a entrada da célula no ciclo de crescimento e divisão celular, assegurando assim a homeostasia do tecido (número de células, assim como a função e a arquitectura tecidular normais) (Hanahan e Weinberg, 2011).
A sinalização mitogénica nas células neoplásicas é bem conhecida (Lemmon e Schlessinger, 2010; Witsh et al., 2010; Hynes e MacDonald, 2009). As células neoplásicas conseguem adquirir a capacidade de manter a sinalização proliferativa de várias maneiras: podem produzir factores de crescimento resultando numa estimulação autócrina, ou podem enviar sinais para estimular células não neoplásicas do estroma tumoral, e estas retribuem fornecendo às células neoplásicas vários factores de crescimento de forma parácrina (Cheng et al.; 2008; Bhowmick et al.; 2004). A sinalização celular também pode ser desregulada elevando os níveis receptores exibidas na superfícies das células neoplásicas, aumentando os níveis de resposta celular ou por alterações estruturais nos receptores facilitando a ligação da sinalização proliferativa. A independência de factores de crescimento pode também derivar da cativação constitutiva das vias de sinalização que operam a jusante desses receptores eliminando a necessidade de estimular essas vias pela activação desse receptor (Hanahan e Weinberg, 2011).
2.1.2 - Evasão dos supressores de crescimento
Em adição à capacidade de indução, sustentação e possivelmente activação de sinais estimuladores de crescimento, as células neoplásicas são capazes de contornar poderosos programas que regulam negativamente a proliferação celular, muitos destes programas dependem da acção de genes supressores de tumores. Os dois genes supressores tumorais protótipos codificam as proteínas RB (proteínas associada ao gene do retinoblastoma) e TP53 (proteínas associada ao gene P53), que actuam como
reguladoras do ciclo celular e governam a decisão de as células proliferarem ou, alternativamente, entrar em senescência ou em apoptose.
A proteína RB integra sinais de diversas fontes intra e extracelulares e, em resposta, decide se a célula deve ou não prosseguir através do seu ciclo de crescimento e divisão. Nas Células neoplásicas com defeitos na via RB a sua função é perdida levando a uma proliferação celular persistente. Enquanto que o RB traduz sinais inibidores do crescimento originários na maioritariamente do meio extracelular, a TP53 recebe sinais de stress e de anormalidade que ocorrem nos sistemas intracelulares: se o grau de danos no genoma é excessivo ou se os níveis de nucleótidos, sinais promotores de crescimento, glicose ou de oxigenação estão abaixo do normal, a TP53 pode por termo à progressão do ciclo celular até que as condições sejam normalizadas. Em alternativa, face aos sinais de alarme indicando danos irreparáveis dos subsistemas, a TP53 pode desencadear a apoptose. Os vários efeitos da TP53 activada são complexos e altamente dependentes do contexto, variando com o tipo de célula assim como da severidade e persistência das condições de stress celular e do dano genómico (Burkhart e Sage, 2008; Deshpande et al.; 2005).
2.1.3 - Resistência á morte celular
O conceito de que a morte celular programada por apoptose serve como barreira natural ao desenvolvimento de neoplasias foi estabelecido por vários estudos nas últimas décadas (Adams e Cory, 2007; Lowe et al,; 2004). A elucidação dos circuitos de sinalização que regem o programa apoptótico revelou como esta é desencadeada em resposta a vários factores de stress fisiológico que as células neoplásicas vivenciam durante a tumorigénese ou em resposta terapia anti-tumoral (Adams e Cory, 2007; Lowe
et al, 2004). A maquinaria apoptótica é composta por reguladores e componentes
efectores (Adams and Cory, 2007). Os reguladores, por sua vez, estão divididos em dois grupos, um que recebe e processa sinais extracelulares indutores de apoptose (programa extrínseco de apoptose) e um outro que detecta e integra vários sinais de origem intracelular (programa intrínseco). Estes culminam na activação de uma protease (caspase 8 e 9, respectivamente), que leva à iniciação de uma cascata proteolítica envolvendo factores de caspases responsáveis pela execução das fases da apoptose em que a célula é progressivamente degradada e consumida pelas células vizinhas ou pelas
células fagocíticas. Normalmente, a via apoptótica intrínseca é considera a maior barreira à patogenia tumoral.
O accionador apoptótico que transmite os sinais entre os reguladores e os efectores são controlados por membros da família de proteínas reguladoras Bcl-2, pró- e anti-apoptóticos (Adams e Cory, 2007). As proteínas Bcl-2 e as proteínas mais próximas (Bcl-X, Bcl-w) são inibidoras da apoptose, ligando-se e suprimindo a acção das duas proteínas pró-apoptóticas (Bax e Bak), esta última é incorporada na membrana mitocondrial externa. Quando libertadas da inibição, as proteínas Bax e Bak afectam a integridade da membrana, causando a libertação de sinais pró-apoptóticos, entre eles o citocromo c, que por sua vez activa uma cascata de caspases que actuam de forma proteolítica induzindo várias alterações celulares associadas com a apoptose (Adams e Cory, 2007).
Embora as condições celulares que desencadeiam a apoptose estejam totalmente enumeradas, vários sensores que desempenham um papel chave no desenvolvimento tumoral foram identificados (Adams e Cory, 2007). O mais notável é o sensor de danos no DNA que funciona através do gene supressor tumoral TP53 (Junttila e Evan, 2009); a TP53 induz a apoptose aumentando a expressão das proteínas Noxa e Puma, em resposta a níveis substanciais de quebras no DNA e outras anomalias cromossómicas. Alternativamente, sinalização insuficiente de factores de sobrevivência (por exemplo, níveis inadequados de interleucina-3 nos linfócitos ou de Factor de crescimento semelhante à insulina 1/2 -“insulin-like growth factor- 1/2 [Igf1/2] nas células epiteliais) pode provocar a apoptose através de uma proteína chamada Bim. No entanto, outra condição pode levar à morte celular pela hiperactivação da sinalização por certas oncoproteinas, como a proteína Myc, que desencadeia apoptose (em parte através da proteínas Bim) a não ser que seja contrabalançado por factores anti-apoptóticos (Junttila e Evan, 2009).
As células neoplásicas envolvem uma série de estratégias para limitar ou circunscrever a apoptose, a mais comum é a perda de função do supressor tumoral TP53, o que elimina um sensor critico de danos durante o ciclo celular; alternativamente, as células neoplásicas podem conseguir o mesmo objectivo aumentando a expressão de reguladores anti-apoptóticos (Bcl-2, Bcl-x) ou de factores de sobrevivência (Igf 1/2), diminuindo a acção de factores pró-apoptóticos (Bax, Bim,
Puma) ou ainda impedindo a morte celular induzida por ligandos extrínsecos. A multiplicidade dos mecanismos de evasão à apoptose presumivelmente reflecte a diversidade de sinais indutores da apoptose que as populações de células neoplásicas encontram durante a sua evolução para o estado maligno (Hanahan e Weinberg, 2011).
2.1.4 - Permissão da imortalidade replicativa
Em 2000, foi aceite que as células neoplásicas requerem um potencial replicativo ilimitado para gerar tumores macroscópicos, capacidade esta que é contrastante com o comportamento da maioria das linhagens celulares do corpo que estão sujeitas a número limitado de ciclos de divisão celular. Esta limitação está associada com duas barreiras distintas à proliferação: a senescência, uma entrada irreversível num estado não-proliferativo mas viável, e a apoptose, que envolve morte celular. Quando as células são propagadas em cultura, ciclos repetidos de divisão celular levam primeiro à indução de senescência e em seguida, para as células que tentam controlar essa barreira, leva à crise, que por sua vez leva à morte das maioria das células da população. Em raras ocasiões, as células podem emergir de uma população em crise e apresentam um potencial replicativo ilimitado, esta transição é chamada de imortalização, uma característica que a maioria das linhas celulares possui em virtude da sua capacidade de proliferar em cultura sem evidenciar qualquer senescência ou crise (Hanahan e Weinberg, 2011).
Existem evidências que indicam que os telómeros protegem as extremidades dos cromossomas e estão centralmente envolvidos na capacidade de proliferação ilimitada (Blasco, 2005). Os telómeros, compostos por múltiplos nucleótidos repetidos em tandem, encurtam progressivamente em células não-imortalizadas propagadas em cultura, eventualmente perdem a sua capacidade de proteger as terminações do DNA cromossómico das fusões “end-to-end”; estas fusões geram cromossomas dicêntricos instáveis, resultando num cariótipo que ameaça a viabilidade celular. Assim, o comprimento do DNA telomérico numa célula determina quantas gerações sucessivas de células conseguem passar antes que os telómeros sejam fortemente degradados e percam a sua função de protecção provocando a entrada em crise.
A telomerase é uma DNA polimerase especializada que adiciona segmentos repetitivos às terminações do DNA telomérico; a telomerase é quase ausente em células não-imortalizadas mas expressa-se a níveis significativos na maioria das células
imortalizadas, incluindo as células neoplásicas. Ao estender o DNA telomérico, a telomerase contraria a progressiva erosão dos telómeros A presença da actividade da telomerase, quer seja espontaneamente em células imortalizadas, quer em células obtidas por engenharia genética para expressar esta enzima, está correlacionada com a resistência à indução de senescência ou crise/apoptose; inversamente a supressão da actividade da telomerase leva ao encurtamento dos telómeros e à activação de barreiras à proliferação (Blasco, 2005).
As duas barreiras à proliferação – senescência e crise/apoptose – foram estabelecidas como duas defesas anti-neoplásicas cruciais. A eventual imortalização das células que precede à formação de neoplasias tem sido atribuída à sua capacidade de manter o DNA telomérico em comprimento suficiente para evitar desencadear a senescência ou a apoptose, alcançada geralmente pelo aumento da expressão da telomerase, ou menos frequente, através de um mecanismo baseado na recombinação alternativa para a manutenção dos telómeros. Assim, os encurtamento dos telómeros passou a ser visto como um dispositivo que determina o potencial limitado de replicação das células e portanto, deve ser superado pelas células neoplásicas (Blasco, 2005).
2.1.5. - Indução da angiogénese
Tal como os tecidos normais, as neoplasias requerem oxigénio e nutrientes, assim como a capacidade de evacuar desperdícios metabólicos e dióxido de carbono; nas neoplasias estas necessidades são asseguradas por uma neovasculatura gerada pelo processo de angiogénese. Durante a embriogénese, o desenvolvimento da vasculatura envolve o surgimento de novas células endoteliais e o seu agrupamento em tubos vasculares (vasculogénese) além do surgimento de novos vasos a partir dos já existentes (angiogénese). No adulto, a angiogénese está activa como parte de processos fisiológicos, como a cicatrização de feridas e o ciclo reprodutivo feminino, mas só transitoriamente; em contraste durante a progressão tumoral a angiogénese está quase sempre activada e permanece assim, causando o aparecimento de novos vasos que ajudam à expansão do crescimento neoplásico (Hanahan e Folkman, 1996) e a disseminação à distância.
Várias provas mostram que a progressão angiogénica é governada por factores pró- ou anti-angiogénicos (Baeriswyl e Christofori, 2009; Bergers e Benjamin, 2003). Alguns destes reguladores angiogénicos são proteínas sinalizadoras que se ligam a
receptores estimuladores ou inibidores da superfície celular das células endoteliais. Os mais conhecidos indutores ou inibidores da angiogénese, são o factor de crescimento endotelial vascular - A (VEGF-A) e a trombospondina – 1 (TSP-1), respectivamente.
O gene VEGF-A codifica ligandos que estão envolvidos no crescimento de novos vasos sanguíneos durante o densenvolvimento embrionário e pós-natal e depois na hemostase das células endoteliais, em situações fisiológicas e patológicas no adulto. A sinalização de VEGF através de três receptores tirosina cinases (VEGFR-1 – 3) é regulada em múltiplos níveis, reflectindo a sua complexidade; assim a expressão do gene VGEF pode ser aumentada por hipóxia e por sinalização por parte dos oncogenes (Ferrara, 2009; Mac Gabhann e Popel, 2008). Além disso, outros sinais pró-angiogénicos, tais como membros da família dos factores de crescimento fibroblástico (FGF), podem estar implicados na manutenção da angiogénese quando a sua expressão é cronicamente aumentada (Baeriswyl e Christofori, 2009).
Os vasos sanguíneos produzidos nos tumores por activação crónica da angiogénese e uma mistura desequilibrada de sinais pró-angiogénicos, são tipicamente aberrantes: a neovasculatura tumoral é marcada pelo surgimento precoce de capilares, excessiva ramificação dos vasos, constituída por vasos distorcidos e alargados, fluxo sanguíneo errático, micro-hemorragias, permeabilidade e níveis anormais de proliferação de célula endoteliais e apoptose (Nagy et al., 2010).
A angiogénese é induzida surpreendentemente cedo durante o desenvolvimento de neoplasias invasoras em modelos animais e humanos; a análise histológica de lesões pré-malignas e não-invasoras, incluindo displasias e carcinomas in situ, decorrentes numa variedade de órgãos revelaram o início precoce da angiogénese (Raica et al., 2009). Historicamente, a angiogénese foi idealizada como sendo importante apenas quando os tumores macroscópicos de rápido crescimento estivessem formados mas, um estudo indicou que a angiogénese também contribui para a fase pré-maligna da progressão neoplásica, consolidando assim a sua importância como marcador oncológico(Raica et al., 2009).
2.1.6. - Activação da invasão e metástases
Em 2000, os mecanismos promotores da invasão e da metastização eram um enigma. Sabia-se que os carcinomas (que derivavam de tecidos epiteliais) progrediam
com graus de agressividade biológica maiores, o que se reflectia em invasão local e metastização à distância. As células neoplásicas carcinomatosas desenvolviam alterações na sua forma, bem como na sua ligação intercelular e à matriz extracelular. A alteração mais bem caracterizada envolve a perda de E-caderina por parte das células carcinomatosas, uma molécula de adesão intercelular chave, que intervém na formação das junções aderentes. A caderina E mantém a adesão das células e promove a diferenciação e manutenção da homeostasia dos tecidos. O aumento da expressão de caderina E foi reconhecida como um antagonista da invasão e da metastização, enquanto que a redução da sua expressão é conhecida por potencializar estes fenómenos. A diminuição da sua expressão e a observação da sua inactivação por mutação em carcinomas humanos, apoiou a ideia de que esta é um regulador chave deste marcador (Caderina E) (Berx e van Roy, 2009; Cavallaro e Christofori, 2004).
Além disso, a expressão de genes que codificam esta e outras moléculas de adesão célula a célula ou célula - matriz extra-celular, está comprovadamente alterada em alguns carcinomas muito agressivos; normalmente a sua expressão está diminuída, mas, por outro lado, moléculas de adesão que normalmente estão associadas com a migração celular que ocorre durante a embriogénese e em estados de inflamação, tem a sua expressão muitas vezes elevada. Por exemplo, a N-caderina, que normalmente é expressa em neurónios e em células mesenquimais durante a organogénese, tem a sua expressão aumentada em muitas células de neoplasias invasoras (Cavallaro e Christifori, 2004).
O processo de invasão e metastização foi esquematizado como uma sequência de vários passos, muitas vezes chamado de cascata de invasão e metastização (Talmadge e Filder, 2010). Este processo prevê uma sucessão de alterações biológicas nas células, começando com a invasão local, seguida da invasão do tecido peritumoral e dos vasos sanguíneos e linfáticos (intravasão), circulação das células (embolia neoplásica) e em seguida, saída das células do lúmen dos vasos para o parênquima dos tecidos distantes, formação de pequenos nódulos de células neoplásicas (micrometástases) e finalmente, o crescimento das lesões micrometastásicas em tumores macroscópicos; a este último passo é chamado de “colonização”.
Um dilema ainda não resolvido em torno da formação de tumores, é o papel do sistema imunológico desempenha na resistência ou em erradicar a formação e progressão de neoplasias incipientes, tumores de estado avançado e em micrometástases. A teoria da vigilância imunológica propõe que as células e tecidos são constantemente monitorizados por um sistema imunológico sempre alerta responsável pelo reconhecimento e eliminação da maioria das células neoplásicas. De acordo com esta lógica, os tumores que surgem, de alguma forma conseguiram evitar a detecção por parte do sistema imunológico ou limitaram a capacidade de destruição do mesmo, fugindo assim à erradicação.
Alterações na actuação do sistema imune foram comprovados pelo aumento de certo tipo de neoplasias em indivíduos imunocomprometidos (Vajdic e van Leuwen, 2009). No entanto, a maioria destas neoplasias são induzidas por vírus, sugerindo que parte do controlo destas neoplasias depende da redução da carga viral nos indivíduos infectados, em parte através da eliminação de células infectadas. Estas observações parecem elucidar um pouco sobre o papel do sistema imunológico em limitar a formação de mais de 80% das neoplasias de etiologia não-viral. No entanto, nos últimos anos, surgiram várias provas tanto em murganhos geneticamente modificados como em neoplasias de epidemiologia clínica, que sugerem que o sistema imunológico funciona como uma barreira para a tumorigénese e progressão tumoral, pelos menos em algumas não-induzidas por vírus.
Em murganhos geneticamente modificados para serem deficientes em vários componentes dos sistema imunológico para o desenvolvimento de tumores induzidos, foi observado que as neoplasias eram mais frequentes e/ou de crescimento mais rápido em murganhos imunodeficientes relativamente ao grupos controlo (imunocompetentes); em particular, deficiências no desenvolvimento ou na função de CD8+, linfócitos T citotóxicos, CD4+ Células T helper ou células Natural Killer (NK), levaram a um aumento considerável da frequência de neoplasias; além disso, murganhos com imunodeficiências combinadas nas células T e NK, foram ainda mais susceptíveis ao desenvolvimento de neoplasias. Os resultados mostraram que, em pelo menos alguns modelos experimentais, tanto o sistema imunitário inato como o adaptativo são capazes de contribuir significativamente para a vigilância imune e para erradicação de neoplasias (Teng et al., 2008; Kim et al., 2007).
A epidemiologia clínica também cada vez mais suporta a existência de resposta imune anti-tumoral em algumas neoplasias humanas, mas a epidemiologia dos indivíduos cronicamente imunodeprimidos não indica o aumento significativo das taxas de incidências de neoplasias não-virais. Isto pode ser tomado como um argumento contra a importância da vigilância imune como uma barreira efectiva contra a tumorigénese e progressão tumoral. De notar que, indivíduos imunodeprimidos por HIV ou por fármacos são predominantemente imunodeficientes em células T e células B e não apresentam deficiência imunológicas multicompetentes como os ratos geneticamente modificados sem células NK e sem linfócitos T citotóxicos, o que levanta a possibilidade que tais pacientes ainda terem uma capacidade imunológica residual associada com células NK e outras do sistema imunológico inato (Bindea et al., 2010; Ferrone e Dranoff, 2010)
Na verdade, a discussão acima simplifica a interacção imunológica tumor-hospedeiro como sendo altamente imunogénica, as células neoplásicas podem muito bem evadir-se ao sistema imune desactivando os componentes do sistema imune que tenham sido enviados para eliminá-los. Por exemplo, as células neoplásicas podem paralisar a acção das células NK e dos linfócitos T citotóxicos, através da secreção de TGF – β (Factor de transformação do crescimento -Transforming growth factor – β) ou outros factores imunosupressores (Yang et al.; 2010; Shields et al., 2010). Outros mecanismos subtis actuam através do recrutamento de células inflamatórias que são imunosupressoras incluindo células T reguladoras e células supressoras derivadas da linha mielóide (myeliod-derived supressor cells – MDSC) que suprimem a acção dos linfócitos citotóxicos (Mougiakakos et al., 2010; Ostrand-Rosenberg e Sinha, 2009).
2.2 - A reprogramação do metabolismo: um marcador emergente
2.2.1 - Visão global do metabolismo
A Bioenergética das células normais está diretamente relacionada com a homeostase da glicose que por sua vez depende do balanço entre a difusão, o armazenamento e o metabolismo da glicose. Este balanço energético está relacionado com pelo menos três vias metabólicas incluindo a glicólise, a lipogénese e o ciclo do ácidos tricarboxilico (ou ciclo de Krebs), que por sua vez estão proximamente ligadas com a biossíntese de aminoácidos e de nucleótidos. Embora as células normais recorram a uma variedade de fontes energéticas como o glicogénio, ácidos gordos e aminoácidos,
a glicose é considerada como a fonte energética chave para o seu crescimento. A glicose é captada por um sistema de transportadores de glicose e é convertida a piruvato através da glicólise (Gatengy e Gillie, 2004). O piruvato é então, na mitocôndria, convertido em acetil-CoA e utilizado como substrato para o ciclo de Krebs; embora a célula gere ATP através da fosforilação oxidativa, um metabolito intermediário, o citrato, é exportado para o citoplasma e convertido em acetil-CoA, que por sua vez é usado como substrato inicial para a geração de ácidos gordos através da lipogénese (Wellen et al, 2009; Vender Heiden et al, 2009). Os ácidos gordos, fornecem energia assim como providenciam componentes chave para a biossíntese membranar e desempenham um papel importante na sinalização celular (Furuta et al, 2010).
2.2.2 - O metabolismo especial das células neoplásicas
As células neoplásicas sintetizam grandes quantidades de macromoléculas para proliferarem e formarem novas células, o que requer uma produção contínua de ATP e de co-factores (NAD+, NADPH, H+) de modo a sustentar as vias sintéticas. Estas células consomem glucose em excesso e grandes quantidades de aminoácidos (em particular a glutamina, derivada da proteólise muscular).
O metabolismo da glicose nas células neoplásicas frequentemente resulta na formação de ácido láctico, mesmo na presença de oxigénio. Este fenómeno é referido como “glicólise aeróbia” ou o “Efeito de Warburg” e tem sido alvo do interesse de vários estudos. Este efeito é o reverso do efeito de Pasteur (inibição da fermentação na presença de O2); a modificações do efeito de Pasteur está ligado, entre outros, ao factor induzido por hipoxia 1 (Hypoxia-inductible factor-HIF 1). O HIF 1 estimula muitos genes codificantes de enzimas glicolíticas, ao passo que bloqueia o uso do piruvato pela piruvato desidrogenase mitocondrial. Este mecanismo leva à troca da fosforilação oxidativa pela glicólise para a produção de ATP. Nas células normais, o efeito de Pasteur é mediado pelo ATP e pelo citrato mas, em condições de hipóxia, a oxidação da glicose é diminuída, resultando na diminuição de ATP e de citrato pela mitocôndria. Por sua vez, o feedback destas moléculas é quebrado, a glicólise é então acelerada, levando à produção de ácido láctico. É importante realçar que a maioria das células dos tumores sólidos são hipóxicas e a glicólise torna-se na forma mais rápida de produzir energia e a melhor maneira de sintetizar vário intermediários metabólicos essenciais para a sua proliferação, mesmo sendo relativamente ineficiente na produção de ATP. É ainda
importante destacar que as células neoplásicas estão mais focadas em manter a suas vias metabólicas de modo a providenciar as moléculas necessárias para a biossíntese do que alcançar a via de produção de energia mais eficiente; deste modo, elas encontram todos os nutrientes necessários em abundancia no seu ambiente (Icard et al, 2012; Feron, 2009; Bristow and Hill, 2008; Simunnet et al, 2003).
Existem várias razões para que o aumento da captação da glicose para a geração de ATP ou de reacções anabólicas constituam vantagens para o crescimento tumoral:
Primeiro, em condições de glicose aeróbia, as células podem viver em condições de tensão de oxigénio flutuantes (devido à inconstante hemodinâmica associada à distância face aos vasos sanguíneos) que podem ser letais para as células que dependem da fosforilação oxidativa para gerar ATP (Pouyssegur et al, 2006).
Segundo, os tumores podem metabolizar a glicose através da via das pentoses fosfato para gerar NADPH, que assegura a defesa anti-oxidante das células contra um microambiente hostil e agentes quimioterápicos (Gatenby e Gillies, 2004). Além disso, o NADPH pode contribuir para a síntese de ácidos gordos.
Terceiro, as células tumorais geram ácido láctico e bicarbónico (o lactato é o principal produto final da glicose aeróbica). Certas condições ácidas do seu ambiente (Koukourakis et al, 2006) favorecem a invasão tumoral (Swietach et al, 2007) e suprimem os efectores imunológicos anti-tumorais (Fischer et al, 2007). O lactato produzido pelas células tumorais pode ser captado pelas células estromais (através dos transportadores monocarboxilados MCT1 e MCT2) para gerar piruvato que pode ser usado para fosforilação oxidativa ou pode ser expulso para reabastecer as células tumorais (KouKourakis et al, 2006). O lactato em excesso, pode ser reconstituído em glicose no fígado através da gliconeogénese; uma das enzimas responsáveis por este processo é a Glucose-6-fosfatase, sub-unidade catalítica (G6pc), enzima-chave na homeostase dos níveis de glicose no sangue, que participa na glicogenólise e na gluconeogénese.
Quarto, e o mais importante, as células tumorais usam os intermediários da via glicolítica para reacções anabólicas (por exemplo, a glucose 6-fosfato para a síntese de glicogénio e ribose 5-fosfato, a diidroxiacetona para a síntese de fosfolpidos e de triglicerídeos e o piruvato para a síntese de alanina e malato) (Gatenby e Gillies, 2004).
Nas células tumorais, o piruvato pode ser importado para a mitocôndria, onde é convertido em citrato, que pode ser integrado no ciclo de Kbrebs ou pode ser expulso para fora da mitocôndria para a síntese de ácidos gordos, colesterol e isoprenoides (Wang et al, 2005). Na mitocôndria, a síntese de citrato é assegurada pela enzima citrato sintase (Cs), encontrada em quase todas as células que são capazes de realizar o metabolismo oxidativo por sua vez, o citrato expulso para o citosol, pode ser utilizado para a síntese de ácidos gordos, envolvendo a ação da enzima acetil-coA carboxilase (Acaca) como catalisadora de um dos passos-chave da via.
O objectivo deste trabalho foi a avaliação da expressão de três genes relacionados com o metabolismo celular num modelo de neoplasia experimental da mama. Foi estudada a variação da expressão génica de Citrato sintase (ciclo de Krebs), Acetil-coA Carboxilase α (sintese de ácidos gordos) e glucose-6-fosfatase (gliconeogénese e Glicogenólise) em quatro grupos histológicos de lesões e no tecido mamário não neoplásico a fim de verificar se existem diferenças revelantes.
A realização do trabalho experimental desta Dissertação decorreu no Serviço de Patologia Experimental da Faculdade de Medicina de Coimbra (Responsável Professor Doutor António Cabrita), no Laboratório de Bioquímica Genética – LBG – do Centro de Neurociências e Biologia Celular (Responsável Professora Doutora Manuela Grazina) e no Laboratório de Histologia e Anatomia Patológica da UTAD (Responsável Professora Doutora Fernanda Seixas Travassos)
Todos os procedimentos experimentais que dizem respeito à indução da patologia, manutenção e sacrifício dos animais foram aprovados pela Direção Geral de Veterinária e seguem o estipulado na Diretiva da União Europeia 2010/63/EU que revê e substitui a Diretiva 86/609/EU. As amostras utilizadas neste estudo foram obtidas no âmbito de outro trabalho que já se encontrava a decorrer no Serviço de Patologia Experimental, devidamente autorizado pelas autoridades competentes.
Os estudos moleculares apresentados neste trabalho foram realizados no LBG.
3.1 Indução Tumoral
Neste estudo foram utilizados 10 fêmeas de rato virgens, da estirpe Sprague-Dawley, provenientes do Biotério Central da Faculdade de Medicina da Universidade de Coimbra. Os animais foram distribuídos por gaiolas com três indivíduos e, antes de iniciar o estudo, todos os animais foram submetidos a uma semana de quarentena. Após este período, administrou-se uma única dose de dimetilbenzantraceno (DMBA), na concentração de 65 mg/kg, usando a metodologia de “gavage”. Após este período, os animais foram mantidos durante 28 semanas nas condições padrão do biotério: temperatura de 25 ºC e 60-65% de humidade, com regime de 12 horas de luz e 12 horas de escuridão.
A vigilância para rastreio de alterações patológicas foi diária, com observação dos seus hábitos alimentares. A pesagem foi realizada semanalmente, coincidindo a última pesagem com a hora do sacrifício.
Previamente à colheita das amostras, todos os animais foram anestesiados, eutanasiados e necropsiados. A anestesia foi efetuada através da aplicação de 50
ml/250g de uma mistura (10:1) de cetamina e clorpromazina. A eutanásia foi provocada por insuficiência cardíaca através da colheita de 5 mL de sangue.
Na necrópsia, foram registados os dados referentes à observação do hábito externo, observação detalhada do hábito interno e fragmentos de tecidos colhidos e respectiva finalidade, em modelo próprio em utilização no Laboratório de Patologia Experimental.
3.2 Colheita e preservação de amostras
Foram recolhidos dois fragmentos, devidamente pesados, de todos as alterações visíveis macroscopicamente, para dois fins: um fragmento foi preservado em etanol a 70%, para histologia clássica (secção 3.3), o outro foi emerso em RNAlater (Quiagen, United States), 10 ul/ mg de tecido, para estudos de transcritos (RNA). O RNAlater é um reagente estabilizador de RNA, evitando a sua degradação e mantendo os padrões de expressão genética. Para os estudos de expressão genética, é essencial que o RNA esteja integro, caso contrário, a análise é inútil.
Foram também recolhidas três amostras de tecido sem qualquer alteração macroscópica visível, para controlo, nas mesmas condições.
3.3 Classificação Histológica
Os fragmentos recolhidos para histologia clássica foram colocados em cassetes histológicas e embebidos em álcool a 70%. Todas as amostras foram sujeitas a um processamento que inclui desidratação em álcool de concentração crescente (70%, 90%, 95%, 100%). Seguidamente, as amostras foram colocadas em xilol, substância miscível com o álcool e com a parafina. As amostras foram sujeitas a impregnação e inclusão com parafina. Foram realizados cortes com o micrótomo com 4 micrómetros de espessura. Depois de cortadas e colocadas em lâminas, as amostras foram coradas com o método clássico Hematoxilina e Eosina (H&E).
A classificação histológica das amostras foi feita tendo em conta os critérios utilizados por Russo e Russo (2000), para a classificação histológica de tumores de glândula mamária de rato.
3.4 Análise de transcritos (RNA)
3.4.1 Tratamento do material
O material utilizado para extração, processamento e armazenamento das amostras foi previamente tratado com Dietil-pirocarbonato (DEPC, Sigma-Aldrich, Alemanha), para eliminar Rnases
3.4.2 Extração de RNA
Para proceder à extração de RNA das amostras obtidas nas necrópsias foi utilizado o kit RNeasy® (Qiagen), de acordo com as instruções do fabricantes. De forma sucinta, usaram-se 30 mg de tecido, a que se adicionaram 600µ L de tampão RLT contendo 6µ L de β-mercaptoetanol para homogeneização com um homogenizador. O lisado foi centrifugado durante 3 minutos à velocidade máxima e o sobrenadante foi transferido para um novo tubo e misturado com etanol a 70%. Procedeu-se à filtração em coluna RNeasy spin, sob centrifugação (8.000xg), usando 50 µL de água sem RNase para eluir o RNA.
3.4.3 Avaliação da Integridade do RNA
Para a avaliação da integridade do RNA, foi utilizado o chip “Agilent RNA 6000 Nano Kit” (Figura 3.4.3.1) da Agilent Technologies (Alemanha) associado ao analisador “2100 Bioanalyser” da Agilent Techologies (Alemanha), em que ocorre uma microelectroforese capilar, que permite calcular a concentração de RNA total e avaliar a integridade do RNA (RIN- RNA Integrity Number) através do quociente de rRNAs 18S/28S, obtendo-se um eletroforetograma.
Fig. 3.4.3.1 – Imagem do chip “Agilent RNA 6000 Nano Kit®” (adaptado de Agilent RNA 6000 Nano Kit guide)
Usou-se 1 µ L de cada amostra e do marcador de peso molecular “RNA Ladder” As amostras que apresentavam baixos valores de RIN ou ainda a presença de impurezas, foram sujeitas a um tratamento com DNase seguido de re-extração, de acordo com o método referido anteriormente.
3.4.4 RT-PCR
Para proceder a transcrição reversa das amostras, foi utilizado o kit “High capacity RNA-to-cDNA” da Applied Biosystems, usando cerca de 2 µg de RNA total por cada 20 µ L de reação.
As reações decorreram num termociclador, a 37ºC durante 60 min, para produzir o cDNA.
3.4.5 PCR em tempo real
Neste trabalho experimental foram estudados três genes que codificam proteínas relacionadas com o metabolismo: a glucose-6-fosfatase - sub-unidade catalítica (G6pc), a citrato sintase (Cs) e a Acetil-coA carboxilase α (Acaca). O gene de referência (também conhecido como gene “housekeeping”) analisado foi o que codifica a β-Actina (Actb).
Para avaliar de forma quantitativa a expressão dos genes-alvo, as amostras foram sujeitas ao protocolo de PCR em tempo real utilizando o kit “QuantiTect SYBR Green PCR Kit®” da Applied Biosystems. O kit “Quantitect primer®” permite uma análise rápida e acessível do RNA. Cada ensaio consiste em primers “foward” e “reverse” específicos derivados da sequência do gene presente na base de dados NCBI. O kit “Quantitect SYBR Green PCR” é uma mistura optimizada, pronta a usar, para um PCR em tempo real usando a química de SYBR Green®. A florescência do corante SYBR Green, na mistura, permite a análise de muitos alvos diferentes sem a necessidade de sintesar sondas marcadas específicas do gene-alvo. Uma combinação equilibrada dos iões K+ e NH4+ no tampão promove o reconhecimento específico do primer, permitindo uma elevada sensibilidade e especificidade do PCR. Além disso, a enzima HotStarTaq Polimerase®, permite um começo restrito, evitando a formação de produtos não específicos. Para cada amostra foram realizadas 3 réplicas. Usaram-se 10 µl de mix por µl de amostra de cDNA.
A reação de PCR em tempo real decorreu no “7500 fast real-time PCR sistem®” da Applied Biosystems, de acordo com o programa: 94ºC 15’’; 55ºC 30’’; 72ºC 30’’.
Terminada a reação de PCR em tempo real, procedeu-se à análise dos resultados.
3.4.6 Avaliação da expressão relativa dos genes-alvo
A quantificação da expressão foi calculada a partir do valor de CT (“threshold cycle”), que é o número do ciclo a que o produto amplificado se acumula de forma exponencial produzindo um sinal detetável. Como a quantificação realizada neste estudo é relativa, o valor de CT do gene-alvo é comparado com o valor de CT do gene referência, obtendo-se assim o valor de ∆CT.
As amostras foram, agrupadas em tipos histológicos: normal, hiperplásico, neoplasias benignas, neoplasias malignas não invasoras e neoplasias malignas invasoras.
Para cada amostra foi calculada a média dos valores de CT das três réplicas, seguidamente foi calculada a média dos valores de CT de cada grupo para os genes-alvo e para o gene de referência.