UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Terapêutica no Cancro do Cólon e Recto:
contribuição da via de sinalização
APC/-catenina/TCF7L2
Neuza Catarina Rocha Gonçalves
Mestrado em Bioquímica
Terapêutica no Cancro do Cólon e Recto II
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Terapêutica no Cancro do Cólon e Recto:
contribuição da via de sinalização
APC/
-catenina/TCF7L2
Dissertação orientada por:
Dra. Cristina Albuquerque, Centro de Investigação de Patobiologia Molecular (IPOFG
EPE)
Prof. Doutora Luisa Cyrne, Faculdade de Ciências da Universidade de Lisboa
Neuza Catarina Rocha Gonçalves
Mestrado em Bioquímica
Terapêutica no Cancro do Cólon e Recto III
Agradecimentos
Gostaria de agradecer a todos aqueles que, de alguma forma, tornaram possível a realização deste trabalho. Em especial gostaria de agradecer:
À Dra. Cristina Albuquerque por ter aceite a minha participação neste trabalho. Obrigada por todo o apoio, compreensão, motivação, paciência, amizade e por todo o conhecimento que me transmitiu, tanto pessoal como profissional.
À Professora Doutora Luísa Cyrne por ter aceite ser orientadora interna e por todo o apoio na realização da presente tese.
Agradeço também à Dra. Inês Francisco e Dra. Patrícia Silva pela paciência, apoio, motivação conhecimento transmitido e por toda amizade.
Às colegas de estágio Sara Pires e Vanda Póvoa agradeço todo o apoio e a ajuda oferecida.
Aos colegas Dr. Bruno Filipe, Dra. Sofia Fragoso, Dra. Patrícia Machado por todo o apoio no laboratório.
Gostaria ainda de agradecer ao Dr. Francisco Caiado e Dr. Jaime Pita pelo apoio no desenvolvimento de algumas técnicas.
Ao CIPM, serviço pertencente ao Instituto Português de Oncologia de Lisboa, Francisco Gentil, E.P.E., quero agradecer as excelentes condições que me proporcionaram e aquisição de conhecimentos por parte dos profissionais que nele trabalham.
Ao serviço de Gastrenterologia, Clínica de risco familiar e ao serviço de Anatomia Patológica do Instituto Português de Oncologia de Lisboa, Francisco Gentil, E.P.E. pela disponibilidade do material biológico utilizado e pela informação clínica relativa aos doentes e dados histológicos dos tumores.
Aos elementos da Consulta de Risco Familiar de Cancro do Cólon e Recto, ao serviço de Farmácia – Citostáticos e ao serviço de Virologia pela cedência de materiais que, de alguma forma, permitiram e realização deste trabalho.
A toda a minha família, em especial aos meus pais e irmão, que tanto se esforçaram para a minha formação e sem eles nada seria possível. Obrigada me terem tornado quem sou hoje.
A todas as minhas amigas pelo apoio e motivação.
Por fim, e não menos importante, gostaria de agradecer ao amigo e namorado Rodolfo Cardoso, pelo apoio, compreensão e motivação ao longo destes anos.
Dedico este trabalho, e toda a minha formação académica, à minha falecida Tia Donzilia que, por ter sofrido de cancro, me motivou a seguir os estudos nesta área, na tentativa de desenvolver conhecimentos que permitissem o combate a esta doença. Por ti, espero ter, com este trabalho, contribuído de alguma forma para o avanço desta área e por ti, espero continuar a desenvolver o meu trabalho nesta área.
Terapêutica no Cancro do Cólon e Recto IV
Resumo
A via Wnt canónica ou via de sinalização APC/-catenina/TCF7L2 encontra-se desregulada na maioria dos cancros do cólon e recto (CCR), assumindo por isso um papel crucial no desenvolvimento tumoral. Em estudos preliminares esta via foi correlacionada com a resposta à terapêutica, tendo sido observada maior resistência ao tratamento para níveis de sinalização mais elevados. Sabe-se que, diferentes mutações no gene APC, que originam a expressão de um número diferente de domínios de regulação da -catenina, resultam em diferentes níveis de sinalização Wnt. Assim, o presente estudo teve como principal objectivo estudar a contribuição de alterações genéticas na via de sinalização APC/-catenina/TCF7L2 para a terapêutica no CCR. Numa primeira fase foram estudadas diferentes mutações no gene
APC e numa segunda fase a perda de função do gene TCF7L2.
Com esta finalidade, foi estudada a citotoxicidade de diferentes concentrações dos fármacos 5-FU, oxaliplatina, irinotecano e diferentes combinações destes nas linhas celulares de CCR HT29 e SW480, as quais apresentam mutações no APC que conduzem a diferentes níveis de activação da via Wnt. Também foi estudado o efeito de três mutações no gene APC, que resultam em proteínas truncadas que retêm três, um ou nenhum domínio de regulação da
-catenina (proteínas truncadas, codões 1-1556, 1-1309 e 1-932, respectivamente), na resistência aos referidos fármacos. O efeito destes foi avaliado através de ensaios de viabilidade e de proliferação celular. Foi ainda estudada a perda de heterozigotia (LOH) do
TCF7L2 em 57 amostras de CCR, a qual foi correlacionada com a presença de metástases
(estadiamento TNM, M0 ou M1). Esta associação foi também analisada, in vitro, através de ensaios de invasão e migração após inibição do TCF7L2.
Os resultados do presente estudo mostraram que o efeito de três mutações no gene
APC, que conferem níveis de sinalização reduzidos, intermédios e elevados, APC 1556,
1-1309 e 1-932, respectivamente, não resulta num aumento da resistência à terapêutica de forma igualmente crescente. De acordo, no tratamento com 5-FU a melhor resposta (diminuição da viabilidade celular) foi obtida para níveis de sinalização intermédios (1-1309), seguida da proteína 1-1556 e, finalmente, da 1-932 que apresentou a pior resposta. No caso da oxaliplatina, as diferenças de viabilidade entre as três proteínas não foram significativas, apesar do comportamento ser diferente entre concentrações reduzidas e elevadas de fármaco. Com o irinotecano, observou-se que a proteína 1-1556 originou uma melhor resposta, seguida da 1-932 e por último da 1-1309. Na segunda fase deste estudo, foi identificada LOH do gene
TCF7L2 em 13/44 (30%) amostras de CCR. A presença de LOH foi significativamente mais
frequente nos tumores de indivíduos que apresentavam metástases (M1) do que nos tumores de indivíduos que não apresentavam metástases (M0) [9/16 (56%) vs. 6/50 (12%), p<10-3]. Adicionalmente, a inibição da expressão do TCF7L2, in vitro, resultou num aumento da invasão e especialmente da migração.
Concluiu-se que, diferentes mutações no gene APC, que conferem diferentes níveis de sinalização da via Wnt, parecem resultar em diferentes respostas ao tratamento do CCR, as quais são dependentes do fármaco e concentração utilizados. Também se concluiu que a LOH do TCF7L2 se encontra significativamente associada à metastização (estadio M1). Sugere-se que a activação do HGF pela inibição do TCF7L2, recentemente descrita, possa tornar a via HGF/MET num alvo terapêutico promissor em doentes com LOH do gene TCF7L2. Em conjunto, este estudo sugere que para além do papel determinante na iniciação tumoral, a via de sinalização Wnt, poderá ser um dos mecanismos determinantes numa fase mais tardia da tumorigénese assim como na resposta à terapêutica.
Terapêutica no Cancro do Cólon e Recto V
Abstract
The Wnt canonic pathway or APC/-catenin/TCF7L2 signaling pathway is deregulated in the majority of colorectal cancers (CRC) and molecular alterations in this pathway are rate-limiting for tumor development. In previous studies this pathway was correlated with the response to chemotherapy, as high signaling activities were associated with an increased resistance to treatment. It is well known that different APC gene mutations, which lead to the expression of truncated proteins retaining different numbers of-catenin regulatory domains, result in different levels of Wnt signaling. Thus, the main aim of the present study was to evaluate the contribution of genetic alterations in the APC/-catenin/TCF7L2 pathway for CRC therapy. This study consisted in two parts, the first was centered in the effect of different APC gene mutations in chemotherapy response and the second was focused in the TCF7L2 loss of function.
The citotoxicity of different concentrations and combinations of the drugs, 5-FU, oxaliplatin and irinotecan, was studied in the CRC cell lines SW480 and HT29, which present
APC mutations leading to different levels of Wnt signaling activation. On the other hand, the
effect, in the resistance to the same drugs, exhibited by three different APC mutations, resulting in truncated proteins that retain three, one or none -catenin regulatory domains (codons 1-1556, 1-1309 and 1-932, respectively), was also studied. The effect of the drugs was evaluated by viability and proliferation assays. The analysis of loss of heterozygosity (LOH) of
TCF7L2 was studied in 57 CRC samples and its correlation with the presence of metastasis (M1
or M0, TNM staging) was also evaluated. This association was also analyzed in vitro by invasion and migration assays following TCF7L2 inhibition.
The results of the present study show that the effect of the three APC gene mutations , leading to reduced, intermediate and high Wnt signaling levels (APC 1556, 1309 and 1-932,, respectively) does not result in an increase of resistance to treatment in the same order. In agreement, when using 5-FU treatment, a better response (reduction of cell viability) was obtained for medium signaling levels (APC 1309), followed by APC 1556 and finally by 1-932. For oxaliplatin, the differences in cell viability between the three truncated proteins were not significant, although some differences were found between low and high drug concentrations. When the cell lines were treated with irinotecan, a better response was observed for APC 1-1556, followed by APC 1-932 and at last by APC 1-1309. In the second part of this study, TCF7L2 LOH was identified in 13/44 (30%) of CCR samples. The presence of LOH was significantly more frequent in the tumors from individuals with metastasis (M1) than in the tumors from individuals that did not presented metastasis (M0) [9/16 (56%) vs. 6/50 (12%), p<10-3]. Additionally, the inhibition of TCF7L2 expression, in vitro, resulted in the increase of invasion and especially of migration.
In conclusion, different APC gene mutations, resulting in different signaling levels in the Wnt pathway, seem to result in different responses to CRC cytotoxic therapy, which are dependent on the concentration and drug used. It was also concluded that the TCF7L2 LOH is significantly associated with the presence of metastasis (M1 stage). Additionally, we suggest that, the recent finding that HGF can be activated by TCF7L2 inhibition, turns the HGF/MET a promising therapeutic target in patients with TCF7L2 LOH. Together, these studies suggest that, besides the determinant role of the Wnt pathway in the initial stages of carcinogenesis, this pathway may also be an important mechanism in the latest stages of colorectal carcinogenesis, as well as in the response to therapy.
Terapêutica no Cancro do Cólon e Recto VI
Índice
Índice Geral
1. INTRODUÇÃO ... 1
1.1. Cancro do cólon e recto ... 1
1.1.1. Sequência adenoma-carcinoma ... 2
1.1.2. Instabilidade genética ... 3
1.2. Via de sinalização Wnt e o CCR ... 3
1.2.1. Gene APC ... 5
1.2.2. -Catenina ... 7
1.2.3. Factores de transcrição TCF/LEF ... 8
1.3. O papel da via de sinalização HGF/MET na invasão e metastização ... 9
1.4. Terapêutica no CCR ... 10 1.4.1. 5-Fluorouracilo (5-FU) ... 10 1.4.2. Oxaliplatina ... 11 1.4.3. Ininotecano ... 12 1.5. Resistência à quimioterapia ... 13 1.5.1. A instabilidade de microssatélites ... 14 1.5.2. A expressão do p21 ... 14
1.6. A via Wnt na resistência à terapêutica... 15
2. OBJECTIVOS ... 16 2.1. Objectivo geral... 16 2.2. Objectivos específicos ... 16 3. MATERIAL E MÉTODOS ... 17 3.1. Material Biológico ... 17 3.1.1. Linhas celulares SW480 e HT29 ... 17
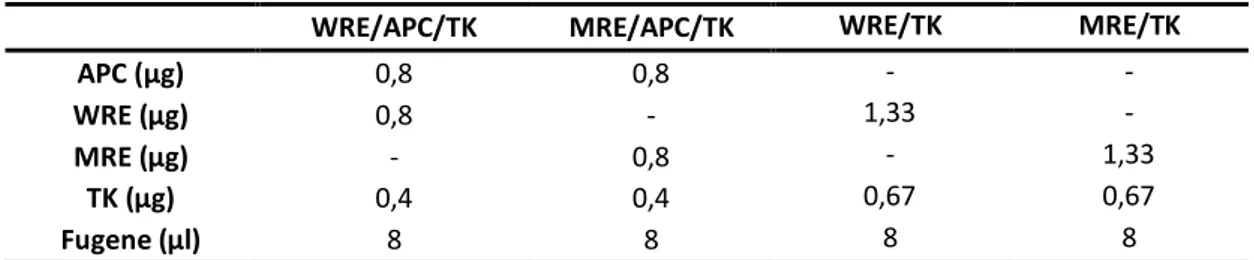
3.1.2. Plasmídeos WRE e MRE... 18
1.1.1. Plasmídeo TK-renilla... 18
1.1.2. Plasmídeos truncantes do gene APC... 19
1.1.3. RNA de interferência ... 19
1.1.4. Amostras de DNA incluídas na análise da perda de heterozigotia do gene TCF7L2 20 1.2. Métodos ... 20 1.2.1. Cultura celular ... 20 1.2.1.1. Descongelação de células ... 20 1.2.1.2. Subcultura... 21 1.2.1.3. Criopreservação ... 21 1.2.1.4. Contagem de células ... 21 1.2.2. Amplificação de plasmídeos ... 22
1.2.3. Extracção de DNA plasmídeo de bactérias transformadas ... 23
Terapêutica no Cancro do Cólon e Recto VII
1.2.5. Quantificação de DNA e RNA... 23
1.2.6. Tratamento das linhas celulares com os fármacos ... 24
1.2.7. Ensaio de viabilidade celular ... 25
1.2.8. Ensaio de proliferação celular ... 26
1.2.9. Transfecção das linhas celulares com DNA plasmídeo ... 26
1.2.10. Quantificação da expressão dos luciferases firefly e renilla ... 27
1.2.11. Amplificação de DNA recorrendo à técnica de polymerase chain reaction (PCR) 28 1.2.12. Controlo da eficiência da reacção de PCR ... 29
1.2.13. Preparação das amostras para análise utilizando o software GeneScan ... 29
1.2.14. Inibição do gene TCF7L2 ... 30
1.2.15. Reacção de transcrição reversa ... 31
1.2.16. Amplificação do cDNA por PCR semi-quantitativo ... 31
1.2.17. Ensaio de invasão e migração ... 32
1.2.18. Fixação e coloração das células invasivas ... 33
1.2.19. Análise estatística ... 33
2. RESULTADOS E DISCUSSÃO – PARTE I ... 34
2.1. Influência de diferentes mutações no gene APC na resistência ao tratamento no CCR 34 2.1.1. Efeito citotóxico dos fármacos 5-FU, oxaliplatina e irinotecano na viabilidade e proliferação celular em linhas celulares com diferentes mutações no gene APC ... 34
2.1.2. Resistência celular ao tratamento com combinação dos fármacos 5-FU, oxaliplatina e irinotecano em linhas celulares com diferentes mutações no gene APC . 38 2.1.3. Resistência celular ao tratamento com 5-FU, oxaliplatina e irinotecano após indução da expressão de proteínas APC truncadas com diferente número de domínios de regulação da -catenina. ... 41
3. CONCLUSÃO – PARTE I ... 46
4. RESULTADOS E DISCUSSÃO – PARTE II ... 47
4.1. Relação entre a perda de função do gene TCF7L2 e a capacidade de metastização no CCR 47 4.1.1. Perda de heterozigotia do gene TCF7L2 em amostras de tumor do cólon e recto 47 4.1.2. Capacidade de invasão e migração celulares após inibição do gene TCF7L2... 49
5. CONCLUSÃO – PARTE II ... 54
6. CONSIDERAÇÕES FINAIS ... 55
7. BIBLIOGRAFIA ... 56
ANEXOS ... 59
Anexo 1. Características clínicas dos indivíduos analisados na LOH do gene TCF7L2 ... 59
Anexo 2. Resultados da análise de LOH do gene TCF7L2 ... 61
Terapêutica no Cancro do Cólon e Recto VIII
Índice de tabelas
Tabela 1 Marcadores preditivos de resposta ao tratamento com 5-FU, oxaliplatina e
irinotecano em estudo ... 14
Tabela 2 Características moleculares das linhas celulares SW480 e HT29. ... 17
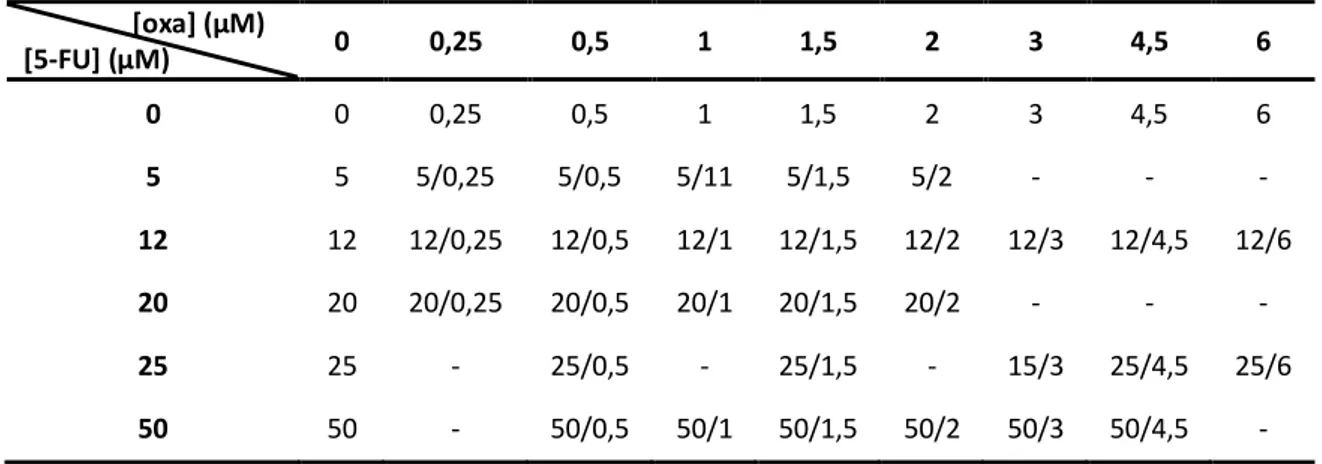
Tabela 3 Concentração das soluções de 5-FU e oxaliplatina (µM) (isoladas e em combinação) utilizadas no tratamento das células no ensaio da resistência celular ... 24
Tabela 4 Concentração das soluções de 5-FU e irinotecano (µM) (isolados e em combinação) utilizadas no tratamento das células no ensaio da resistência celular. ... 25
Tabela 5 Resumo da composição das misturas de transfecção preparadas para a quantificação da sinalização da via Wnt. ... 27
Tabela 7 Características dos primers utilizados na amplificação dos marcadores microssatélite do gene TCF7L2 e condições utilizados nas reacções de PCR. ... 28
Tabela 6 Programa de PCR. ... 29
Tabela 8 Características dos primers utilizados na quantificação dos genes TCF7L2, HGF e GAPDH e número de ciclos efectuados no programa de PCR. ... 31
Tabela 9 Programa de PCR semi-quantitativo. ... 32
Tabela 10 Valores de IC50 calculados após 72h de tratamento com os fármacos 5-FU, oxaliplatina e irinotecano. ... 35
Tabela 11 Resultados obtidos para a análise de LOH do gene TCF7L2 e correlação com a presença ou ausência de metástases... 48
Tabela 12 Resumo das características clínicas dos indivíduos analisados na perda de heterozigotia do TCF7L2. ... 59
Tabela 13 Resultados obtidos na análise de perda de heterozigotia do gene TCF7L2 para cada marcador microssatélite utilizado. ... 61
Índice de Figuras
Figura 1 Eventos moleculares que caracterizam a transição do epitélio cólico normal a adenocarcinoma. ... 2Figura 2 Representação esquemática da via de sinalização Wnt ... 4
Figura 3 Representação da estrutura da proteína APC e seus domínios funcionais. ... 6
Figura 4 Representação da actividade transcripcional regulada pelo TCF7L2 ... 6
Figura 5 Diagrama ilustrativo da interdependência entre os dois eventos genéticos necessários à perda de função do gene APC. ... 7
Figura 6 Representação esquemática da proteína TCF7L2. ... 8
Figura 7 Esquema do metabolismo do 5-FU. ... 11
Figura 8 Representação da formação dos aductos de platina no DNA pela oxaliplatina ... 12
Figura 9 Representação esquemática dos mecanismos de acção do irinotecano.. ... 13
Figura 10 Representação dos plasmídeos WRE e MRE ... 18
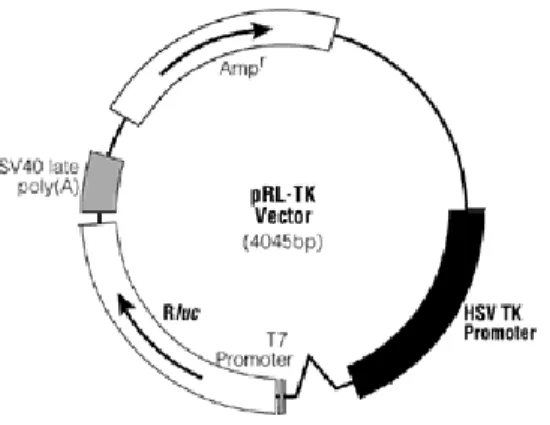
Figura 11 Representação esquemática do plasmídeo pRL-TK ... 19
Figura 12 Representação esquemática dos plasmídeos truncantes do gene APC. ... 19
Figura 13 Ilustração da grelha de contagem do hemacitómetro Neubauer improved... 22
Figura 14 Representação da quantificação de células viáveis e em proliferação após tratamento com os fármacos 5-FU, oxaliplatina e irinotecano, para cada a linha celular SW480 e HT29... 35
Terapêutica no Cancro do Cólon e Recto IX Figura 15 Representação do efeito dos fármacos (5-FU, oxaliplatina e irinotecano) na viabilidade e proliferação para as linhas celulares SW480 e HT29. ... 37 Figura 16 Representação da quantificação da viabilidade celular em resposta ao tratamento com os fármacos 5-FU, oxaliplatina e com a combinação destes. ... 38 Figura 17 Representação da quantificação da viabilidade celular em resposta ao tratamento com os fármacos 5-FU, irinotecano e com a combinação destes. ... 39 Figura 18 Quantificação da expressão dos genes TCF7L2, p21 e HGF por PCR semi-quantitativo e utilizando o software ImageJ. ... 40 Figura 19 Transcrição induzida pelo TCF7L2 na linha SW480 e HT29. ... 41 Figura 20 Transcrição induzida pelo TCF7L2 na linha SW480 após indução da expressão das três formas truncantes do gene APC (1-1556, 1-1309 e 1-932). ... 42 Figura 21 Quantificação da expressão dos genes TCF7L2, p21 e HGF sem transfecção (não Transf.) e após transfecção das formas truncantes do APC na linha SW480, por PCR semi-quantitativo e utilizando o software ImageJ. ... 43 Figura 22 Representação da viabilidade celular após tratamento com os fármacos 5-FU, oxaliplatina e irinotecano nas células transfectadas com as formas truncantes do gene APC 1-1556, 1-1309 e 1-932. ... 44 Figura 23 Imagem da membrana de PET das placas de invasão após coloração com violeta de cristal das células invasivas.. ... 50 Figura 25 Representação do número de células com capacidade invasiva ou com capacidade de migração após transfecção com o inibidor do gene TCF7L2 e inibidor controlo... 51 Figura 24 Imagem da membrana de PET das placas de migração após coloração das células invasivas com violeta de cristal. ... 50 Figura 26 Quantificação da expressão dos genes TCF7L2 e HGF após transfecção do inibidor do
TCF7L2 e do inibidor controlo , por PCR semi-quantitativo e utilizando o software ImageJ ... 52
Figura 27 Quantificação da expressão dos genes TCF7L2, HGF e p21 por PCR semi-quantitativo nas células SW480 sem tratamento e após tratamento com os fármacos 5-FU, oxaliplatina e irinotecano. ... 53
Terapêutica no Cancro do Cólon e Recto X
Abreviaturas
5-FU 5-Fluorouracilo
-TrCP beta-transducin repeat containing
ABC transportadores ATP-binding site APA ácido carboxilico aminopentona
APC adenomatous polyosis coli
ATP adenosine trifosfato
BRAF v-raf murine sarcoma viral oncogene homolog B1
BrdU 5’ – bromo – 2’ - desoxiuridina
BSA Bovine serum albumin
Caspase cysteinyl aspartate-specific protease
CCR cancro do colon e recto
c-DNA DNA complementar
CES carboxilesterase
c-Myc v-myc myelocytomatosis viral oncogene homolog
CTNNB1 catenina (protein associada à caderina)
CYP citocromo P450
DEPC dietilpirocarbonato
DHFU monofosfato de fluorodesoxiuridina DMEM dulbecco’s modified eagle medium
DMSO dimetil sulfóxido
DPBS dulbecco’s phosphate buffered saline
DNA ácido desoxiribonucleico
dNTP desoxirobonucleósido de trifosfato DPD dihidropirimidina desidrogenase
Dsh Dishevelled
dTMP desoxitimidina de monofosfato dUMP desoxiuridina de monofosfato dUTP desoxiuridina de trifosfato
EB1 receptor do estrogénio
ERCC excision repair cross-complementing rodent repair deficiency
FBS fetal bovine serum
ELISA enzyme linked immuno sorbent assay
FOLFIRI combinação terapêutica dos fármacos 5-FU e irinotecano FOLFOX combinação terapêutica dos fármacos 5-FU e oxaliplatina FdUMP fluorodesoxiuridina de monofosfato
FdUTP fluorodesoxiuridina de trifosfato FUTP fluorouridina de trifosfato
Fz receptor Frizzled
GAPDH glyceraldehyde-3-phosphate dehydrogenase
GSK3 glycogen synthase kinase 3 beta
hDLG homólogo humano da proteína de Drosophila HNPCC hereditary nonpolyposis colorectal cancer
Terapêutica no Cancro do Cólon e Recto XI
HGF hepatocyte growth factor
IC50 concentração de fármaco necessária à redução para 50% de células
kDa kilodalton
KRAS gene homólogo do vírus do sarcoma de Kirsten LB meio de cultura Luria-Bertani
LBamp meio LB com ampicilina LOH perda de heterozigotia
LRP low-density lipoprotein receptor
MCR mutation cluster region
MET hepatocyte growth factor receptor
MLH1 primeiro gene humano homólogo do MUTL bacteriano
MMP7 metaloproteinase 7
MMP9 metaloproteina 9
MMR mismatch repair
mRNA RNA mensageiro
MSH2 segundo gene humano homólogo do MUTL bacteriano MSH6 sexto gene humano homólogo do MUTL bacteriano MSI instabilidade de microssatélites
MSS microssatélites estáveis
MUTYH gene homólogo do MUY bacteriano NER nuclear excision repair
NrCAM molécula de adesão de células neuronais
NPC 7-etil-10[4-(1-piperidino)-1-amino]-carboniloxicamptotecina p21 cyclin dependent kinase inhibitor 1A
p53 proteína 53
PAF polipose adenomatosa familiar
PAM polipose adenomatosa associada ao gene MUTYH
PCR polymerase chain reaction
PET tereftalato de polietireno
PFA paraformaldeído
pH simétrico do logaritmo decimal da concentração hifrogeniónica PMS2 human posmeiotic segregation 2
POD peroxidase
primer oligonucleótido iniciador
RNA ácido ribonucleico
RNases ribonucleases
RNAi RNA interferência
RT transcrição reversa
SL sindroma de Lynch
SLC31A1 solute carrier family 31 copper transporters
SN-38 composto activo do irinotecano (7-etil-10-hidroxicamptotecina) SN-38G SN-38 glucoronide
TBE tampão tris-borato-EDTA
TE tampão tris-EDTA
Terapêutica no Cancro do Cólon e Recto XII TCF3 T-cell factor 3
TCF7L2 transcription factor 7 like 2
TCF/LEF T-cell factor/Lymphoid enhancing factor
TS timidilato sintase
TP timidina fosforilase
TP53 tumor protein p53
UDG glicosilase de uracilo
UGT uridina difosfato glicosil transferase VEGF vascular endothelial growth factor
Wnt homólogo humano da proteína wingless na Drosophila
Wt wild type
% (p/v) percentagem expressa em peso por volume % (v/v) percentagem expressa em volume por volume
Terapêutica no Cancro do Cólon e Recto 1
1. Introdução
1.1. Cancro do cólon e recto
O cancro do cólon e recto (CCR) é uma entidade etiológica multifactorial e um dos maiores problemas oncológicos, sendo a terceira causa de morte mais frequente, relacionada com cancro, na população dos países ocidentais [1, 2]. A incidência de CCR é mais elevada na Europa, com aproximadamente 412 900 novos casos em 2006 [1], seguindo-se os Estados Unidos da América (EUA), com 148 810 novos casos em 2008. A frequência de CCR na Ásia, África e em zonas da América do Sul é baixa [3], o que apoia o papel de factores, como o estilo de vida e dieta, no desenvolvimento de CCR. Uma dieta rica em fibras e vegetais pode reduzir até 40% o risco de aparecimento de CCR, enquanto que, a ingestão de carnes vermelhas e o consumo de álcool podem aumentar o risco [3]. A actividade física também poderá estar associada à diminuição do risco de desenvolvimento de CCR, devido ao seu efeito no trânsito intestinal, no metabolismo dos ácidos biliares e no sistema imunitário [4].
Em Portugal, a taxa de incidência em 2006 foi de 58.9/100 000 (número de casos por 100 000 habitantes) no homem e de 30.9/100 000 na mulher e a taxa de mortalidade foi de 30.2/100 000 no homem e de 17.5/100 000 na mulher [1].
Os casos de CCR podem ser divididos em dois grupos principais, os de origem esporádica e os de origem familiar. Os primeiros, apresentam desenvolvimento de CCR tardio, sem história familiar e podem estar associados aos factores ambientais descritos anteriormente. Pelo contrário, os casos de CCR familiar têm um risco de desenvolvimento associado a factores genéticos, existindo história familiar. Estes representam cerca de 20-25% dos casos de CCR. Dentro dos casos familiares estão incluídos os sindromas hereditários de elevada penetrância como a polipose adenomatosa familiar (PAF), causada por mutações germinais no gene APC, a polipose associada ao gene MUTYH (PAM) e o cancro do cólon e recto hereditário não associado a polipose (HNPCC- hereditary nonpolyposis colorectal cancer), onde se insere o sindroma de Lynch (SL), causado por mutações germinais nos genes de reparação do DNA do tipo mismatch como o MSH2, MLH1, MSH6 e PMS2. Apesar de já existir um enorme conhecimento molecular em relação a estes sindromas hereditários, estes representam apenas 5% dos casos de CCR familiar, o que indica que a maioria dos factores genéticos que predispõem para o desenvolvimento de CCR não são ainda conhecidos e muito permanece por identificar [5].
Como na maioria dos tumores sólidos, o CCR é classificado com base no estado de invasão do local primário e pela presença de metástases nos nódulos linfáticos e órgãos distantes. Os dois sistemas de classificação mais comuns são o Dukes e o TNM. No primeiro sistema, os tumores são classificados de Dukes A, quando o tumor invade apenas a mucosa do epitélio cólico; de estadio Dukes B quando o tumor invade o músculo; de estadio Dukes C quando o tumor atinge pelo menos um dos nódulos linfáticos e por fim, de Dukes D quando existem metástases do tumor. A classificação TNM descreve o tamanho do tumor (T), a presença de células cancerígenas nos nódulos linfáticos (N) e a migração do cancro para outras zonas do corpo (metástases) (M). Assim, o tumor pode ser considerado T1, quando o tumor atinge apenas a membrana interna do epitélio cólico; de T2, quando o tumor atinge o músculo;
Terapêutica no Cancro do Cólon e Recto 2 T3, quando invade a membrana externa do epitélio cólico ou atinge órgãos ou estruturas próximas do intestino; ou de T4, quando o tumor cresce para outros órgão do corpo (presença de metástases). O estadio N é classificado de N0, que significa que não existem células cancerígenas em nenhum nódulo linfático; N1, quando existem 1 a 3 nódulos, próximos do intestino, com presença de células cancerígenas e de N2, quando há presença de células cancerígenas em mais de quatro nódulos linfáticos com células cancerígenas. Por fim, a classificação M caracteriza os tumores aos quais estão associados a presença (M1) ou ausência (M0) de metástases noutros órgãos [6].
1.1.1. Sequência adenoma-carcinoma
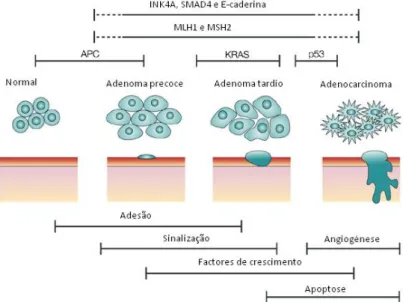
Em 1990, o estudo da carcinogénese colorectal registou um grande avanço através da publicação de Fearon e Vogelstein que propuseram um modelo genético e histológico capaz de explicar a evolução das células do epitélio cólico para carcinoma. Assim, a evolução das células do epitélio cólico normal para carcinoma segue, normalmente, um modelo de progressão com múltiplos passos definidos por estadios histológicos e alterações genéticas e epigenéticas características. Molecularmente, esta sequência, designada por adenoma – carcinoma (Figura 1), é iniciada com uma alteração genética no gene APC. Alternativamente, em tumores sem mutação no APC podem ocorrer mutações nos genes CTNNB1 ou AXIN2 (genes que tal como o
APC, pertencem à via de sinalização Wnt). A progressão de adenoma precoce para adenoma
tardio ocorre com a alteração da via de sinalização RAS-RAF-MAPK, responsável pela modulação do crescimento e sobrevivência celular. Em cerca de 50% dos casos de CCR, esta alteração tem origem na activação do oncogene KRAS e em cerca de 20% dos casos ocorre com activação do gene BRAF. Por fim, a progressão para carcinoma surge com a inactivação bi-alélica do gene TP53 que codifica para o factor de transcrição p53, responsável pela inibição do crescimento celular e pela activação da morte celular quando é induzido stress celular [7].
No decorrer do desenvolvimento tumoral, diversos processos celulares são alterados como a adesão celular, sinalização, angiogénese, factores de crescimento e apoptose. No final, as células apresentam as características necessárias à formação do tumor, adquirindo um crescimento desregulado [8].
Figura 1 Eventos moleculares que caracterizam a transição do epitélio cólico normal a adenocarcinoma. (Adaptado de [8])
Terapêutica no Cancro do Cólon e Recto 3
1.1.2. Instabilidade genética
A taxa de mutação numa célula normal não deverá ser suficiente para permitir a acumulação das alterações genéticas necessárias ao desenvolvimento do tumor. Assim, será necessária a aquisição de uma forma de instabilidade genética, para que possa ocorrer a iniciação e progressão tumoral. Em geral, são observadas duas formas de instabilidade genética no CCR: instabilidade de microssatélites (MSI – microsatellite instability) e instabilidade cromossómica (CIN – chromosomal instability). A MSI é observada em aproximadamente 15% dos casos de CCR e resulta da perda da capacidade de reparação de erros no DNA do tipo mismatch (MMR) pelos genes deste sistema. Estes genes são responsáveis pela reparação de inserções, delecções ou emparelhamento incorrecto de bases que podem ocorrer durante a replicação ou recombinação. As sequências de microssatélites representam uma pequena parte do genoma humano e correspondem a pequenas sequências repetitivas, normalmente de 1 a 6 nucleótidos. Estas são regiões extremamente informativas quando os genes do sistema MMR se encontram mutados, uma vez que, na falta destes, as sequências de microssatélites são alteradas. Assim, definiram-se marcadores de moleculares que permitem avaliar a instabilidade de microssatélites (MSI) em determinadas regiões do genoma, permitindo definir o tumor como estável (MSS) ou instável. A este conjunto de marcadores de microssatélites deu-se o nome de marcadores de Bethesda [4, 7].
A CIN caracteriza-se pela ocorrência de perdas ou ganhos nos cromossomas, total ou parcialmente, originando alterações estruturais e numéricas referidas como aneuploidia. Esta forma de instabilidade verifica-se em cerca de 75-85% dos casos de CCR e origina a desregulação de diferentes genes responsáveis pelo controlo e assistência na formação do fuso mitótico; pelo checkpoint do ciclo celular; pela expressão do telomerase e encurtamento dos telómeros; pela regulação do número de centrossomas e reparação de quebras na cadeia dupla de DNA [4, 7, 9]. Os marcadores moleculares da instabilidade de microssatélites também são bastante informativos deste tipo de instabilidade, pois permitem aferir quanto à perda de heterozigotia (LOH) de um determinado gene, quando são estudadas sequências de microssatélites flanqueadoras ou próximas do gene em estudo.
1.2. Via de sinalização Wnt e o CCR
A via de sinalização Wnt, também denominada de via de sinalização APC/ -catenina/TCF7L2, encontra-se desregulada na maioria dos casos de CCR. O termo Wnt deriva da fusão dos nomes de dois genes ortólogos: Wingless, gene de um segmento de polaridade da Drosophila, e Int-1, um proto-oncogene de rato. Wnts são uma família de glicoproteinas com papel na especificação do destino celular, padronização do sistema nervoso central e divisão celular assimétrica. A expressão destas proteínas é regulada ao longo do desenvolvimento de uma forma temporal e espacial [10].
A sinalização intracelular da via Wnt manifesta-se em pelo menos três formas: a via canónica, ou via APC/-catenina; a via de polaridade celular-planar e a via Wnt/Ca2+. A via canónica (Figura 2) é a mais estudada e fortemente implicada no desenvolvimento de CCR.
Terapêutica no Cancro do Cólon e Recto 4 Figura 2 Representação esquemática da via de sinalização Wnt canónica na forma inactiva (A) e na forma activa (B). Na ausência de sinalização Wnt a -catenina é marcada para degradação (-catenina degradation) pelo complexo formado pela -catenina, APC, axina, GSK3- e Dsh. Na sua forma activa verifica-se a ligação das proteínas Wnt ao receptor Frizzled, ocorre a destabilização do complexo de degradação da -catenina, o que possibilita a sua translocação para o núcleo, onde juntamente com os factores de transcrição TCF/LEF promove a activação de genes alvo da via Wnt (Wnt target genes) (Adaptado de [11])
Na ausência das proteínas Wnt (forma inactiva da via Wnt), a -catenina é marcada para degradação por um complexo multiproteico formado pela axina, APC, GSK3- e Dsh. A axina terá a função de estabilizar estruturalmente o complexo mantendo-o ligado e, em conjunto com o APC, facilita a eficiência da fosforilação da -catenina nos quatro resíduos N-terminal, pelo GSK3-. A fosforilação marca a -catenina para ligação à proteína -TrCP e subsequente ubiquitinação e degradação no proteossoma. Ao ser degradada não ocorre a sua translocação para o núcleo e consequente activação dos genes alvo dos factores de transcrição TCF/LEF (T-cell factor/lymphoid enhancing factor), para os quais a -catenina é co-factor (Figura 2A) [12].
A sinalização, na sua forma activa, inicia-se pela ancoragem do ligando Wnt ao receptor Frizzled (Fz) e co-receptores LRP-5 e LRP-6, membros da família de receptores de lipoproteinas de baixa densidade (LRP- low-density lipoprotein receptor). Uma vez formado o complexo ligando-receptor, a proteína axina é translocada até à membrana e interage com a extremidade intracelular do LRP-5, o que destabiliza o complexo de degradação. Na ausência deste é favorecida a acumulação da -catenina no citoplasma que, posteriormente, é translocada para o núcleo, onde se irá ligar aos factores de trancrição TCF/LEF, activando os genes alvo da via Wnt envolvidos em vários processos celulares como a apoptose, proliferação e diferenciação (Figura 2B) [13-15].
A identificação dos genes alvo dos factores de transcrição TCF/LEF, pode explicar o papel da via de sinalização Wnt no cancro. De entre estes, destacam-se os reguladores da progressão do ciclo celular, como o c-Myc, que ao inibir a expressão génica do p21 activa o complexo ciclina/Cdk e permite a progressão do ciclo celular da fase G1 para a fase S, e a
Terapêutica no Cancro do Cólon e Recto 5 proteases capazes de degradar a matriz extracelular, como o matrilisina/MMP7 e MMP-26; e as moléculas de adesão celular como o CD44 e NrCAM, que parecem implicar a via Wnt na regulação da invasão e metastização
.
A via de sinalização Wnt regula também a proliferação celular, pela indução da expressão de factores de crescimento e seus receptores, como o tirosina cinase MET e a apoptose pela sobre-regulação de proteínas anti-apoptóticas, como o inibidor das caspases, survivina. A sobre-regulação do VEGF (vascular endothelial growthfactor), confere-lhe a capacidade de estimular a angiogénese [16].
Alterações na via de sinalização Wnt originam a estabilização anormal da -catenina o que leva à activação constitutiva da via de sinalização APC/-catenina/TCF7L2 [17]. De entre as alterações genéticas encontradas nos casos de CCR, são destacadas as do gene APC, -catenina e factores de transcrição TCF/LEF.
1.2.1. Gene APC
As mutações germinais no gene APC são responsáveis pelo desenvolvimento da PAF e, na maioria (85%) dos casos esporádicos de CCR são detectadas mutações somáticas neste gene. Este é um gene que está implicado no desenvolvimento do epitélio normal em adenoma precoce na sequência adenoma - carcinoma. Este gene codifica para uma proteína de 2843 aminoácidos, responsável por manter a renovação do tecido normal e assegurar o aumento de proliferação em caso de danos no tecido, sendo considerado o gatekeeper do epitélio cólico. Apesar disso, a principal função de supressor tumoral deste gene aparenta estar relacionada com a regulação dos níveis de -catenina intracelular [18].
A proteína APC (Figura 3) é estruturalmente composta por diversos domínios que permitem a interacção com várias proteínas. A região N-terminal é formada por um domínio de oligomerização e uma região Armadillo. Na zona central, encontram-se repetições de 15 e 20 aminoácidos e locais de ligação à axina, que contêm na sua estrutura a sequência de aminoácidos característica SAMP. A região C-terminal é composta pelo domínio básico e locais de ligação às proteínas EB1 e hDLG (human disc large) [19].
O domínio de oligomerização permite a formação de dímeros entre proteínas APC, tanto wild type (wt) como proteína truncada. Este é um processo que poderá ser responsável pelo efeito dominante negativo da proteína truncada, ou seja, esta pode-se associar à proteína wt interferindo com a regulação da -catenina. A região Armadillo liga-se à proteína fosfatase 2A (PP2A), enzima que também se liga à axina. Na região central da proteína existem três repetições de 15 aminoácidos e sete repetições de 20 aminoácidos, as quais são responsáveis pela ligação e regulação da -catenina, respectivamente. O domínio básico, assim denominado devido à sua composição em resíduos de aminoácidos básicos (arginina e lisina), tem a função de promover a interacção entre a proteína APC e a tubulina. Na região terminal estão localizados ainda, os domínios de ligação hDLG e EB1, o último dos quais tem uma função bastante importante na segregação cromossómica [19].
A grande maioria das mutações neste gene são nonsense ou frameshift (tipicamente inserções ou delecções) e originam proteínas truncadas com um número variável de repetições de domínios de regulação e ligação da -catenina. A maioria das mutações somáticas (60%) no gene APC ocorrem entre os codões 1286 e o 1513, região esta denominada de mutation cluster region (MCR) e resultam numa proteína truncada sem locais de ligação à axina, e com apenas um ou dois domínios de regulação da -catenina [19-21].
Terapêutica no Cancro do Cólon e Recto 6 Figura 3 Representação da estrutura da proteína APC e seus domínios funcionais. Os asteriscos a rosa representam os domínios de ligação à axina. (Adaptado de [19])
A delecção de domínios de ligação e de regulação da -catenina impede a regulação negativa dos níveis de -catenina, o que resulta na activação da via Wnt. No entanto, encontra-se demonstrado que proteínas truncadas que apresentam três domínios de regulação da -catenina ainda apresentam alguma capacidade de regulação negativa da -catenina. O mesmo tem vindo a ser demonstrado para proteínas truncadas com um ou dois domínios de regulação, apesar da capacidade de regulação ser ainda menor. Na Figura 4 encontra-se representada a actividade transcripcional regulada pelo factor de transcrição TCF7L2, a qual é traduzida na actividade da via de sinalização Wnt, em diferentes linhas celulares de CCR com diferentes mutações no gene APC, as quais originam a expressão de um número diferente de domínios de regulação da -catenina. Observa-se que, quanto menor o número de domínios de regulação da -catenina no gene APC, maior a actividade de sinalização Wnt, devido à menor regulação e consequente degradação da -catenina [22].
Figura 4 Representação da actividade transcripcional regulada pelo TCF7L2 em linhas celulares de CCR que expressam proteínas truncadas com diferentes números de domínios de regulação da -catenina (adaptado de[22]).
Terapêutica no Cancro do Cólon e Recto 7 Sendo o gene APC um supressor tumoral, segundo a hipótese de Knudson, são necessários dois eventos genéticos para haver a perda de função deste [23]. No entanto, foi demonstrado que o gene APC não parece seguir completamente este modelo, uma vez que, existe interdependência entre os dois eventos. Assim, nos pólipos de PAF a mutação germinal determina a posição e o tipo do segundo evento genético, relação esta, que também é observada entre as duas mutações somáticas nos casos esporádicos de CCR. Deste modo, foi proposto um modelo – modelo just right signaling – que explica esta interdependência entre os dois eventos no gene APC, o qual se baseia na combinação entre a mutação germinal nos indivíduos com PAF a as mutações somáticas detectadas nos respectivos adenomas [24].
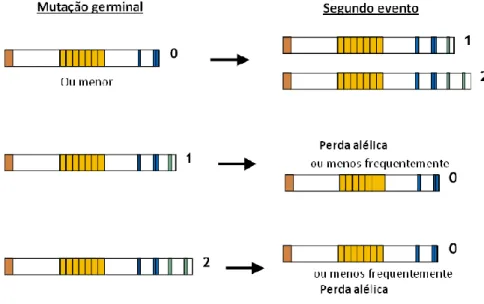
Tendo em conta o número de domínios de regulação da -catenina resultantes da mutação germinal, o segundo evento genético pode originar três cenários diferentes (Figura 5). Quando a mutação germinal resulta numa proteína truncada sem domínios de regulação da
-catenina, o segundo evento tende a originar uma proteína truncada com um, ou menos frequentemente, com dois domínios de regulação. Caso o primeiro evento genético leve à formação de uma proteína truncada com apenas um domínio de regulação, a perda da proteína wt, traduzida pela perda de heterozigotia, tende a ser o segundo evento observado. Por fim, quando a mutação germinal resulta numa proteína que apresenta pelo menos dois domínios de regulação da -catenina, o segundo evento tende a ser uma mutação truncante que origina uma proteína APC sem domínios de regulação ou então, menos frequentemente, a perda de heterozigotia [24].
Figura 5 Diagrama ilustrativo da interdependência entre os dois eventos genéticos necessários à perda de função do gene APC, de acordo com o número de domínios de regulação da -catenina, (a vermelho encontra-se representado o domínio de oligomerização, a amarelo as repetições armadillo, a azul as repetições de 15 aminoácidos e a verde as repetições de 20 aminoácidos)[24].
1.2.2.
-Catenina
O gene que codifica para a proteína -catenina (CTNNB1) encontra-se mutado em cerca de 10% dos casos esporádicos de CCR e em aproximadamente 30% dos CCR no contexto
Terapêutica no Cancro do Cólon e Recto 8 de sindroma de Lynch [25]. A maioria das mutações neste gene ocorre nos resíduos de serina e treonina, e aminoácidos adjacentes a estes, da região N-terminal, que são essenciais à marcação para degradação através da fosforilação. Estas mutações anulam a interacção entre a -catenina e o -TRCP, componente de um E3 ubiquitina ligase (componente do proteossoma) [13]. Na ausência de degradação da -catenina, esta acumula-se no núcleo, havendo a activação anormal dos genes alvo dos factores de transcrição TCF/LEF, como o
c-Myc e ciclina D1. Alterações na expressão normal destes genes levam a um aumento da taxa
de proliferação, o que favorece a carcinogénese [26, 27].
1.2.3. Factores de transcrição TCF/LEF
A família de factores de transcrição TCF/LEF é composta por quatro membros: TCF1, LEF1, TCF3 e TCF4 (ou TCF7L2). Estes, constituem uma grande família de reguladores transcripcionais que contêm o domínio HMG (high mobility group) de ligação ao DNA bastante conservado. Os TCF são os mais estudados como efectores da sinalização Wnt, nomeadamente o TCF7L2. O gene que codifica para a proteína TCF7L2 (esquematizada na Figura 6) é composto por 17 exões. A proteína é formada por diversos domínios, entre eles o domínio de ligação à -catenina, localizado na região N-terminal, os domínios de ligação aos co-repressores Groucho
related gene (Grg) e Carboxy terminal binding protein (CtBP), o domínio HMG de ligação ao
DNA, o local de sinalização nuclear (NLS) e o domínio CRARF (C- cisteína; R- arginina; A- alanina; F- fenilalanina) (também denominado de domínio C-Clamp) que tem a função de auxiliar a ligação ao DNA [28].
Figura 6 Representação esquemática da proteína TCF7L2. Estão indicados os domínios de ligação à -catenina (-cat), aos co-repressores Grg e CtBP, a HMG-box e o NLS. O motivo CRARF também se encontra representado a vermelho (motivo C-clamp) (adaptado de [29]). O gene TCF7L2 tem diversos locais sujeitos a splicing alternativo que podem originar diversas isoformas da proteína. Estas isoformas podem agrupar-se em quatro grupos: a isoforma TCF4E que contém todos os elementos representados na Figura 5; a TCF4N, composta apenas pelo local de ligação à -catenina, não contendo por isso o domínio HMG e todos os elementos localizados a jusante; a TCF4M que não contém o local de ligação à CtBP e pode ter ou não o motivo C-clamp; e por fim, a isoforma TCF4S que é igual à TCF4M com excepção do motivo C-clamp que está incompleto [27, 30].
Na presença de -catenina no núcleo dá-se a formação do complexo -catenina:TCF7L2 e como resultado, o co-repressor transcripcional GrG é deslocado do seu domínio de ligação e os co-activadores são recrutados, levando assim, à activação dos genes alvo da via de sinalização Wnt [27, 28, 30].
Os tumores colorectais com instabilidade de microssatélites apresentam mutações no gene TCF7L2. Estas caracterizam-se, maioritariamente, pela deleção de um par de bases na repetição de nove adeninas localizada na região 3´ do gene. Estas são mutações frameshift que aparentam afectar a proporção das diferentes variantes do TCF7L2. As mutações neste gene, existem na ausência, e também na presença, de mutações no APC ou CTNNB1, o que indica
Terapêutica no Cancro do Cólon e Recto 9 que mutações no TCF7L2 não substituem as mutações APC/-catenina, mas podem actuar de uma forma aditiva no desenvolvimento do cancro [31].
Os estudos quanto à função do TCF7L2 no desenvolvimento tumoral têm sido controversos, uma vez que, foram descritas as funções de supressor tumoral, assim como, de oncogene. Estudos realizados em ratinhos descrevem que a deficiência do gene TCF7L2 resulta na morte pré-natal, devido à ausência do compartimento de proliferação celular no epitélio intestinal [32]. A expressão de uma forma dominante negativa do TCF1 ou TCF7L2 induz a paragem do ciclo celular em linhas celulares de CCR. Estas observações estão de acordo com a função oncogénica do TCF7L2 na cascata de sinalização da via Wnt. No entanto, outros estudos descrevem que, a isoforma TCF4E tem propriedades únicas de transactivação dos genes AXIN2 e Cdx1, que contribuem para a supressão tumoral [33-35].
Assim, e após o desenvolvimento de técnicas mais sensíveis na determinação da actividade transcripcional regulada pelo TCF7L2, as funções opostas de manutenção da proliferação celular e supressão tumoral foram clarificadas por Hecht A. et al [27, 30], que verificou que as diferentes isoformas do gene TCF7L2 regulam diferentes grupos de genes, podendo actuar como activadores ou como repressores.
Recentemente [36], foi observado que a inibição do gene TCF7L2 induz o aumento de expressão do HGF (hepatocyte growth factor). Estes resultados, aliados ao facto de a via de sinalização HGF/MET ter a função de regulação da invasão e metastização tumoral, apoiam a hipótese de o TCF7L2 estar associado à supressão tumoral, por inibição da metastização e invasão.
1.3. O papel da via de sinalização HGF/MET na invasão e
metastização
O crescimento do tumor pode ser acompanhado do desenvolvimento de metástases (migração das células neoplásicas para órgãos adjacentes) que resistem às terapêuticas convencionais, sendo esta a maior causa de morte por cancro.
O tirosina cinase MET é o receptor do HGF e a formação do complexo ligando:receptor activa diversas vias de sinalização intracelular que estão envolvidas em vários processos celulares, nomeadamente, aumento da invasão e metastização. Alguns estudos realizados mostram a associação entre a elevada expressão do MET e HGF e a capacidade do tumor de desenvolver metástases. Por exemplo, a implantação de células com sobre-expressão do HGF ou MET, em ratinhos com o sistema imunitário deficiente (nude mice), leva à formação de tumores que progridem no sentido do desenvolvimento de metástases. Resultados semelhantes foram obtidos num estudo em linhas celulares de cancro do pulmão, onde a inibição do MET induziu significativamente a inibição do crescimento celular. De facto, a sobre-expressão do MET encontra-se descrita em vários tumores sólidos [37].
Assim, o MET e o seu ligando HGF parecem ser responsáveis por iniciar o processo de metastização em alguns tipos de cancro, o que os torna por excelência num alvo para o desenvolvimento de terapêuticas anti-cancerígenas. As estratégias a adoptar, como possíveis terapêuticas, são: moléculas antagonistas do HGF, que previnem a ligação deste ao receptor; inibidores dos cinases e moléculas competitivas na ligação ao receptor [37].
Terapêutica no Cancro do Cólon e Recto 10 Tendo em conta o aumento de expressão do HGF em resultado da inibição do gene
TCF7L2, como foi demonstrado recentemente [36], é importante o estudo do papel deste
último, no desenvolvimento de metástases.
As mutações no gene TCF7L2 parecem associar-se a pior sobrevivência dos doentes com CCR, mas não ao desenvolvimento de metástases. Por outro lado, a LOH do gene TCF7L2, que é um mecanismo genético de perda de função, encontra-se em alguns carcinomas (resultados obtidos pelo grupo de Gastrenterologia do CIPM). No entanto, a associação da LOH deste gene com o desenvolvimento tumoral ainda não está definida. A correlação, recentemente descrita [36], entre a inibição do TCF7L2 e a sobre-expressão do HGF, torna a LOH num possível mecanismo genético que pode contribuir para a invasão e metastização tumoral.
1.4. Terapêutica no CCR
O aparecimento de tratamentos mais eficazes, e o diagnóstico precoce do CCR, têm contribuído para o aumento da sobrevivência ao longo das últimas décadas. No entanto, mais de 50% dos novos casos diagnosticados irão, eventualmente, progredir no desenvolvimento de metástases, e morrer da doença. Por isso, é importante o aparecimento de terapêuticas eficazes no tratamento de CCR, assim como, a identificação de marcadores de prognóstico e preditivos da resposta à terapêutica [38].
Até 1985, o 5-FU era o único fármaco disponível no tratamento de CCR. O tempo médio de sobrevivência era de 9-11 meses tendo, contudo, aumentado a partir do ano 2000 sendo agora superior a 16 meses, com a combinação do 5-FU com novos fármacos como a oxaliplatina (FOLFOX) e o irinotecano (FOLFIRI). Estas combinações são ainda hoje as mais utilizadas no tratamento do CCR [39]. A terapêutica no CCR apresenta duas abordagens, o tratamento paliativo e o ajduvante. A primeira consiste no tratamento do doente após a cirurgia e a segunda abordagem é realizada em cancro metastático de forma não curativa.
1.4.1. 5-Fluorouracilo (5-FU)
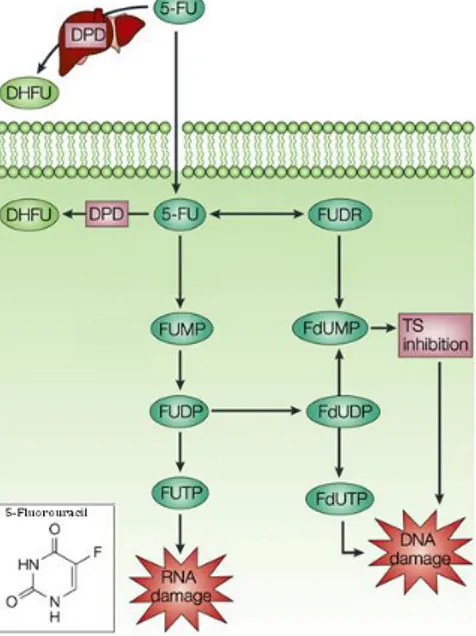
O fluoropirimidina 5-FU, é um análogo das moléculas de pirimidina do DNA e RNA e é um dos fármacos utilizados no tratamento dos tumores colorectais, da mama e do tracto digestivo. O seu mecanismo de acção consiste na inibição do timidilato sintase (TS) e incorporação no DNA e RNA. O passo limitante do catabolismo do 5-FU é controlado pelo enzima dihidropirimidina desidrogenase (DPD), que é responsável pela catálise da reacção de conversão do 5-FU em dihidrofluorouracilo (DHFU) (Figura 4). Mais de 80% do 5-FU administrado é normalmente catabolisado no fígado, onde o DPD é expresso abundantemente [40].
Na célula, o 5-FU é convertido em três metabolitos com efeito farmacodinâmico, o fluorodesoxiuridina monofosfato (FdUMP), responsável pela inibição do TS; o fluorodesoxiuridina trifosfato (FdUTP) e o fluorouridina trifosfato (FUTP) (Figura 7).
O TS catalisa a reacção de metilação do dUMP em dTMP, sendo o folato (5,10-metilenotetrahidrofolato – CH2THF) o dador do grupo metilo. Esta é uma proteína dimérica de 36kDa, em que cada subunidade contém um local de ligação ao nucleótido e outro de ligação ao folato. O metabolito do 5-FU, FdUMP, liga-se ao local de ligação ao nucleótido do TS formando um complexo ternário estável com o enzima e o folato. A formação deste complexo impede a ligação do nucleótido ao TS, o que inibe a síntese do dTMP resultando num
Terapêutica no Cancro do Cólon e Recto 11 desequilíbrio das razões de desoxinucleótidos e consequentemente em defeitos na síntese e na reparação do DNA e RNA. Os níveis de dUTP, em resultado da inibição da síntese do dTMP, aumentam, e assim como o metabolito FdUTP, podem ser incorrectamente incorporados no DNA. Este tipo de erros poderá ser reparado pelo enzima de excisão de nucleótidos, DNA glicosilase de uracilo (UDG) o que origina ciclos de incorporação incorrecta, excisão e reparação que poderão levar a quebras na cadeia de DNA. O metabolito FUTP é extensivamente incorporado no RNA, causando um mau processamento e função deste, o que origina efeitos severos no metabolismo celular e viabilidade [40, 41].
Figura 7 Esquema do metabolismo do 5-FU. Os três principais metabolitos activos do 5-FU são o FdUMP, o FdUTP e o FUTP. O primeiro actua pela inibição do timidilato sintase (TS), enquanto que os outros dois actuam pela incorporação incorrecta no DNA e RNA, respectivamente. Estão representados os metabolitos 5-FU (5-Fluorouracilo), dihidropirimidina desidrogenase (DPD), dihidrofluorouracilo (DHFU), fluorodesoxiuridina (FUDR), fluorodesoxiuridina monofosfato (FdUMP), fluorodesoxiuridina difosfato (FdUDP), fluorodesoxiuridina trifosfato (FdUTP), fluorouridina monofosfato (FUMP), fluorouridina difosfato (FUDP), fluorouridina trifosfato (FUTP). (Adaptado de [40]).
1.4.2. Oxaliplatina
A oxaliplatina é um fármaco derivado de platina, da mesma família que a cisplatina e a carboplatina. Este fármaco é indicado para o tratamento de carcinoma avançado do cólon ou recto e no tratamento adjuvante de doentes em estadio Dukes C de CCR com recessão completa do tumor primário. Normalmente, é utilizado em combinação com o 5-FU [42].
O mecanismo de acção da oxaliplatina consiste na formação de aductos de platina na cadeia de DNA (Figura 8) o que origina ligações inter e intra-cadeia que inibem a transcrição e a replicação do DNA. O influxo do composto activo do fármaco para a célula é regulado pelo transportador solute carrier family 31 copper transporters (SLC31A1) e o seu efluxo pelos transportadores ATP-binding cassete (ABCC2 e ABCG2). A formação dos aductos de platina no DNA provoca a paragem do ciclo celular e, posteriormente, a apoptose. Por isso, alterações nos genes envolvidos no sistema MMR, tais como o MSH6 e MLH1, uma vez que induzem a
Terapêutica no Cancro do Cólon e Recto 12 resistência à apoptose, diminuem a sensibilidade da célula ao fármaco. Para além destes, alterações nos genes envolvidos no mecanismo de reparação do DNA por excisão de nucleótidos (NER- nucleotide excision repair) afectam a resposta do doente ao tratamento com oxaliplatina. Exemplo disso, são os enzimas ERCC1 e ERCC2, componentes essenciais da via de reparação NER, cujos níveis de expressão aparentam estar inversamente relacionados com a resposta à terapêutica nos cancros colorectal e gástrico [38, 43, 44].
Figura 8 Representação da formação dos aductos de platina no DNA pela oxaliplatina (Adaptado de [44]).
1.4.3. Ininotecano
O irinotecano é um composto análogo à camptotecina que inibe o topoisomerase I, o que provoca quebras na dupla cadeia de DNA e a consequente morte celular, especificamente, na fase S do ciclo celular. O fármaco apresenta resultados satisfatórios no tratamento dos cancros: colorectal metastático, esófago, pulmão, leucemia, linfoma e gliomas malignos do sistema nervoso central. No caso do tratamento de CCR é utilizado em combinação com o 5-FU (FOLFIRI) [45].
O irinotecano é convertido na sua forma activa SN-38 (7-etil-10-hidroxicamptotecina), tanto no interior como no exterior da célula, pelo carboxilesterase (CES). Ambas as formas do fármaco, irinotecano e SN-38, são transportadas para o interior e exterior da célula (Figura 9A) pelas proteínas ABC (ATP-binding cassete), como a ABCB, ABCC e ABCG. Quando se encontra no interior da célula, na sua forma activa, o irinotecano tem a função de inibir o topoisomerase I, o que pode resultar em dois tipos de dano no DNA, na cadeia simples e na cadeia dupla. No primeiro caso, os danos não são tóxicos, uma vez que, são reversíveis e rapidamente reparados assim que o fármaco é removido. Os danos na cadeia dupla, pelo contrário, são letais e irreversíveis e, ocorrem quando, no processo de síntese de DNA, o complexo topoisomerase I – DNA – irinotecano encontra o garfo replicativo (Figura 9B). A citotoxicidade do irinotecano é específica da fase S do ciclo celular, uma vez que é nesta fase que ocorre a duplicação do conteúdo genómico. Alternativamente, o irinotecano e o SN-38 podem ser inactivados pelos enzimas do citocromo P450 (CYP) ou pelo uridina difosfato glicosiltransferase (UGT), respectivamente. As formas inactivas são, no caso do irinotecano, o APA (ácido carboxílico aminopentona) e o NPC (7-etil-10[4-(1-piperidino)-1-amino]-carboniloxicamptotecina), e o SN-38 é convertido em SN-38G (SN-38 glucuronide)(Figura 9B) [38, 45, 46].
Terapêutica no Cancro do Cólon e Recto 13 Figura 9 Representação esquemática dos mecanismos de acção do irinotecano. A-Metabolismo do irinotecno. De notar os componentes representados: irinotecano (CPT-1), forma activa do irinotecano (SN-38), carboxilesterase (CES), proteínas ABC (ATP-binding
cassete) (ABCB, ABCG, ABCG), topoisomerase I (TOPI), uridina difosfato glicosiltransferase
(UGT1A1). B- danos provocados no DNA pelo complexo SN-38 – topoisomerase I (Adaptado de [47] e [48]).
1.5. Resistência à quimioterapia
A quimioterapia é utilizada como forma de tratamento de diversos cancros, no entanto, em muitos casos, a eficiência do tratamento não é a desejada, devido ao aparecimento de resistência por parte do tumor. Este é um processo que pode ser intrínseco, quando já existe antes da administração do tratamento, ou adquirido, quando se desenvolve durante o tratamento. Os mecanismos intracelulares de resistência são vários, entre eles as alterações no influxo e efluxo do fármaco, que originam diferenças na concentração intracelular deste; alterações no metabolismo do fármaco, o que pode interferir com a quantidade activa do fármaco no interior da célula; alterações no alvo do fármaco, que podem ser modificações na expressão ou mutações; modificação da capacidade das células tumorais para a reparação de erros no DNA, que pode ser um mecanismo de resistência quando são utilizados fármacos que induzem danos no DNA e por fim, a alteração na regulação da morte celular, que pode fazer com que a indução da morte celular pelo fármaco utilizado não seja bem sucedida [49].
Com a identificação de marcadores de resposta à terapêutica, é possível prever quais os doentes que são intrinsecamente resistentes e os que reagem melhor a determinado fármaco. Assim, pode ser aumentado o número de casos que beneficiam dos tratamentos existentes, uma vez que se pode saber à partida quais os fármacos mais adequados para um determinado perfil/doente. No entanto, esta é uma área pouco estudada e alguns dos marcadores identificados tornam-se, por vezes, dúbios dada a incoerência entre a comunidade científica. Alguns dos marcadores estudados, embora ainda não utilizados como marcadores clínicos estão resumidos na Tabela 1 [38, 46].
Terapêutica no Cancro do Cólon e Recto 14 Tabela 1 Marcadores preditivos de resposta ao tratamento com 5-FU, oxaliplatina e irinotecano em desenvolvimento (Adaptado de [46]).
Fármaco Marcador Comentário
5-FU
TS A sobre-expressão do gene TS
está associada à resistência. TP
Níveis reduzidos de expressão do TP estão associados à resposta e sobrevivência dos doentes.
Oxaliplatina ERCC1
A elevada expressão do ERCC1 e TS são marcadores preditivo de baixa resposta nos doentes tratados com FOLFOX.
Irinotecano Topo I
Poderá existir uma relação positiva entre a actividade do Topo I e a sensibilidade celular ao tratamento com irinotecano, mas ainda não foi provado.
Legenda: 5-FU, 5-Fluorouracil; TS, timidilato sintase; TP, timidina fosforilase; DPD, dihidropirimidina desidrogenase;
TopoI, topoisomerase I.
Existem outras alterações moleculares que, apesar de não se utilizarem ainda como marcadores de resposta ao tratamento, por não ser consensual a sua utilização, têm sido associadas a uma maior resistência à terapêutica. Entre alguns exemplos encontram-se a MSI e a expressão de p21.
1.5.1. A instabilidade de microssatélites
A instabilidade de microssatélites é um marcador de prognóstico já identificado. A presença deste tipo de fenótipo está associada a melhor prognóstico, a maior sobrevivência e menor recidiva do tumor [46]. No entanto, a utilização de quimioterapia em doentes com tumores MSI tem sido controversa. Alguns estudos têm demonstrado que um número significativo destes doentes não parece beneficiar da terapêutica convencional ao contrário do que acontece com os tumores MSS [50, 51]. Uma explicação para este facto parece residir na resistência à apoptose, a qual é uma das consequências da deficiência no sistema de reparação de erros no DNA MMR. Por isso, quando são analisadas as respostas ao tratamento com determinado fármaco, é necessário ter em conta que os doentes com tumores MSI poderão responder pior que os que apresentam tumores MSS, e dever-se-á separar os dois tipos de tumores.
1.5.2. A expressão do p21
O p21 é uma das proteínas reguladas pela via de sinalização Wnt, uma vez que, é regulada negativamente pela proteína c-Myc, que é um dos alvos principais desta via [52]. Por isso, uma maior activação da via Wnt induz a diminuição da expressão do p21. Esta é uma proteína de 21kDa que pertence à família Cip/Kip de inibidores de cinase dependentes de ciclina (CKI) e que pode ser considerado como um supressor tumoral ou como oncogene, dependendo do background genético. Uma das funções do p21, como gene supressor tumoral,
Terapêutica no Cancro do Cólon e Recto 15 é a indução da paragem do ciclo celular pela ligação ao complexo ciclina/Cdk, impedindo assim, a progressão da fase G1 para a fase S. Um dos mecanismos de regulação do p21 é via p53. Esta, embora não seja necessária para a transcrição do p21, é bastante importante na sua regulação, na presença de lesões no DNA. Assim, a ocorrência de erros no DNA desencadeia a indução da expressão do p53 que, consequentemente, leva à acumulação da expressão do p21. Este é responsável pela inibição dos complexos ciclina/Cdk, induzindo a paragem do ciclo celular da fase G1 para a fase S o que permite a correcção dos erros presentes no DNA, prevenindo assim a apoptose[53-55].
O p21 tem também a função de regular a apoptose, podendo, em determinadas situações, ser anti- ou pro-apoptótico (oncogene ou supressor tumoral, respectivamente). O seu papel anti-apoptótico está relacionado com a sua ligação à pro-caspase-3, que impede a activação da forma madura da caspase-3, originando a resistência à apoptose. Por outro lado, o p21 é clivado proteoliticamente pela caspase-3, durante a apoptose, o que permite a sua libertação do complexo ciclina/Cdk e resulta na elevada actividade deste, na fase inicial da apoptose [56]. Dado o seu papel na regulação do ciclo celular e apoptose, o p21 assume um papel importante na resposta à terapêutica e é por isso, estudado como marcador preditivo de resposta ao tratamento. Por exemplo, a elevada expressão de p21 foi associada à pior sensibilidade ao 5-FU, devido ao consequente aumento de expressão do TS [57, 58]. No entanto a supressão do p21 pelo c-Myc foi demonstrada como benéfica no tratamento com fármacos que induzem a apoptose [59], o que é apoiado pelos resultados obtidos pelo grupo de Gastrenterologia do CIPM, onde foi associada a reduzida expressão do p21 com a maior sobrevivência em doentes com CCR esporádico MSS submetidos a quimioterapia.
Dada a função do p21 na regulação do ciclo celular e na apoptose, este assume um papel bastante importante na terapêutica. Embora seja conhecido o papel da regulação do p21 pela proteína p53 na resposta à terapêutica e no desenvolvimento tumoral, ainda não é conhecida a regulação da expressão do p21 pela via Wnt na resposta ao tratamento, o que torna a via de sinalização Wnt num potencial alvo de estudo na resistência à terapêutica.
1.6. A via Wnt na resistência à terapêutica
Dois estudos recentes relacionam directamente a activação da via de sinalização Wnt com a resposta à terapêutica. Num primeiro estudo, analisando doentes com carcinoma hepatocelular, foi demonstrado que a maior activação da via de sinalização Wnt/β-catenina está associada à resistência ao tratamento com IFN-α/5-FU [60]. Num segundo estudo, em linhas celulares de CCR, foi igualmente demonstrado que, a maior resistência celular ao tratamento com irinotecano e 5-FU está associada às células com maior activação da via de sinalização Wnt [61]. Após estes estudos, e visto que, diferentes mutações do gene APC conferem diferentes níveis de sinalização da via Wnt devido à expressão de um número diferente de domínios de regulação da -catenina, coloca-se a hipótese de mutações distintas no gene APC se associarem a diferentes respostas à terapêutica.
![Figura 4 Representação da actividade transcripcional regulada pelo TCF7L2 em linhas celulares de CCR que expressam proteínas truncadas com diferentes números de domínios de regulação da -catenina (adaptado de[22])](https://thumb-eu.123doks.com/thumbv2/123dok_br/15594345.1051073/18.892.148.740.110.309/representação-actividade-transcripcional-celulares-expressam-proteínas-diferentes-regulação.webp)
![Figura 8 Representação da formação dos aductos de platina no DNA pela oxaliplatina (Adaptado de [44])](https://thumb-eu.123doks.com/thumbv2/123dok_br/15594345.1051073/24.892.245.658.259.492/figura-representação-formação-aductos-platina-dna-oxaliplatina-adaptado.webp)

![Figura 10 Representação esquemática dos plasmídeos WRE (A) e MRE (B). A vermelho estão ilustrados os locais de ligação ao TCF7L2 que no caso do WRE estão na sua forma wt e no MRE estão mutados (Adaptado de [62])](https://thumb-eu.123doks.com/thumbv2/123dok_br/15594345.1051073/30.892.270.669.437.604/figura-representação-esquemática-plasmídeos-vermelho-ilustrados-ligação-adaptado.webp)