2018
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
Characterization of Chronic Lymphocytic Leukemia by
aCGH/MLPA
Diana Cristina Antunes Candeias Adão
Mestrado em Biologia Molecular e Genética
Dissertação orientada por:
Professora Doutora Isabel Maria Marques Carreira
Professor Doutor Manuel Carmo Gomes
I Agradecimentos
Começo por agradecer ao CIMAGO e à ACIMAGO por todo o apoio prestado no âmbito do desenvolvimento deste trabalho, tanto a nível logístico como financeiro. Agradeço também ao Laboratório de Citogenética e Genómica e ao Laboratório de Oncologia e Hematologia da FMUC, pelo fornecimento dos equipamentos e bens necessários à realização deste projeto.
À Professora Doutora Isabel Marques Carreira, muito obrigada por me permitir desenvolver o meu projeto de tese de mestrado no Laboratório de Citogenética e Genómica da FMUC e por orientar este trabalho. Agradeço tudo o que me ensinou, o apoio e, acima de tudo, o exemplo que é como profissional na área da Genética Humana, que em muito contribuiu para o meu desenvolvimento e crescimento a nível académico e profissional.
Ao Professor Doutor Manuel Carmo Gomes, pela sempre célere e incondicional orientação durante este ano. Mostrou-se sempre disponível para responder às minhas dúvidas e questões, e por isso demonstro o meu agradecimento.
À Professora Doutora Ana Bela Sarmento Ribeiro, pela proposta que me apresentou para trabalhar no âmbito da Leucemia Linfocítica Crónica, pelo apoio nas correções necessárias, e pela possibilidade que me deu de realizar alguns passos do meu trabalho nas instalações do Laboratório de Oncologia e Hematologia da FMUC.
Ao Miguel Pires, por todos os ensinamentos ao nível do trabalho laboratorial, pela enorme disponibilidade e apoio na realização desta tese, bem como pela amizade e conselhos que me deu.
À Doutora Ana Cristina Gonçalves, que sempre me ajudou na correção de conteúdos da tese e nas questões relacionadas com as amostras a estudar. Muito obrigada pelo apoio.
À Susana Ferreira, Ilda Ribeiro, Mariana Val, Joana Jorge e Raquel Alves, pelo apoio ao nível de aplicação de técnicas e interpretação de resultados, bem como pela amizade.
À Drª Amélia Pereira e ao Dr. José Pedro Carda, do Serviço de Medicina do Hospital Distrital da Figueira da Foz e Departamento de Hematologia Clínica dos CHUC, respetivamente, pela disponibilização de amostras para este estudo.
Um especial agradecimento a todos os doentes a serem seguidos nos serviços referidos, que aceitaram fornecer as suas amostras e contribuir para o desenvolvimento deste estudo.
À equipa técnica da Unidade de Gestão Operacional Citometria do Serviço de Patologia Clínica dos CHUC, que se mostrou sempre prestável na preparação das amostras disponíveis. Apresento um especial agradecimento ao coordenador deste serviço, Professor Doutor Artur Paiva.
À restante equipa do Laboratório de Citogenética e Genómica da FMUC, Professora Doutora Joana Barbosa de Melo, Alexandra Marques, Ana Jardim, Cláudia Pais, Lúcia Simões, Patrícia Paiva, Nuno Lavoura, Marta Pinto, Carla Henriques e Sónia Pereira, pela forma como me receberam, por tudo o que me ensinaram ao nível do trabalho deste laboratório e pela amizade.
II
Às minhas colegas Alexandra Oliveira, Inês Gonçalves, Inês Tavares, Laura Silvério, Luísa Esteves e Mariana Tomás que sempre estiveram disponíveis para discutir ideias, pontos de vista e dar conselhos. Obrigada pelo companheirismo e pelos bons momentos que passámos.
À minha colega de Licenciatura Ana Gaspar, pela companhia nas sessões de escrita da tese, pelo apoio e motivação, pela partilha das preocupações e conquistas deste ano e, principalmente, pela amizade que mantemos.
Ao Zé Miguel, por me ter ajudado a manter a motivação para a tese até ao fim, e por ouvir sempre os meus relatos sobre o dia-a-dia do laboratório!
Por último, quero agradecer a toda a minha família pelo apoio que me deram, especialmente aos meus pais e à minha irmã, pois sempre me motivaram e acreditaram que eu seria capaz de chegar onde cheguei. Para vocês, um beijinho grande!
III Resumo
Palavras-chave: Leucemia Linfocítica Crónica; CNVs; iFISH; aCGH; MLPA
A Leucemia Linfocítica Crónica (LLC) é o tipo de leucemia mais frequente na população adulta de países ocidentais, cuja idade média ao diagnóstico varia entre os 67 e os 72 anos. É caracterizada pela expansão clonal e acumulação de linfócitos B neoplásicos no sangue periférico e na medula óssea, resistentes à apoptose e imunologicamente incompetentes.. Estas células têm também a capacidade de infiltrar outros órgãos como o baço ou o fígado.
Os indivíduos com LLC apresentam diversas alterações genómicas características, que estão associadas a diferentes subtipos da doença com diferentes comportamentos biológicos e clínicos. Os dois principais subtipos de LLC são caracterizados pela ausência ou presença de genes mutados na região codificante da cadeia pesada da região variável das imunoglobulinas (IGHV), sendo que o estado não mutado destes genes é o que constitui a situação de doença mais agressiva. A sobre-expressão dos genes CD38 e ZAP70 é um parâmetro que auxilia na estratificação de doentes em grupos com diferentes prognósticos que, possivelmente, necessitam de diferentes estratégias terapêuticas. Para além destes marcadores, podem ocorrer alterações epigenéticas; mutações somáticas nos genes MYD88, NOTCH1, SF3B1 e TP53; variações no número de cópias (CNVs) de segmentos de DNA; trissomias 9, 12, 18 e 19, e translocações que envolvam o locus IGH. Uma CNV é um segmento de DNA com pelo menos 1 kilobase (Kb) que apresenta variação no número das suas cópias (perda ou ganho) relativamente a um DNA de referência de um indivíduo saudável. Uma translocação recíproca é uma troca de posições entre dois segmentos de DNA, entre cromossomas não homólogos. Ao longo das últimas décadas estas alterações têm vindo a ser detetadas, na prática clínica, pela técnica de Hibridização Fluorescente In Situ aplicada a células em interfase (iFISH). A iFISH permitiu realizar uma melhor estratificação destes doentes em diferentes grupos, aos quais são associados diferentes prognósticos. Estes grupos baseiam-se na presença de: (i) deleção em 13q como única alteração; (ii) nenhuma alteração; (iii) trissomia 12; (iv) deleção em 11q e (v) deleção em 17p (grupos apresentados por ordem crescente de pior prognóstico previsto), sendo (i) o grupo de melhor prognóstico e (v) o de pior prognóstico. Posteriormente, foram adicionadas a este painel sondas para a deteção da deleção em 6q e translocações do gene IGH. No entanto, devido à heterogeneidade biológica e clínica desta doença, existe consciência da importância de procurar novas CNVs com possível valor como marcadores de prognóstico. De facto, nem todas as terapias desenvolvidas são aplicáveis a doentes com diferentes tipos de alterações. Um exemplo disso é o caso dos doentes com a del(17p) e/ou mutação do gene TP53. Neste grupo de doentes, a quimioimunoterapia é altamente desaconselhada devido ao facto de esta recorrer a agentes dependentes da atividade da proteína p53 que, por estar ausente e/ou mutada nos doentes com del(17p) e/ou mutação do gene TP53, impede que esta terapêutica conduza a resultados favoráveis. Assim sendo, os doentes com estas alterações são candidatos a terapêutica dirigida recorrendo a inibidores da via BTK, como o ibrutinib.
Com este trabalho piloto, o nosso grupo realizou a caracterização de uma coorte de 20 doentes com LLC, ao nível do seu conteúdo em CNVs. Para tal, aplicámos as técnicas de Amplificação Multiplex de Sondas Dependente de Ligação (MLPA) e arrays de Hibridização Genómica Comparativa (aCGH), após extração de DNA a partir de células mononucleares do sangue periférico (PBMCs), e tendo em conta o genoma de referência GRCh37/hg9 (Genome Reference Consortium
Human Build 37/human genome 19). Após a obtenção de resultados, fizemos um estudo de
sobrevivência global ao longo de um período médio de follow-up de 114 meses, aplicando o método estatístico de Kaplan-Meier. Com base nos resultados de MLPA, dividimos as amostras em duas categorias: um grupo com doentes que tinham ≥2 alterações e outro com doentes que tinham <2 alterações. Interpretámos, também, a distribuição de algumas das alterações que detetámos em maior
IV
frequência: del(11q), del(14q), e mutação no gene NOTCH1, em doentes agrupados de acordo com o seu estadiamento pelos sistemas Binet e Rai (sistemas de classificação de estadiamento específicos para LLC). Para tal, aplicámos o teste qui-quadrado de Pearson.
Relativamente à análise de sobrevivência, apesar de os nossos resultados não terem apresentado significância estatística, estes sugerem que existe uma associação entre o aumento no número de alterações genómicas que um doente tem e a diminuição na taxa de sobrevivência. Quanto às prevalências das alterações del(11q), del(14q) e mutação do gene NOTCH1, verificámos que estas são significativamente mais altas em doentes que se encontram num estadiamento intermédio/alto de risco de doença do que em doentes com estadiamento de baixo risco, o que sugere a sua importância como marcadores de mau prognóstico. Isto é algo que já foi previamente descrito para a del(11q) e para a mutação em NOTCH1 mas, tanto quanto é do nosso conhecimento, é algo que não foi ainda visto, por outros grupos, para doentes com a alteração del(14q).
Para além das conclusões tiradas relativamente às alterações genómicas em doentes com esta neoplasia, foi-nos possível fazer algumas comparações entre as técnicas aplicadas. Apresentamos, também, uma comparação crítica entre a aplicação destas tecnologias e a utilização da técnica de diagnóstico iFISH, tanto ao nível da prática clínica como no âmbito da investigação científica.
O MLPA e o aCGH demonstraram ser técnicas apropriadas para realizar a caracterização genómica da LLC, uma vez que estas foram capazes de detetar CNVs. A técnica de aCGH permitiu-nos detetar um total de 134 alterações, das quais a maior parte foi vista em regiões que não são estudadas pelo iFISH. Isto provou o seu valor do aCGH como técnica aplicável na investigação de alterações genómicas na LLC. Realizámos, de seguida, a validação dos resultados de aCGH pela técnica de MLPA, o que nos permitiu chegar à conclusão de que todas as alterações detetadas por ambas as técnicas são reais. Concluímos também que o MLPA é uma técnica candidata a substituir a aCGH no âmbito da investigação, principalmente por ser menos dispendiosa e menos morosa. Num contexto de prática clínica, o MLPA poderia eventualmente substituir o iFISH pois também fornece informação sobre a presença das alterações mais comuns: del(11q), trissomia 12, del(13q) e del(17p), bem como sobre a presença de outras alterações características da LLC, mas menos prevalentes: ganho em 2p, del(6q), perda em 8p, ganho em 8q, del(9p21) e trissomia 19. Para além disso, deteta também possíveis mutações somáticas nos genes NOTCH1, MYD88 e SF3B1, que podem ocorrer nesta doença. Contudo, o iFISH é a única destas três técnicas que é capaz de detetar mosaicismos de baixa expressão e a ocorrência de translocações na LLC, tais como: t(14,18)(q32,q24), t(14,18)(q32,q21) e t(14,19)(q32,q13). Estas translocações ocorrem em até 5% dos casos diagnosticados.
As nossas perspetivas para trabalhos futuros consistem em aumentar o número de amostras estudadas bem como o tempo de seguimento destas com o objetivo de, numa próxima análise de sobrevivência, alcançar uma maior confiança nos resultados obtidos. Para além disso, temos também o objetivo de reavaliar a importância da del(14q) como possível marcador de mau prognóstico, num grupo maior de doentes. Finalmente, esperamos ser capazes de conhecer melhor a distribuição das alterações que encontrámos com elevada prevalência (del(5q13.2), del(8q24.23), del(22q11.22), del(Xq21.1), dup(4p16.3), dup(6p25.3), dup(8p11.1), and dup(10q11.22)) numa coorte de maior de doentes. Esperamos conseguir identificar, de entre estas alterações, possíveis marcadores de doença, algo que será apenas possível por comparação com uma população controlo. Com o nosso trabalho, esperamos incentivar não só a procura de novos alvos terapêuticos como também a descoberta e avaliação de novas terapias. Por último, propomos a continuação de estudos de caracterização genómica da leucemia linfocítica crónica, destacando o aCGH e MLPA como técnicas preferenciais para esse fim.
V Abstract
Keywords: Chronic Lymphocytic Leukemia; Copy Number Variation; aCGH; iFISH; MLPA
Chronic lymphocytic leukemia (CLL) is one of the most common hematological malignancies, being the most recurrent type of leukemia in the adult population in western countries. It is characterized by a clonal expansion and accumulation of neoplastic B lymphocytes on peripheral blood and in the bone marrow.
There are some characteristic genomic alterations in these individuals, linked to different subtypes of CLL with different biological and clinical behaviors. The two main types of CLL are characterized by either the lack or the presence of mutated genes at the coding region of the immunoglobulins heavy chain’s variable region (IGHV), being the unmutated state the one leading to a more aggressive disease course. Overexpression of the molecular markers CD38 and ZAP70 are also parameters that determine disease stage and help to stratify patients into groups with different prognosis and possible different therapy strategies. Furthermore, there are some CLL characteristic genomic alterations: epigenetic alterations; somatic mutations in MYD88, NOTCH1, SF3B1, and TP53; copy number variations (CNVs) of some DNA regions; trisomies 9, 12, 18, or 19; and some translocations involving the IGH
locus. Over the last decades, CLL reported CNVs have been evaluated with the standard technique of
interphase Fluorescent In Situ Hybridization (iFISH), in order to stratify patients into different groups of predicted outcomes. Those groups are based on the presence of: (i) 13q deletion as sole alteration; (ii) no alterations; (iii) Trisomy 12; (iv) 11q deletion; and (v) 17p deletion. These groups are presented in order of best (i) to worse disease outcome (v). Subsequently, probes for the detection of 6q deletion and IGH translocations have been added. However, due to the clinical and biological heterogeneity of this disease, it is currently acknowledged that it is important to search for other CNVs that may become new disease markers.
Our team has characterized, in this pilot study, a cohort of 20 CLL patients, regarding their CNVs content, in agreement with the Genome Reference Consortium Human Build 37/human genome 19 (GRCh37/hg9). We have applied Multiplex Ligation-dependent Probe Amplification (MLPA) and array Comparative Genomic Hybridization (aCGH) technologies. We conducted an overall survival analysis of patients presenting either ≥2 alterations or <2 alterations detected by MLPA, based on a median follow-up time of 114 months. Moreover, we have interpreted the distribution of some of the common alterations found: del(11q), del(14q), and NOTCH1 mutation, in patients grouped according to their disease staging by Binet and Rai CLL staging systems.
As for the survival analysis, despite the lack of statistical significance, our results show an association between the increasing number of alterations in a patient and a shorter overall survival. Finally, the prevalence of del(11q), del(14q), and NOTCH1 mutation in our samples are significantly higher in patients with intermediate/high risk stages than in low risk disease stage patients, suggesting their value as bad prognosis markers. That is something already known for del(11q) and NOTCH1 mutation but appears to be a new information for patients with del(14q).
We conclude that MLPA and aCGH are competent techniques for the genomic characterization of CLL. The aCGH technique has detected additional alterations in regions other than the ones evaluated by the standard diagnosis method iFISH, proving its value as a research tool in CLL. The aCGH results regarding reported CLL CNVs were positively validated by MLPA, showing that MLPA is a good substitute of the aCGH technique on day-to-day research. Moreover, MLPA has the advantages of being a less expensive and less time-consuming technique. In terms of clinical practice, MLPA could substitute iFISH for delivering the same results on del(11q), trisomy 12, del(13q), and del(17p) assessment, and the additional information on the other CNVs it also evaluates (2p gain, del(6q), 8p loss, 8q gain, del(9p21), and trisomy 19), as well as the detection of NOTCH1, MYD88, and SF3B1
VI
somatic mutations. However, iFISH is the only one of these three techniques capable of detecting low expression mosaicism and the presence of translocations, a type of alteration that occurs in up to 5% of patients, making it still irreplaceable.
In the future, all our results should be re-evaluated using a larger cohort of patients. Additionally, the survival analysis must be repeated when a larger follow-up time is reached, and the number of samples analyzed is greater. It is also important to re-assess del(14q) value as a possible bad prognosis marker. Additionally, we intend to better characterize the prevalence of del(5q13.2), del(8q24.23), del(22q11.22), del(Xq21.1), dup(4p16.3), dup(6p25.3), dup(8p11.1), and dup(10q11.22) in a larger cohort of CLL patients. Among these alterations, it would be interesting if we could identify possible new markers of diseases something only possible after comparison with a control groups.
With our work, we expect to stimulate further research on new therapeutic targets and new treatment strategies. At last, we encourage the continuation of CLL genomic characterization, with the use of both aCGH and MLPA.
VII Index Agradecimentos ... II Resumo ... IV Abstract ... VI Index ... VIII List of figures and tables ... X Abbreviations ... XI
Chapter 1. Introduction...1
1.1 Chronic Lymphocytic Leukemia: General characterization ...1
1.1.1 CLL diagnosis, staging, and prognostic assessment ...1
1.1.2 Prognostic markers in CLL ...2
1.1.3 Monoclonal B cell lymphocytosis: a previous state of CLL ...3
1.1.4 CLL treatment and the advantages of targeted therapy ...3
1.1.5 Possibly altered pathways in CLL ...4
1.1.6 CLL genomic alterations ...4
1.1.6.1 Chromosomal abnormalities: reported CNVs and translocations in CLL ...4
1.1.6.1.1 Genomic alterations as prognostic markers in CLL ...5
1.1.6.1.2 Deletions at 13q ...5
1.1.6.1.3 Trisomy 12 ...5
1.1.6.1.4 Deletions at 11q ...6
1.1.6.1.5 Deletions at 17p and TP53 mutations ...6
1.1.6.1.6 Deletions at 6q, 14q, 8p losses, and 2p and 8q gains ...7
1.1.6.2 Somatic mutations and SNPs in CLL ...7
1.1.6.3 Epigenetic alterations in CLL: altered methylation profiles ...8
1.1.6.4 Clonal evolution in CLL ...8
1.2 CLL genomic characterization: comparing the effectiveness of conventional and molecular cytogenetic techniques ...9
1.2.1 Applying aCGH for copy number analysis ...11
1.2.2 Applying MLPA for copy number analysis ...12
1.3 Objectives ...13
Chapter 2. Materials and methods ...14
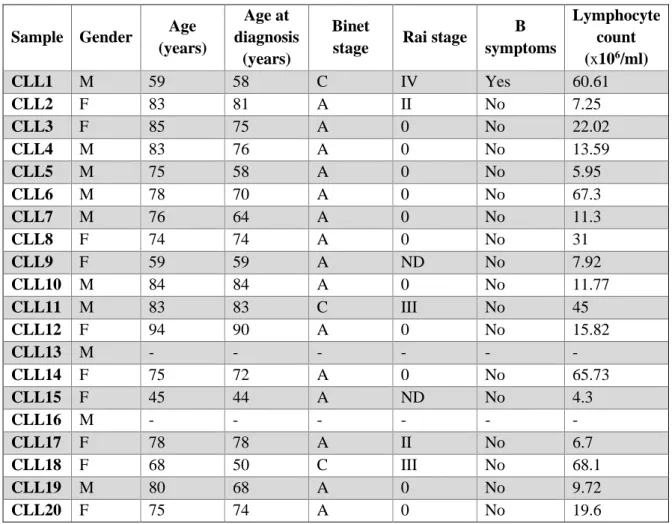
2.1 Selection and characterization of the study population ...14
2.2 Samples collection and preparation ...15
VIII
2.2.2 DNA extraction and purity determination ...15
2.3 Genomic analysis by aCGH and MLPA ...16
2.4 Interpretation of aCGH results ...16
2.5 Statistical analysis ...17
Chapter 3. Results and discussion ...18
3.1 General findings by aCGH and MLPA ...18
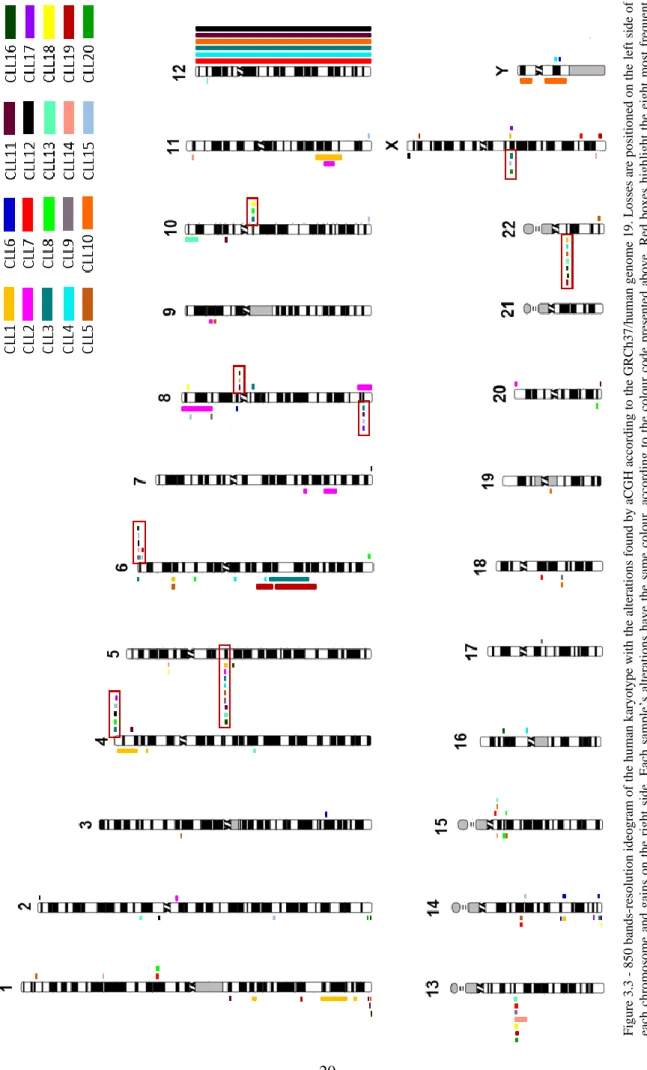
3.2 aCGH results ...19
3.2.1 Chromosome 13q deletions ...21
3.2.2 Trisomy 12 ...21
3.2.3 Chromosome 11q deletions ...22
3.2.4 Deletions at 6q, 9p, 14q, losses at 8p, and 2p and 8q gains ...22
3.2.5 other common CNVs found by aCGH ...23
3.2.5.1 dup(4p16.3) ...24
3.2.5.2 del(5q13.2) ...24
3.2.5.3 dup(6p25.3) ...25
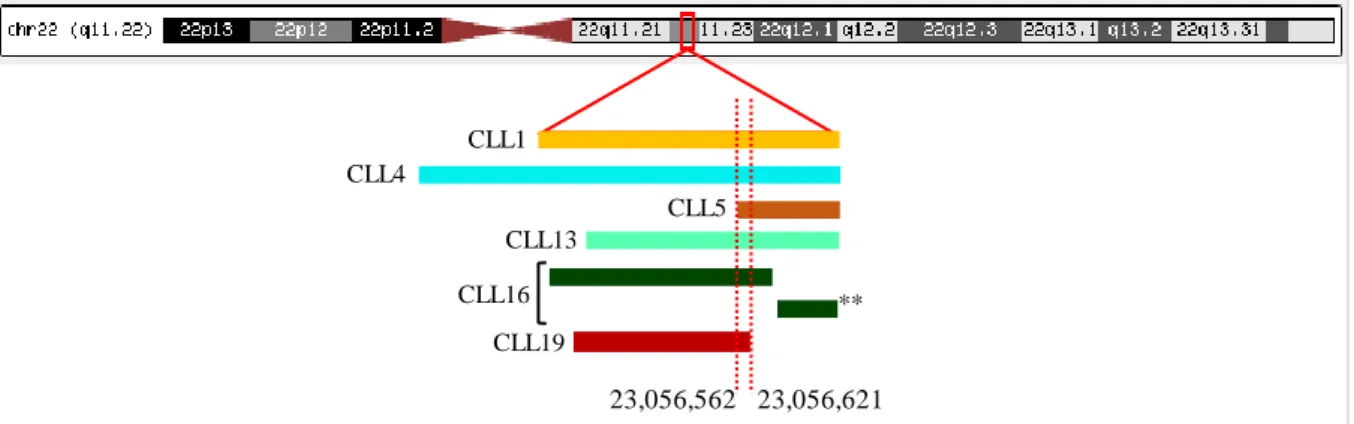
3.2.5.4 del(22q11.22) ...25
3.2.5.5 dup(8p11.1), del(8q24.23), dup(10q11.22), and del(Xq21.1) ...26
3.3 MLPA results ...26
3.3.1 Chromosomes 11q and 13q deletions and trisomy 12 ...26
3.3.2 Losses at 6q, 9p, 14q, losses at 8p, gains at 8q, and NOTCH1 mutation ...26
3.4 Comparison between aCGH and MLPA results ...27
3.5 Survival analysis for patients with two or more CNVs ...28
Chapter 4. Conclusions and future perspectives ...29
Chapter 5. References...30
IX List of figures and tables
Figure 1.1 – Origins of B-CLL cells. ...2
Figure 1.2 (a-c) – Schematic representation of aCGH labelling. ...12
Figure 1.3 (a-e) – Schematic representation of the preparation of a MLPA reaction. ...13
Table 2.1 – Clinical data obtained for the 20 CLL samples studied. ...14
Figure 2.1 (A and B) – Result of a density gradient centrifugation, using Ficoll-Paque. ...15
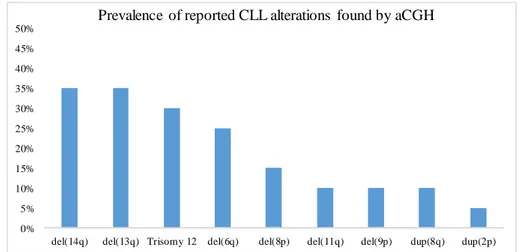
Figure 3.1 – Prevalence of reported CLL alterations found by aCGH. ...18
Figure 3.2 – Prevalence of reported CLL alterations found by MLPA. ...18
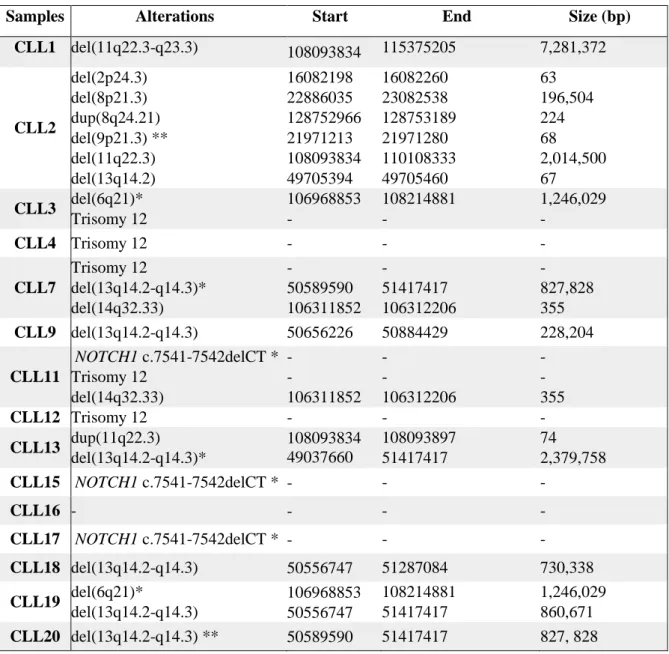
Table 3.1 – Alterations found by MLPA in CLL samples studied. ...19
Figure 3.3 – 850 bands-resolution ideogram of the human karyotype with the alterations found by aCGH. ...20
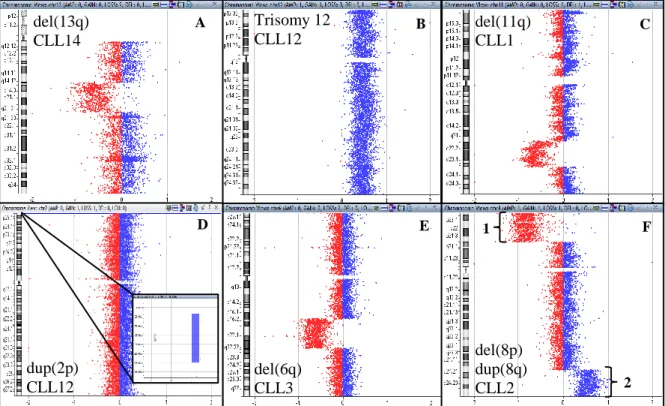
Figure 3.4 (A-F) – Chromosome view of characteristic CLL genomic alterations we found in our samples, using aCGH. ...21
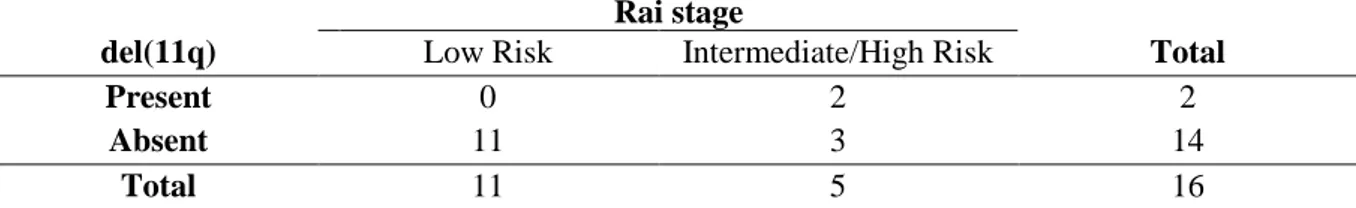
Table 3.2 – Contingency table in which the qui-squared test was based on, for the evaluation of the prognosis value of del(11q). ...22
Table 3.3 – Contingency table in which the qui-squared test was based on, for the evaluation of the prognosis value of del(14q). ...23
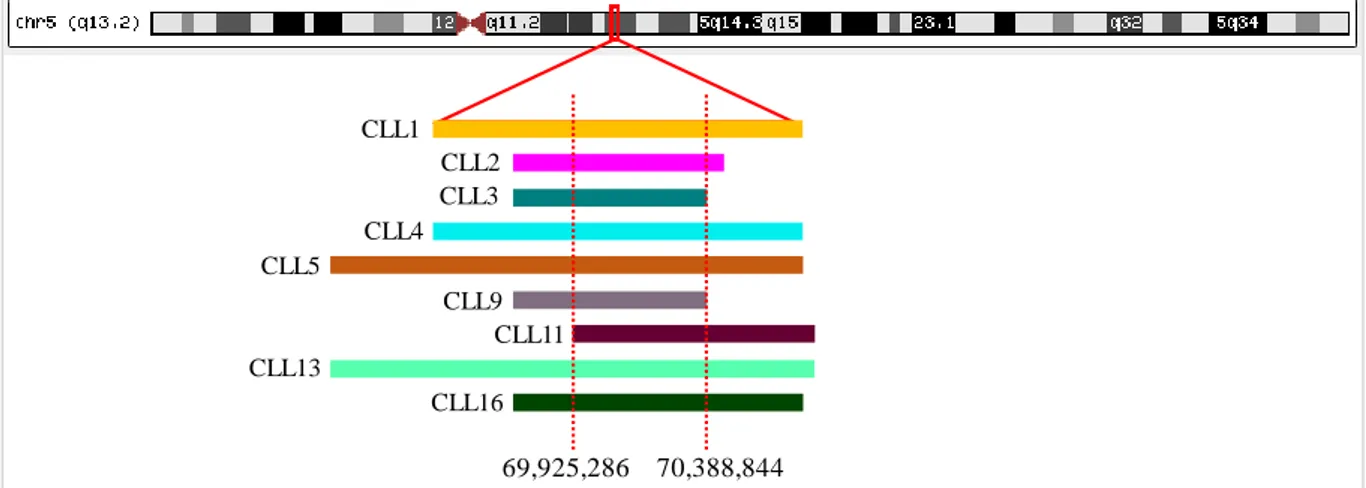
Figure 3.5 Deletions detected at 5q, using aCGH. ...24
Figure 3.6 Deletions detected at 22q, using aCGH. ...25
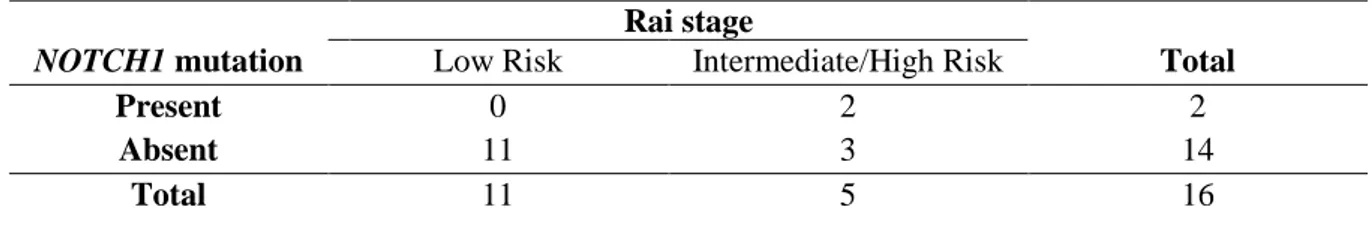
Table 3.4 – Contingency table in which the qui-squared test was based on, for the evaluation of the prognosis value of NOTCH1 mutation. ...27
Figure 3.7 – Electropherogram for sample CLL11. ...28
Figure 3.8 – Survival curve obtained by the Kaplan-Meier method for OS analysis. ...28
X Abbreviations
ACAT – Acetyl-CoA Acetyltransferase 1
aCGH – Array Comparative Genomic Hybridization amp – Amplification
ARID1A – AT-rich interactive domain-containing protein 1A ATM – ATM serine/threonine kinase
ATP1B2 – ATPase Na+/K+ Transporting Subunit Beta 2
BAC – Bacterial artificial chromosome
BCL2 – B-cell CLL/lymphoma 2 BCL2L1 – BCL2-like 1
BCL3 – B-cell CLL/lymphoma 3
BCR – B-cell receptor
BIRC2 – Baculoviral IAP Repeat Containing 2 BIRC3 – Baculoviral IAP Repeat Containing 3
bp – Base pair
BTK – Bruton’s tyrosine kinase
C11orf53 – 11 Open Reading Frame 53 CASP1 – Caspase 1 CASP4 – Caspase 4 CASP5 – Caspase 5 CASP12 – Caspase 12 CCND1 – Cyclin D1 CCND3 – Cyclin D3
CD5 – T-cell surface glycoprotein CD5 CD10 – Neprilysin
CD19 – B-lymphocyte antigen CD19 CD20 – B-lymphocyte antigen CD20 CD22 – B-cell receptor CD20 CD27 – CD27 Antigen
CD38 – ADP-ribosyl cyclase/cyclic ADP ribose hydrolase 1
CD79b – B-cell antigen receptor complex-associated protein beta chain CD200 – Cell surface glycoprotein CD200 receptor 1
CDK4 – Cyclin-dependent kinase 4
CDKN1B – Cyclin-dependent kinase inhibitor 1B CDKN2A – Cyclin-dependent kinase inhibitor 2A CDKN2B – Cyclin-dependent kinase inhibitor 2A
CGH – Comparative Genomic Hybridization
CHD2 – Chromodomain Helicase DNA Binding Protein 2
Chr – Chromosome
CHUC – Centro Hospitalar e Universitário de Coimbra CK – Complex Karyotype
ClinGen – Clinical Genome Resource CLL – Chronic Lymphocytic Leukemia
CLLU1 – Chronic Lymphocytic Leukemia up-regulated protein 1
CNV – Copy Number Variation
CUL5 – Cullin-5
XI Cy5 – Cyanine 5
del – Deletion
DGV – Database of Genomic Variants
DLC1 – Deleted in Liver Cancer 1
DLEU2 – Deleted in Lymphocytic Leukemia 2 DLEU7 – Deleted in Lymphocytic Leukemia 7
DMSO – Dimethyl Sulfoxide DNA – Deoxyribonucleic Acid DSB – Double-Strand Break dup – Duplication
DUSP22 – Dual Specificity Phosphatase 22 E2F1 – E2F Transcription Factor 1
EDTA – Ethylenediaminetetraacetic acid
FAM110C – Family with Sequence Similarity 110 Member C
FISH – Fluorescence in situ Hybridization
GATA2 – GATA Binding Protein 2
GRCh37/hg9 – Genome Reference Consortium Human Build 37/human genome 19
GTF2H2 – General Transcription Factor IIH Subunit 2 HIP1R – Huntingtin Interacting Protein 1 Related
iFISH – Interphase Fluorescence in situ Hybridization Ig – Immunoglobulin
IgA – Immunoglobulin A IgD – Immunoglobulin D IgG – Immunoglobulin G
IGH – Immunoglobulin Heavy Chain
IGHV – Immunoglobulin Heavy Chain Variable Region
IgM – Immunoglobulin M
IKZF3 – IKAROS Family Zinc Finger 3 IRF4 – Interferon Regulatory Factor 4 KDELC2 – KDEL Motif Containing 2 KRAS – KRAS Proto-Oncogene, GTPase
LCR – Low-Copy Repeats LDH – Lactate Dehydrogenase
LEF1 – Lymphoid Enhancer Binding Factor 1
LPO – Left Probe Oligonucleotide
mir15a/16-1 locus – MicroRNA 15a and MicroRNA 16-1 locus
miRNA – MicroRNA
miRNA-34a – MicroRNA 34a
MBL – Monoclonal B lymphocytosis
MDM2 – MDM2 Proto-Oncogene
MDR – Minimal Deleted Region
MGA – MGA, MAX Dimerization Protein
MI – Methylation Index
MIR650 – MicroRNA 650
MLPA – Multiplex Ligation-Dependent Probe Amplification
MRE11 – MRE11 Homolog, Double Strand Break Repair Nuclease
mRNA – Messenger RNA
XII
MYC – MYC Proto-Oncogene, BHLH Transcription Factor MYCN – MYCN Proto-Oncogene, BHLH Transcription Factor MYD88 – Myeloid Differentiation Primary Response 88 MYF6 – Myogenic Factor 6
NAHR – Nonallelic Homologous Recombination
NAIP – NLR Family Apoptosis Inhibitory Protein
NF-Kb – Nuclear factor κ-light-chain-enhancer of activated B cells pathway NFKBIE – NFKB Inhibitor Epsilon
NFKB2 – Nuclear Factor Kappa B Subunit 2
NGS – Next-Generation Sequencing
NOTCH1 – Translocation-Associated Notch Protein TAN-1 NPAT – Nuclear Protein, Coactivator of Histone Transcription NRAS – NRAS Proto-Oncogene, GTPase
OMIM – Online Mendelian Inheritance in Man OS – Overall Survival
p53 – Tumor Protein 53
PAX5 – Paired Box 5
PBMC – Peripheral Blood Mononuclear B Cell PCR – Polymerase Chain Reaction
PDGFRL – Platelet Derived Growth Factor Receptor Like
PFS – Progression-Free Survival
PRAME – Preferentially Expressed Antigen in Melanoma PTEN – Phosphatase and Tensin Homolog
PTPN11 – Protein Tyrosine Phosphatase, Non-Receptor Type 11
qPCR – Quantitative Polymerase Chain Reaction
RB1 - RB Transcriptional Corepressor 1
RCN – Relative Copy Number
RDX – Radixin
RFU – Relative Fluorescent Units RPO – Right Probe Oligonucleotide
RUNX1 – Runt Related Transcription Factor 1 SERF1A – Small EDRK-Rich Factor 1A SF3B1 – Splicing Factor 3b Subunit 1
SHM – Somatic Hypermutation SLL – Small Lymphocytic Lymphoma
SMARCA2 – SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin,
Subfamily A, Member 2
SMN1 – Survival of Motor Neuron 1, Telomeric SMN2 – Survival of Motor Neuron 2, Centromeric
SNP – Single-Nucleotide Polymorphism
ssDNA – Single-Strand DNA
SYK – Spleen Associated Tyrosine Kinase
TAL1 – TAL BHLH Transcription Factor 1, Erythroid Differentiation Factor TERT – Telomerase Reverse Transcriptase
TP53 – Tumor Protein P53
trp – Triplication
TTFT – Time to First Treatment
XIII WGS – Whole Genome Sequencing
WHO – World Health Organization
XPO1 – Exportin 1
ZAP70 – Zeta Chain of T Cell Receptor Associated Protein Kinase 70
ZMYM3 – Zinc Finger MYM-Type Containing 3 ZNF280A – Zinc Finger Protein 280A
ZNF280B – Zinc Finger Protein 280B ZNF292 – Zinc Finger Protein 292 ZNF595 – Zinc Finger Protein 595 ZNF718 – Zinc Finger Protein 718
1 Chapter 1. Introduction
1.1 Chronic Lymphocytic Leukemia: General characterization
Chronic lymphocytic leukemia (CLL) is the most common type of leukemia and one of the most common lymphoid malignancies, being biologically and clinically very heterogeneous [1]. It affects mainly adults and western countries’ populations, with a median age at diagnose between 67 and 72 years old. Life expectancy for these patients ranges from months to decades [2].
There is a twofold increased risk for men to develop this malignancy compared to women, and increased age also seems to be a risk factor for CLL development [1]. CLL patients’ first-degree relatives have an 8.5 times increased risk of developing the disease, in comparison to the general population [1, 3], suggesting that family history is a risk factor for this condition [4, 5]. Also monoclonal B cell lymphocytosis (MBL) and small lymphocytic lymphoma (SLL) constitute risk factors for the development of this disease [1]. Although the driving causes for CLL are unknown, it is possible to characterize CLL as a hematological malignancy presenting clonal expansion of neoplastic B cells that accumulate in peripheral blood and infiltrate the bone marrow and lymphoid tissues such as lymph nodes, the liver, and the spleen [6, 7]. This accumulation is due to these cells’ inability to suffer apoptosis, sometimes because of the overexpression of BCL2 gene, an anti-apoptotic gene located at 18q21.33 [8, 9]. B-CLL cells differentiation might be blocked, also due to genetic alterations such as the mutation of an enhancer of PAX5 gene, located at 9p13. The PAX5 expression product is a transcription factor involved in B cell differentiation that suffers under-expression when the referred enhancer is mutated [1]. This suggests the need for the assessment of CLL driver alterations also present at the non-coding genome [7]. Although it was thought cell cycle arrest of neoplastic B lymphocytes to be the main cause for these B cell’s accumulation in CLL, it has been unveiled that this it occurs also due to the proliferation of some neoplastic cells that are able to multiply: between 0.1-1% neoplastic CLL lymphocytes duplicate, per day [2]. In fact, the disease course depends on the balance between these two types of cells[10-12].
1.1.1 CLL diagnosis, staging, and prognosis assessment
CLL is diagnosed through blood smears, cell counting, and immunophenotyping. An initial counting of >5,000 clonal lymphocytes/µl of peripheral blood, that remains for at least three months, and small mature lymphocytes with small cytoplasm and dense nucleus are CLL features detected at diagnosis. In advanced stages, cell counting can reach up to ≥10,000 lymphocytes/µl [6]. CLL patients may be stratified according to Rai and Binet staging systems, that acknowledge three prognostic types: poor, intermediate, and good prognosis. They are suitable helpers for disease risk determination and in the choice of patients who do or do not need immediate treatment at the time of diagnosis. Rai staging system has five risk stages, ranging from 0-IV: stage 0 and I – low risk, II – intermediate risk, and III and IV – high risk [13, 14]. The Binet staging system has three risk stages: A – low risk, B – intermediate risk, and C – high risk [15]. The Rai system defines low risk disease as the presence of leukemic cells and lymphocytosis; intermediate-risk disease by the presence of lymphocytosis and splenomegaly and/or enlarged lymph nodes; and high-risk disease as having the conditions from the first two stages, and also disease-related anemia and/or thrombocytopenia [6]. As for the Binet staging system, it stratifies patients based on the number of anatomical affected regions, like the lymph nodes from the neck, groins, axillae, spleen, and liver [6]. However, these staging systems do not take into account the molecular alterations and cannot predict the evolution course of the disease and so it is important to consider informative clinical markers for disease progression, enabling outcome prediction [16].
2
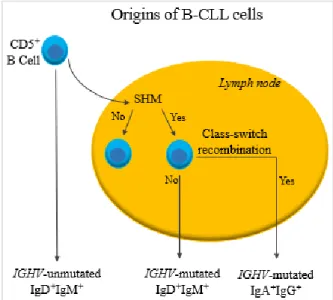
Figure 1.1 – Origins of B-CLL cells. Cancer cells in CLL are CD5+ B cells that either remain in the blood stream or enter
lymph nodes, experiencing SHM and, in both cases, presenting surface expression of IgM and IgD. Inside lymph nodes and after SHM, B cells may suffer apoptosis after the selection of low affinity cells or may be presented to antigens. That leads to the exit, to the blood stream, of IgM+IgD+ IGHV-mutated cells or of IGHV-mutated cells that underwent class-switch
recombination. (Adapted from Kipps et al. 2017).
1.1.2 Prognostic markers in CLL
B-CLL cells immunophenotypic analysis shows cell-surface expression of CD5, CD19, CD200 [16], generally reduced expression of CD20 [17, 18], CD22, CD79b, and lack of CD10, compared to healthy B cells [2, 19]. The CD20 level of expression is variable amongst CLL patients, depending on the cytogenetical abnormalities present. Supporting this is the finding of Tam et al. [17], that detected a significant increase on CD20 expression in patients with trisomy 12 and a significant decreased expression in patients with del(11q). The expression levels of CD38 and ZAP70 are informative about CLL subtype. ZAP70 overexpression indicates a more aggressive disease [8, 20], as well as CD38 overexpression [21-23]. ZAP70 overexpression is known to be an aggressive disease marker once it is linked to increased B Cell receptor (BCR) signaling, a feature that leads to an aggressive phenotype as well as to the modulation of other signaling pathways [8, 24]. As a matter of fact, the discovery that these two genes’ overexpression coexist with unmutated immunoglobulin heavy chain variable region (IGHV), a bad prognosis marker, has simplified patients’ prognosis assessment since IGHV state evaluation is a complex and dispendious process [10, 25]. However, after that discovery, CD38 overexpression has been determined to be an independent prognostic factor from IGHV mutational state because its expression levels may vary during the course of disease [10, 26]. ZAP70 consists, then, in a more secure source for the assessment of IGHV mutational state once it maintains a stable expression level during the course of disease [10, 27, 28]. Still, its evaluation is not yet applied for clinical practice purposes.
CLL has a heterogeneous clinical course, being divided in two subtypes characterized by the lack
or presence of somatic hypermutation (SHM) of the IGHV of B cell receptors. B lymphocytes with mutated IGHV result from B cells that went to lymph node’s germinal center and differentiated, whereas B lymphocytes with unmutated IGHV result from naive B cells that did not suffer SHM (Figure 1.1). Thus, B cell differentiation determines the behavior of the disease. It is more aggressive when B lymphocytes are less mature, which means that patients with unmutated IGHV present a more preoccupying situation [1]. Knowing cell-surface immunoglobulins (Ig) is also important to characterize CLL because it gives information about the origin of anomalous B cells [8, 29]. IgM and IgD are mainly present in IGHV-unmutated B cells, whilst IgA and IgG are characteristic of more mature cells that underwent class-switch recombination, the IGHV-mutated B cells (Figure 1.1) [8].3
1.1.3 Monoclonal B cell lymphocytosis: a previous state of CLL
Monoclonal B cell lymphocytosis is characterized by the presence of fewer than 5,000 monoclonal B cells/µl, a subset of B cells of which the clinical significance is unknown. In 75% of MBL cases, these cells present the B-CLL cells phenotype [4], leading to the so-called CLL-phenotype MBL, but are characterized by the absence of any other CLL symptoms [30]. MBL might present either a low or high count of monoclonal B cells: <500 cells/µl and >500 cells/µl, respectively [1].
The fact that this phenotype is detected in about 3% of the healthy adult population suggests the possibility of this being the previous stage of CLL [4, 8]. A study from Goldin et al. [5] reported the development of MBL in first-degree relatives of CLL individuals in a higher frequency than in the general population, suggesting that MBL is a candidate marker for CLL development [4, 5]. Hence, detecting this condition is a valuable step to identify patients for regular follow-ups and treat possible future CLL situations from an early stage.
A follow-up study was conducted by Rawstron et al. [30] with the intent of determining if CLL chromosomal abnormalities also occur in CLL-phenotype MBL. This study also evaluated if this phenotype may lead to a CLL situation requiring chemotherapy. The main results were that individuals with the CLL-phenotype MBL presented del(13q14) and trisomy 12 in the same frequencies as described for CLL (48% and 20%, respectively) and an IGHV mutation frequency of 87% (higher than the frequency found in the literature for IGHV-mutation frequency in CLL, that ranges from 60-65%). They also found that 15% of CLL-phenotype MBL individuals evolved to CLL, suggesting that, in fact, exists a relation between CLL and CLL-phenotype MBL. Some of these patients ended up needing chemotherapy but, having all the results in consideration, it was possible to conclude that to predict the outcome of CLL-phenotype MBL individuals is a difficult task [30].
More recently, genetically modified mice with both mir15a/16-1 locus and DLEU2 gene deleted (both located at 13q), ended up developing monoclonal B-cell lymphocytosis-like disorder and, ultimately, CLL. This supports the hypothesis that both deleted elements influence CLL leukemogenesis, possibly enhancing the appearance of a pre-CLL monoclonal B-cell lymphocytosis that will result in CLL development [6, 31].
1.1.4 CLL treatment and the advantages of targeted therapy
To decide on the course of CLL treatment, some clinical aspects should be considered: age, staging of the disease, and the presence or absence of del(17p), and of TP53 mutations. The main therapies are based on drugs that lead to different outcomes depending on patient’s phenotype. Fludarabine, cyclophosphamide, and rituximab are the first-line strategies for CLL patients, contributing to an increased progression-free survival (PFS) and overall survival (OS), and often results in a remission period of more than 10 years. However, these strategies are not advised for patients that have del(17p) and/or BIRC3 and TP53 mutation [32-34]. When present, these alterations lead to a decreased response to chemoimmunotherapy. Thus, therapeutic alternatives based on drugs which action is independent of p53 (the expression product of TP53) such as ibrutinib, were developed [35, 36]. It is an irreversible inhibitor of bruton’s tyrosine kinase (BTK) and is recommended for patients with relapsed disease [32, 37, 38] and for disease control in patients with high-risk characteristics such as del(11q) and unmutated IGHV [32].
Targeted therapy has been extensively studied and developed over the last years, regarding CLL. The main advantage is that it is designed to act upon a specific target, which therefore guarantees increased security in the resulting effect. The Chronic Lymphocytic Leukemia Treatment publication by the National Cancer Institute mentions one of the possible targeted therapies as being monoclonal antibody therapy [39]. New anti-CD20 monoclonal antibodies are an example of antibodies used for
4
this purpose, which are used in obinutuzumab, ofatumumab, and rituximab therapies [7, 37], three strategies that target B-CLL cells’ CD20 [6, 40]. Despite the variety of treatments available, in some cases there is no need for them, as about one third of CLL patients can survive for twenty or more years without receiving any treatment [10, 41].
1.1.5 Possibly altered pathways in CLL
Several genetic and epigenetic alterations culminate in dysfunctions of lymphoid cell’s signaling pathways such as Notch, NF-kB, BCR, DNA damage repair, cell cycle regulation, apoptosis, chromatin modification, and RNA metabolism [42]. This results in CLL development and progression, being some genetic variants associated with familial CLL [1]. For instance, the NF-kB and BCR signaling pathways are commonly altered, being constitutively activated, resulting in an increased expression of anti-apoptotic genes such as BCL2 and BCL2L1 [2, 7]. The activation of the NF-kB signaling pathway is thought to be a mechanism of resistance to disease treatment, in CLL [43, 44], and may be abnormally activated due to the mutation or deletion of the BIRC3 gene, a negative regulator of this pathway [44, 45].
1.1.6 CLL genomic alterations
1.1.6.1 Chromosomal abnormalities: reported CNVs and translocations in CLL
Chromosomal abnormalities such as copy number variations (CNVs), trisomies, and translocations are detected, by Interphase Fluorescent In Situ Hybridization (iFISH), in about 80% of diagnosed CLL cases [2]. A CNV is a segment of DNA with 1Kb or more, that presents a variation (deletion or amplification) in the number of its copies, in comparison to a reference healthy DNA [46]. Some CNVs are harmful for the individual, and others are not, and that is why it is possible to stratify CNVs as: benign, likely to be benign, likely to be pathogenic, pathogenic (due to overlapping with microdeletions or microduplications related to known syndromes), or with unknown clinical significance [47, 48]. Considering CLL CNVs, the common known alterations are: the gains at 2p and 8q; deletions at 6q, 11q, 13q, 14q, 17p, and losses at 8p. Moreover, trisomy 12 is also commonly found in CLL patients [35]. The 2p and 8q gains, and 8p loss appear in 2-5% of CLL cases, del(6q) in ~6%, del(17p) in 3-8%, del(14q) in ~8%, trisomy 12 and del(11q) appear in 10-20% of cases, and del(13q) in 40-60% of cases, representing the most common CNV in CLL [49].
Chromosomal translocations might be found in 32-42% of patients [35, 50]. As an example of CLL translocations, and showing up in less than 5% of patients, are the translocations between chromosomes 14 and 18: t(14,18)(q32,q24); t(14,18)(q32,q21); or between chromosomes 14 and 19: t(14,19)(q32,q13) [35, 51]. These translocations lead to the relocation of the IGH locus, located at 14q32.33 [10, 52]. Although some studies have shown that the prognostic significance of translocations involving this locus depends on the other chromosome involved, their prognostic interpretation is controversial. In t(14,19)(q32,q13), the prognostic seems to be poor once it involves the BCL3 locus, located at 19q13.32, which regulates the expression of NF-kB target genes [53], and also for being associated to trisomy 12 and unmutated IGHV [35, 54]. As for t(14,18)(q32,q21), the resulting fusion gene is IGH-BCL2, and it has also been related to poor outcome [51, 55].
It is possible to find complex karyotypes (CK) in approximately 16% of CLL cases [35, 50]. CK usually involves three or more chromosomal abnormalities at the same time, causing an aggressive course of disease with short patient survival [10, 56]. It is known to be caused by impaired DNA double-strand break response, due to altered function of ATM, TP53, and RB1 genes, since they have a role in cell cycle checkpoint and apoptosis regulation [35, 57]. In CLL, this complexity seems to be
5
related to worse outcomes since it shortens the time to first treatment (TTFT), OS, and leads to worse response to therapy [21, 35, 56]. Besides CNVs’ number, also their size is a parameter for genomic complexity evaluation. CLL patients with poor prognosis abnormalities have shown to be the main group of patients with the largest CNVs, suggesting that genomic complexity might be itself a disease progression marker [21].
1.1.6.1.1 Genomic alterations as prognostic markers in CLL
The 13q deletion, when found as a sole alteration, is associated with favorable prognosis. Trisomy 12 is considered to be an intermediate risk alteration, and del(11q) and del(17p) are associated with a poor prognosis [42, 58]. Despite the fact that all of these alterations contribute to CLL pathogenicity, it is common to consider gains as being less harmful than losses [59]. In this disease, it is more common to find deletions than amplifications [48, 60].
1.1.6.1.2 Deletions at 13q
Deletions in the 13q region might involve RB1, a tumor suppressor gene that is also involved in cell cycle regulation, promoting G0/G1 transition [61]. RB1 gene deletion is associated with short TTFT and short OS [35, 62], contributing to accelerated disease progression [16, 63]. Single-Nucleotide Polymorphisms (SNP) microarray analysis of del(13q) cases have shown deletions of RB1 in only some cases, while the miRNA15a/16-1 locus was always deleted [49], which seems to support the idea that the absence of this last locus is a cause for CLL pathogenicity. It is located at 13q14.3 and downregulates anti-apoptotic genes such as BCL2 by targeting their mRNA [9, 10, 31, 64]. With this said, in case of del(13q), the absence of this locus leads to the upregulation of BCL2, increasing its expression, resulting in an enhanced anti-apoptotic function of the BCL2 gene product [9, 10]. Despite that, deletions involving 13q are considered low-risk abnormalities when detected as the only abnormality in the patient but lead to increased poor outcome when coexisting with other alterations [21, 58].
Bigger 13q deletions also lead to a worse outcome [21, 65]. In fact, del(13q) may occur with variable breakpoints, with a minimum size of 300Kb and a maximum of 70 megabases (Mb) [62]. The minimal deleted region (MDR) in 13q has been demarcated to begin in a distal position relatively to
RB1, encompassing DLEU2 gene and the miRNA15a/16-1 locus [66, 67]. Ouillette et al. [62]
classified the 13q deletion as type I del(13q), also known as “short” del(13q), or as type II del(13q). Whereas type I 13q deletions have <2Mb, type II del(13q) or “large” del(13q), often includes the RB1 gene, in contrast to type I [68, 69]. It is known that this alteration can occur in heterozygosity in about 76% of cases and in homozygosity in 24% of patients, being the homozygous state a characteristic later event in CLL development [10, 22, 23]. Moreover, Hernandez et al. [70] have seen that the increased burden of cancer cells with del(13q) is positively correlated with worse outcome, a discovery that made possible the stratification in two prognostic groups: one group with worse prognosis and short TTFT due to high percentage of del(13q) cells, and a group of better outcome with low percentage proportion of del(13q) cells [10].
1.1.6.1.3 Trisomy 12
Trisomy 12, related to an intermediate risk prognosis, frequently appears associated with other chromosomal abnormalities such as deletions on 11q, 13q, 14q, 17p, and trisomies 18 and 19 [35, 58]. Some trisomy 12 cases specifically with mutated-IGHV have already shown the tendency to acquire trisomy of chromosome 19 [35, 71]. This alteration occurs mostly in early stages of CLL and it seems
6
to be a driver for gene mutations in the transcription factor NOTCH1 and tumor suppressor gene TP53, located at 9q34.3 and 17p13.1, respectively [35, 72, 73]. From this aneuploidy results the upregulation of genes such as CDKN1B, CDK4, HIP1R, MYF6, MDM2 [35, 74] and CLLU1 [10, 75]. CLLU1, located at 12q22, has been proposed as a prognosis marker once its elevated burden of expression has been connected to shorter OS [49, 75]; the MDM2 gene is located at 12q15 and its upregulation leads to cell cycle deregulation [35, 49, 76]; and CDK4 expression product is a kinase that regulates the activity of the transcription factor E2F1, involved in cell proliferation, meaning that CDK4 upregulation leads to E2F1 overexpression. Hence, CDK4 upregulation might cause the over-proliferation of B cells with trisomy 12. This examples help explaining the pathogenesis of trisomy 12 [35, 77].
1.1.6.1.4 Deletions at 11q
Deletions at 11q can be variable in sizes, most of the times being larger than 20Mb [35, 78, 79]. The minimal deleted region spans from 11q22.3-q23.1, including the ATM gene (11q22.3) in about 30% of patients [10, 80] [81]. The ATM gene is a tumor suppressor gene also involved in the DNA damage repair pathway, being responsible for the induction of apoptosis when a damage is not repairable, or in the restoration of double-strand breaks (DSB) that might occur in the DNA [10, 82]. Other possibly deleted genes in this region are: RDX, CUL5, ACAT, NPAT, KDELC2, MRE11, and
BIRC3 [35]. Marasca et al. [83] have detected an association between del(11q), genomic instability,
and increased complexity once it is regularly accompanied by other CNVs, such as del(17p).
In conclusion, the prognostic information given by this CNV is of poor outcome once it leads, almost in every case, to disease progression whit short TTFT, OS, and remission duration [35, 84].
1.1.6.1.5 Deletions at 17p and TP53 mutations
Deletions in 17p region are less prevalent than the anomalies described on the last sections, but may rise up to 40% in patients undergoing chemotherapy, suggesting that these deletions may be triggered after treatment [44, 85]. It is already known that the standard chemoimmunotherapy treatments based on fludarabine, cyclophosphamide, and rituximab are not effective on del(17p) and/or TP53-mutated patients, or even in BIRC3 and IKZF3-mutated patients, since these alterations confer an advantage to tumor cells [7, 35, 86]. Therefore, del(17p) is usually classified as a bad prognosis marker and the treatment of patients with this genomic alteration should not include chemoimmunotherapy.
This deletion can appear as an early event or a secondary alteration acquired during clonal evolution, and that difference determines patient’s median OS: 4 to 5 years and 1 to 1.5 years, respectively [35, 87]. It is common to detect TP53 mutation in ~75% of del(17p) cases, on the remaining allele [35]. Even if del(17p) exists without this mutation on the other allele, or if TP53 mutation occurs in absence of del(17p), the disease prognosis is still bad [35, 88]. In fact, del(17p) is always associated with a very aggressive disease behavior, leading to short OS, due to unresponsiveness to therapy [10, 58, 89, 90]. TP53 expression might also be dysfunctional due to
MDM2 overexpression caused by trisomy 12 [35, 76]. MDM2 overexpression leads to cell cycle
deregulation since, in healthy situations, its product regulates p53 degradation. Enhanced degradation of p53 will affect p53 dependent genes and miRNAs. For example: miRNA-34a, a miRNA involved in the regulation of cell cycle arrest and apoptosis, is underexpressed in case of abnormal p53 degradation, leading to a more aggressive disease and therapy resistance [35, 91]. It is then easy to understand why fludarabine based therapy has no effect on these individuals: it is a p53 dependent
7
drug, and the absence of a functional TP53 gene incapacitates fludarabine’s required conditions to function properly [10, 22].
Concerning the size of this alteration, it is usual to find the short arm almost entirely deleted [10]. The minimal deleted region has 34Kb and encompasses ATP1B2 and TP53 genes, both located at 17p13.1 [92]. With the loss of this segment, genes such as TP53, CCND1, CCND3, BCL2, ATM, and
SYK become downregulated, being TP53 the most significantly underexpressed, whereas other genes
suffer upregulation (e.g. MYC, located at 8q). Altogether, these altered genes lead to increased genetic instability of del(17p) patients [77, 93]. Other alterations that, in some studies, have been associated with del(17p) are: del(4p), 8p loss, del(18p), del(20p), and 8q gain [21, 35, 94]. For instance, 8p loss and 8q gain have been identified in 80% and 44% of del(17p) cases, respectively [35, 95, 96]. Patients with del(17p) are also immunophenotypically atypical, presenting overexpression of CD79b, CD38, ZAP70, and unmutated IGHV [35, 97].
1.1.6.1.6 Deletions at 6q, 14q, 8p losses, and 2p and 8q gains
Concerning 2p gains, they occur only in ~5% of CLL cases, and may encompass the MYCN gene, located at 2p24.3 [98]. With that alteration, MYCN undergoes overexpression, suggesting its pathogenicity in CLL because of its known involvement in transcriptional misregulation in cancer and apoptosis [35, 99]. In fact, this gain appears as a secondary alteration in about 28% of untreated CLL patients, indicating a poor outcome prognosis [100]. The association between this alteration and del(11q) encompassing ATM gene is indicative of an even worse outcome since ATM absence is related to rapid disease progression [16, 101].
The deletions found at 6q occur in 3 to 7% of cases [35]. They show high heterogeneity, which difficult the determination of the minimal deleted region and consequently restrain knowledge of the genes which are responsible for the pathogenicity associated with this alteration [35, 102]. However, it is commonly considered as an intermediate-risk alteration [10, 103]. As for the gains and losses usually found in chromosome 8 (8p loss and 8q gain), their prevalence in CLL ranges from 2 to 5% of patients, and are known as valuable prognosis markers [35, 95]. The gains in 8q, when encompassing the MYC gene, are indicative of an aggressive phenotype since this gene’s overexpression is associated with CLL progression and high risk clinical stage [104]. Deletions at 14q occur with a prevalence of ~8% of CLL cases [49]. Their physiological consequences are not well known yet, making it difficult to determine their prognostic value. Breakpoints may occur in centromeric or telomeric regions [105].
1.1.6.2 Somatic mutations and SNPs in CLL
Among the most frequent somatic mutations in CLL are: IGHV mutations (60-65% of cases); TP53 gene mutation (4-12% of untreated patients); ATM gene mutation (12% of cases, and in 30% of patients with del(11q)) [10, 106, 107]; NOTCH1 gene mutations (10% of patients); SF3B1 gene mutation (5-10% of cases); BIRC3 gene mutation (4% of cases), and MYD88 gene mutation (3-5% of cases) [49]. Some patients may even show germline ATM mutations, which is a risk factor for the development of CLL [10, 108]. NOTCH1 is involved in cell growth, differentiation, and self-renewal through the regulation of several genes such as MYC, TAL1, GATA2, RUNX1. NOTCH1 mutations are linked to short survival, short treatment resistance, and disease progression [49], frequently appearing in trisomy 12 cases that have a poor outcome prognosis [35]. The mutations in this gene lead to the constitutive activation of the Notch signaling pathway, enabling cell’s resistance to apoptosis and steering altered expression of the genes that depend on this pathway [10, 109]. The majority of
NOTCH1 mutation cases in CLL occur simultaneously with unmutated IGHV, indicating a poor
8
occurring in del(11q) cases, and also both in IGHV-mutated and IGHV-unmutated patients [10, 111]. Other possibly mutated genes are: BIRC3, NFKBIE, NFKB2 (involved in NF-kB signaling pathway);
MYD88 and IKZF3 (involved in BCR signaling); CDKN1B and CDKN2A (involved in cell cycle
regulation); CHD2, ARID1A and ZMYM3 (involved in chromatin modification); SF3B1, MGA and
XPO1 (involved in RNA metabolism); and other genes such as KRAS, NRAS, PTPN11, ZNF292, and SMARCA2 [42, 49]. BIRC3 and SF3B1 gene mutations have been found to occur in patients that
underwent fludarabine treatment, suggesting the refractory effect of this drug in those patients. In fact, previous reports have found that BIRC3 mutations are detected in 24% of fludarabine-refractory CLL patients [44, 85]. These mutations might therefore account for the ineffectiveness of fludarabine when used in patients with BIRC3 and SF3B1 mutations, suggesting that some therapeutic adjustments have to be made [10, 112].
Finally, genetic variants resulting from SNPs in genes such as IRF4, LEF1, BCL2, TERT or, for example, SNPs on the miRNA15a/16-1 locus might be found, being related with familial CLL [1].
1.1.6.3 Epigenetic alterations in CLL: altered methylation profiles
Epigenetic alterations such as hypomethylation of BCL2 gene [113] and/or hypermethylation of
CD38 gene may also occur in CLL [10], resulting in a heterogeneous methylation profile [1]. The CD38 gene, located at 4p15.32, is involved in apoptosis induction [114, 115]. If hypermethylated, CD38 is underexpressed, consequently resulting in reduced apoptosis. As for the BCL2 gene, as
previously mentioned in section 1.1, it is an anti-apoptotic gene. This means that, when hypomethylated, BCL2 is overexpressed.
CLL patient’s genome is usually globally hypomethylated [10, 116-118], but can also present some tumor suppressor gene’s promoters hypermethylated [10, 114, 119]. Yu et al.[120] quantified methylated DNA through methylation index (MI) analysis in CLL patients with different Rai stages, and it has shown to be useful and informative on CLL progression: most Rai 0-I patients have shown low MI; clinically stable disease patients had an even lower MI; and Rai IV patients presented the highest MI values. Elevated MI values suggest the hypermethylation of tumor suppressor gene’s promoters, which relate to a poor outcome [10, 120].
1.1.6.4 Clonal evolution in CLL
Clonal evolution should be taken into account since it determines the development of this disease, both in treated and untreated patients. According to some follow-up studies using Fluorescence In Situ Hybridization (FISH), clonal evolution occurs in up to 43% of CLL patients [21, 23, 121] and most of the times it arises as a late event [16]. However, each patient has a different probability of developing clonal evolution, based on age and CLL baseline features such as IGHV mutational state and lactate dehydrogenase (LDH) level [122]. Clonal evolution is characterized by the appearance of sub-clones with newly acquired genetic abnormalities [21], a phenomenon that depends on: 1) selective pressures from the microenvironment in which CLL B cells are located [35, 73]; 2) genetic instability driven by genetic and epigenetic alterations [16, 123]; 3) and by the interacting immune system cells present, such as T cells [8, 124]. The study of clonal evolution has been proven to be important for prognosis determination at the time of diagnosis. For example, the presence of various clones in early stages is indicative of a potentially more aggressive disease in the future [16, 23]. Hence, an initial assessment on clonal diversity has the potential to help predicting the disease course and consequently to help stratify patients according to their risk level. Clonal evolution has been linked to disease progression, and most of the times to a poor outcome due to the presence of unmutated IGHV and ZAP70 and CD38 overexpression [21-23, 121]. In fact, Berkova et al. [121] succeeded to positively correlate the
9
presence of these three markers with the development of clonal evolution. Also in 2011, and using genomic SNP microarray analysis in a follow-up study, Gunnarsson et al.[21] found an increased occurrence of clonal evolution in IGHV-unmutated samples relatively to IGHV-mutated samples. Even among IGHV-mutated samples, it was possible to detect a different incidence of clonal evolution,
being that untreated IGHV-mutated individuals did not shown any novel genomic alteration in the follow-up study, whereas treated IGHV-mutated individuals did show some novel alterations. In conclusion, more aggressive B cell phenotypes seem to be a risk factor for CLL clonal evolution and disease progression.
CLL genomic alterations such as del(13q), trisomy 12, NOTCH1 mutation, or MYD88 mutation usually occur as early events [35, 73]. It has been reported that this disease might be originated by large genomic losses or gains [6, 86]. It has also been found that del(13q) can result from clonal evolution since it was found to be the most prevalent newly-acquired abnormality in the CLL follow-up study previously referenced [22]. Loss of the miRNA15a/16-1 locus (located at 13q) seems to be one of the triggering events of CLL [8, 66]. There is also the possibility that CLL is driven by the recognition of autoantigens or bacterial compounds such as lipid A [8, 125]. On the other hand, abnormalities such as ATM, TP53, or SF3B1 mutations occur in more advanced stages, clearly resulting from clonal evolution [35, 73]. Alterations as del(11q) and del(17p) might occur both as early or late events [35, 87, 126], but appear mostly in later stages of disease [12, 58].
Regarding patients undergoing chemotherapy, the events of clonal evolution are triggered by the selection of specific sub-clones, mainly with TP53 and/or SF3B1 mutations, that represent driver mutations, and that proliferate and accumulate until they substitute the other existing sub-clones [35, 73].
1.2 CLL genomic characterization: comparing the effectiveness of conventional and molecular cytogenetic techniques
Over the last decades, several techniques such as conventional cytogenetics through chromosome banding; iFISH; Multiplex Ligation-dependent Probe Amplification (MLPA); Comparative Genomic Hybridization (CGH); and microarray comparative genomic hybridization technologies like bacterial artificial chromosomes (BAC) arrays, SNP microarrays, and oligonucleotide microarrays, have enabled clinicians and researchers to characterize CLL genomic alterations. All these techniques have contributed to the study of CLL patients since they detect the unbalanced chromosomic alterations that recurrently occur in this pathology. However, it is important to bear in mind the pros and cons of the different techniques mentioned. Therefore, on the next paragraphs, they will be briefly discussed and compared.
Cytogenetic abnormalities are important prognostic markers in CLL, making the genetic characterization of CLL patients extremely important in clinical practice since the treatment of each patient depends on detailed knowledge of his/hers genomic abnormalities [1]. The detection of chromosomal alterations by conventional cytogenetics using G-banding cytogenetics (GTG) enables the study of chromosome number and structure but it is conditioned by the characteristic cell cycle arrest between phases G0/G1 that occurs on this disease, making it difficult to obtain an acceptable mitotic index needed for analysis [10, 127]. The alterations described in section 1.1.6 are considered valuable prognostic markers since their detection and the determination of the affected genes enables the development of specific personalized therapies [128-130]. For example, patients with del(13q) usually have a good prognosis whereas patients with del(17p) have poor prognosis and short survival. This knowledge supports the importance of a more precise genomic characterization of CLL diagnosed patients, in order to apply personalized therapeutic strategies and even to identify new risk patient groups and therapeutic targets. Some improvements have been achieved concerning medium