www.jped.com.br
ORIGINAL
ARTICLE
Microarray-based
comparative
genomic
hybridization
analysis
in
neonates
with
congenital
anomalies:
detection
of
chromosomal
imbalances
夽
Luiza
Emy
Dorfman
a,
Júlio
César
L.
Leite
b,
Roberto
Giugliani
a,b,
Mariluce
Riegel
a,b,∗aProgramadePós-graduac¸ãoemGenéticaeBiologiaMolecular,UniversidadeFederaldoRioGrandedoSul(UFRGS),
PortoAlegre,RS,Brazil
bServic¸odeGenéticaMédica,HospitaldeClínicas,PortoAlegre,RS,Brazil
Received27February2014;accepted28April2014 Availableonline6September2014
KEYWORDS
Birthdefects; Congenital anomalies; Newbornselective screening; Chromosomal abnormalities; Molecular cytogenetics; Array-CGH
Abstract
Objective: Toidentifychromosomalimbalancesbywhole-genomemicroarray-based compara-tivegenomichybridization(array-CGH)inDNAsamplesofneonateswithcongenitalanomalies ofunknowncausefromabirthdefectsmonitoringprogramatapublicmaternityhospital. Methods: Ablindgenomicanalysiswasperformedretrospectivelyin35storedDNAsamplesof neonates bornbetween Julyof2011andDecember of2012.AllpotentialDNA copynumber variationsdetected (CNVs)were matchedwiththosereportedinpublicgenomicdatabases, andtheirclinicalsignificancewasevaluated.
Results: Outofatotalof35samplestested,13genomicimbalancesweredetectedin12/35 cases(34.3%).In4/35cases(11.4%),chromosomalimbalancescouldbedefinedaspathogenic; in5/35(14.3%)cases,DNACNVsofuncertainclinicalsignificancewereidentified;andin4/35 cases(11.4%),normalvariantsweredetected.Amongthefourcaseswithresultsconsidered causally related totheclinicalfindings, twoofthefour (50%)showed causativealterations alreadyassociatedwithwell-definedmicrodeletionsyndromes.Intwoofthefoursamples(50%), thechromosomalimbalancesfound,althoughpredictedaspathogenic,hadnotbeenpreviously associatedwithrecognizedclinicalentities.
Conclusions: Array-CGHanalysisallowedforahigherrateofdetectionofchromosomal anoma-lies, andthisdeterminationisespecially valuableinneonates withcongenitalanomalies of unknownetiology,orincasesinwhichkaryotyperesultscannotbeobtained.Moreover,although theinterpretationoftheresultsmustberefined,thismethodisarobustandprecisetoolthat
夽
Pleasecitethisarticleas:DorfmanLE,LeiteJC,GiuglianiR,RiegelM.Microarray-basedcomparativegenomichybridizationanalysisin neonateswithcongenitalanomalies:detectionofchromosomalimbalances.JPediatr(RioJ).2015;91:59---67.
∗Correspondingauthor.
E-mail:[email protected](M.Riegel).
http://dx.doi.org/10.1016/j.jped.2014.05.007
canbeusedinthefirst-lineinvestigationofcongenitalanomalies,andshouldbeconsideredfor prospective/retrospectiveanalysesofDNAsamplesbybirthdefectmonitoringprograms. ©2014SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
PALAVRAS-CHAVE
Defeitoscongênitos; Anomalias
congênitas; Triagemseletivade recém-nascidos; Anomalias cromossômicas; Citogenética molecular; CGH-array
Hibridizac¸ãogenômicacomparativabaseadaemmicroarranjosemneonatoscom anomaliascongênitas:detecc¸ãodedesequilíbrioscromossômicos
Resumo
Objetivo: Identificardesequilíbrioscromossômicospormeiodahibridizac¸ãogenômica compa-rativabaseadaemmicroarranjos(CGH-array)emamostrasdeDNAdeneonatoscomanomalias congênitasdecausadesconhecidadeumprogramademonitoramentodedefeitoscongênitos emumamaternidadepública.
Métodos: Uma análise genômica cega foi realizada retrospectivamente em 35 amostras armazenadasde DNAdeneonatos nascidosentrejulhode2011edezembro de2012.Todas aspossíveisvariac¸õesnonúmerodecópias(CNVs)deDNAforamcomparadascomasrelatadas embasesdedadosgenômicospúblicas,esuarelevânciaclínicafoiavaliada.
Resultados: Deumtotalde35amostrastestadas,foramdetectados13desequilíbriosgenômicos em12/35casos(34,3%).Em4/35casos(11,4%),osdesequilíbrioscromossômicospoderiamser definidoscomopatogênicos;em5/35(14,3%)delesforamidentificadasCNVsdeDNAde relevân-ciaclínicaincerta;e,em4/35(11,4%),foramdetectadasvariac¸õesnormais.Dentreosquatro casoscomresultadosconsiderados relacionadoscausalmenteaosachadosclínicos,2/4(50%) apresentaramalterac¸õescausaisjárelacionadasasíndromesdemicrodelec¸ãobemdefinidas. Em2/4amostras(50%),osdesequilíbrioscromossômicosencontrados,emborapreditivoscomo patogênicos,nãoestavamrelacionadosanteriormenteaentidadesclínicasreconhecidas. Conclusões: AanálisedeCGH-arraypermitiumaiortaxadedetecc¸ãodeanomalias cromossômi-cas,eessadeterminac¸ãoévaliosaprincipalmenteemneonatoscomanomaliascongênitasde etiologiadesconhecidaouemcasosemqueosresultadosdocariótiponãopodemserobtidos. Alémdisso,emboraainterpretac¸ãodosresultadosdevaserrefinada,essemétodoéuma fer-ramentarobustaeprecisaquepodeserusadanainvestigac¸ãodeprimeiralinhadeanomalias congênitasedeveserconsideradaemanálisesfuturas/retrospectivasdeamostrasdeDNApor programasdemonitoramentodedefeitoscongênitos.
©2014SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduction
Although Mendelian, chromosomal, and environmental causeshave beenestablishedfor manycongenital anoma-lies and dysmorphic syndromes, the precise etiology of severalsuchconditionshasnotyetbeenidentified.Etiologic investigations of congenital anomalies suggest that 6% of birth defects arerelated to chromosomal abnormalities.1
However,theproportionofchromosomalanomaliesinbirth defects may be higher. Some individuals with congenital anomaliesmayhavegenomicimbalancesbelowthe resolu-tion(>5Mb)ofstandardchromosomeanalysis.Inthelast decade, significant developments in themolecular detec-tionofchromosomal imbalanceshave occurred, andtheir causal relationship to congenital anomalies and mental disabilities has increased. The considerable gap between theresolution for detectionof chromosomeabnormalities with light microscopy and molecular gene analysis was bridged with the introduction of molecular approaches, suchasmicroarray-based comparative genomic hybridiza-tion(array-CGH).Array-CGHiscurrentlyapowerfulmethod forthesimultaneousdetectionofchromosomalimbalances
and the most prevalent chromosome abnormalities. It allowsforthedetectionoftrisomiesandlargechromosomal anomalies (already recognized by standard karyotype analysis) as well as smaller submicroscopic chromosomal imbalances (deletions, duplications, or triplications of any chromosomalregion, few of which arerecognized by fluorescence in situ hybridization [FISH]) that result in copy-numbervariations(CNVs).Severalstudieshaveshown that the use of array-based technologies increases the detection rate of chromosomal abnormalities to approxi-mately14%to18%,comparedwitharateofapproximately 3% (excluding trisomy 21) using standard cytogenetic approaches in individuals with developmental delays, intellectual disabilities,learningdifficulties,multiple con-genitalabnormalities (MCAs), autisticspectrum disorders, schizophrenia, and other neuropsychiatricdisorders.2 The
overallfrequencyofunbalancedchromosomeabnormalities was reported in neonates as 0.43%, according to recent reports.3,4 Therefore, the introduction of genome-wide
chromosomal abnormalities consistent with a genetic/ genomicdisorder.
Therefore, the aim of this study was toidentify chro-mosomal imbalances using a retrospective whole-genome array-CGHanalysisinstoredDNAsamplesofneonateswith congenital anomalies of unknown cause. In addition, this study evaluated the contribution of array-CGH as a first-line diagnostictoolinneonates withcongenitalanomalies evaluatedbyabirthdefectsmonitoringprogramatapublic maternityhospitalinSouthernBrazil.
Methods
Sampleselection
Thisretrospectivestudywasperformedusingde-identified DNAsamplesextracted fromthebloodofneonates,which wereobtained fromthe biorepositoryofthe Programade Monitoramento de Defeitos Congênitos (PMDC) of Hospi-tal de Clínicas de Porto Alegre (HCPA), Brazil. Subjects were less than 30 days of age, presenting a wide range ofcongenital anomaliesof unknown causeandinwhom a chromosomalabnormalitywassuspected.Theclinical indi-cationsforcytogeneticanalysisatthetimeofreferralwere taken from the clinical and laboratory data collected at birthandavailableinhospitalrecords,anddidnotinclude follow-upinvestigationsandinformationaboutdisease out-comes.Cases withoutenoughclinicaldatawereexcluded, as were cases where mothers had clinical or laboratory suspicion of infectious/parasitic diseases or a history of use/abuseofillicitdrugs/alcoholduringpregnancy. Accord-ingtothesecriteria, atotalof45sampleswereselected, but ten were excluded because they did notachieve the optimalDNAqualityneededforthearray-CGHanalysis,and thus,thestudywasconductedwith35samples.Theresults ofpreviouschromosomeanalyseswereobtainedin32cases. Conventionalcytogenetictestingatthe500-550bandlevel resolutionwasinitiallynormalforallcases,butinonecase areportofanabnormalkaryotypewasprovidedlater.This studywasapprovedbytheInstitutionalReviewBoardofthe HCPAandwasconductedinaccordancewithcurrent institu-tionalethicsrulesregardingtheuseofbiologicalmaterials frombiorepositories.5
Whole-genomeArray-CGH
Oligonucleotide array-based CGH wasperformed using an 8×60k whole-genome platform (design 021924, Agilent Technologies,SantaClara,USA),whichhasanaverage spac-ing of 40kb between probes. Genomic DNA was isolated from the peripheral blood (provided by the PMDC-HCPA) of35 neonatalindividualsand subsequently analyzed.For each experiment, a gender-mismatched normal reference (Promega Corp. Madison, WI, USA) was used. The exper-iments were performed according to the manufacturer’s protocol.Imagesofthearraysweretakenusinga microar-rayscanner (design G2600D, Agilent, California,USA) and processedusingFeatureExtractionsoftware(designv9.5.1, Agilent,California,USA),bothfromAgilent.Theraw data were analyzed by Agilent Cytogenomics (design v2.7.8.0, Agilent, California, USA) software with the statistical
algorithmADM-2,usingathresholdof6.0andafour-probe minimumaberrationcall.Subsequent software normaliza-tionofthedatawasperformed fortheverificationofDNA copy number changes. The p-values for each probe were calculated,providingadditionalobjectivestatistical crite-riatodeterminewhetherthedeviationofeachprobefrom zerowasstatisticallysignificant.6Allexperimentsincluded
twoarray hybridizationspersample, andtheresultswere recordedandcompared.Onlygenomicimbalancesdetected inbothdye-swapexperimentswerereported.
DataAnalysis
Whole-genomearray-CGHdataanalyseswereperformedina blindedfashion;sampleswerereceived,de-identified,and investigatorswhoperformed thearray-CGH analyseswere notawareofthepreviousclinicalandlaboratoryinformation related to each sample. The DNA copy number varia-tions(CNVs)detected werecomparedwithCNVs reported in publicly available online resources and databases of chromosomal abnormalities and variants.7---13 The CNVs
(gains/duplicationsorlosses/deletions)wereclassifiedinto differentcategories:benignCNV(normalgenomicvariant); CNVofuncertainclinicalrelevance(variantofuncertain sig-nificance [VOUS]); and CNV of possible clinical relevance (pathogenic variant). In thisstudy, the pathogenic abnor-malitiesincludedthedetectionofCNVsinknownpathogenic regions,deletion/duplication>3Mbinsize,orvisibleby G-bandedkaryotypethathavenotbeenreportedinthenormal population,anddeletionsorduplications<3Mbpreviously reported as pathogenic. Benign deletions or duplications includedvariantswelldocumentedinthenormalpopulation orpreviouslyreportedasbenign.Deletionsorduplications were classified asbeing VOUS when insufficient evidence wasavailabletoconcludeiftheCNVwaseitherpathogenic orbenign.
Results
Thedataof the35neonates withcongenitalanomalies of unknowncause,bornbetweenJulyof2011andDecemberof 2012,whoseDNAsampleswereanalyzedbywhole-genome array-CGH, are presented in Table 1. The maternal age ranged from 16 to41 years of age. This study identified 12(34.3%)caseswithDNAcopynumbervariations(CNVs). Fromthosecases,7/12(58.3%)weremaleand5/12(41.7%) werefemale.Thedetailsofthearray-CGHresultsfromthe caseswithgenomicimbalancesaresummarizedinTable2. Thirteen CNVs were identified in 12 individuals. Overall, duplicationswereverifiedin6/35(17%)anddeletionswere verifiedin7/35(20%)ofthecases.In6/35(17%)ofthecases, onlyadeletionwasidentified;5/35cases(14.3%)onlyhad aduplication,and1/35(2.8%)hadadeletionanda dupli-cation. Additionally, a FISH test confirmed the array-CGH resultsinonedeletioncase(case14)fromwhichstoredcells wereavailable(datanotshown).
A
B
chr1 chr4 chr6 chr10 chr17
C
D
E
2
1
0
–1
–2
2
1
0
–1
–2
2
1
0
–1
–2
2
1
0
–1
–2
2
1
0
–1
–2
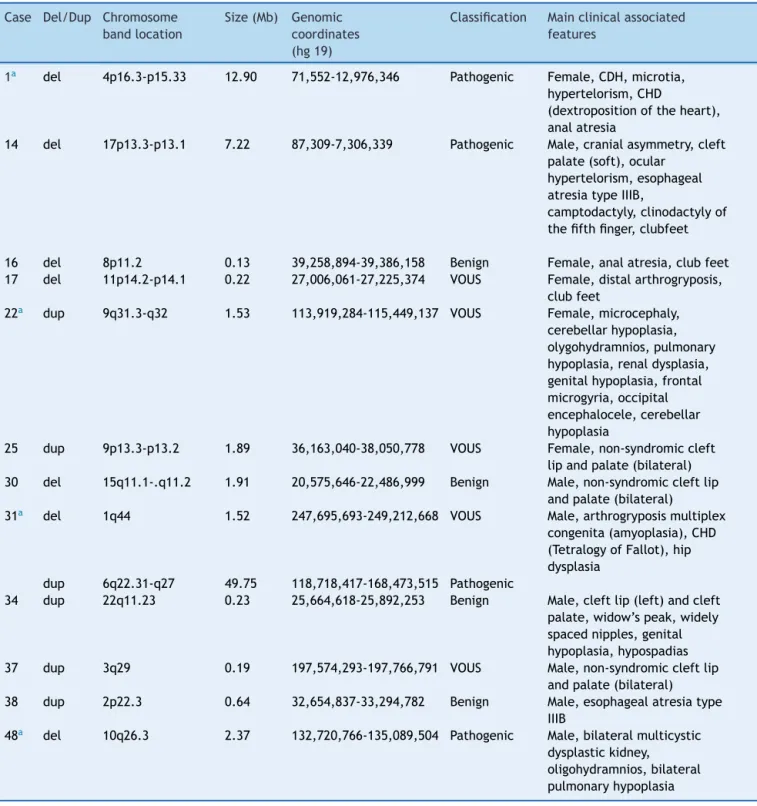
Figure1 Array-CGHratioprofilesofthechromosomesinfourneonateswithpathogenicchromosomalimbalancesusinggenomic
DNAfromtheneonatesastest(inred)andDNAfromnormalsubjectsasreference(inblue).Thetest/referenceratiodataforeach chromosomeareshown.Eachdotrepresentsasingleprobe(oligo)spottedonthearray.Thelogratioofthechromosomeprobes isplottedasafunctionofchromosomalposition.Copynumberlossshiftstheratiototheleft(valueofapproximately-1x).Copy numbergainshiftstheratiototheright(valueofapproximately+1x).Theideogramofeachchromosome(leftmargin)showsthe locationofeachprobe.Theprobelog2ratioswereplottedaccordingtogenomiccoordinates(basedontheUCSCGenomeBrowser, February2009,NCBIBuild37referencesequence). A:A∼1.5Mbterminaldeletionatchromosome1q44(blue line)incase31.
B:A∼12.9Mbterminaldeletionatchromosome4p16.3-p15.33 (bluebox)incase1.C:A ∼49.7Mbinterstitialduplicationat
chromosome6q22.31-q37(bluebox)incase31.D:A∼2.37Mbterminaldeletionatchromosome10q26.3(bluebox)incase48.E:
A∼7.2Mbterminaldeletionatchromosome17p13.3-p13.1(bluebox)incase14.
knownMiller-Diekersyndrome (MDS) region. FISHanalysis confirmedthedeletionofthechromosome17p13.3region (datanotshown).Theotherfourcaseswithoralfacialclefts showedCNVsthatwereclassifiedasbenignorasVOUS.Of thetwocaseswitharthrogryposismultiplexcongenital(17 and31),inoneindividualaninterstitialduplicationofthe longarmofchromosome6atband q22.31-q27wasfound, aswell asaterminaldeletionof thelongarm of chromo-some1atbandq44.Thepreviouskaryotypeanalysisshowed theidentificationofachromosomalabnormalityofunknown origininvolvingthelongarmofchromosome6,butnotthe chromosomalimbalancethatinvolvedchromosome1.This infantdiedat theageof35 days.Of fiveadditionalcases with MCAs (1, 16, 22, 38, and 48), this study identified clinicallysignificant chromosomal imbalances or potential
pathogenic CNVs in threecases (1,31, and 48).The sub-jectsdied at theage of 2days,5hours,and3 daysafter birth,respectively.Overall,thedeletionswereclassifiedas pathogenicinthreecases(1,14,and48),asbenignintwo cases(16 and30), andasVOUSin twocases(17 and31). The duplicationswereclassifiedaspathogenicinonecase (31), asbenign in twocases (34 and38), and asVOUS in threecases(cases22,25,and37).Examplesofarray-CGH graphicaloverviewsareshowninFig.1.
Discussion
Table1 Summaryoftheclinicalindicationsfromthe35 samplesatthetimeofreferralforchromosomalanalysis.
Case Mainclinicalassociatedfeatures
1a Female,CDH,microtia,hypertelorism,CHD
(dextropositionoftheheart),analatresia 2 Male,CHD(tetralogyofFallot)
4 Male,omphalocele,microcephaly
5 Male,omphalocele,limbagenesis,ambiguous
genitalia,analatresia,bladderdysfunction
10 Female,gastroschisis
12a Male,CDH
13 Male,unilateralphocomelia,hipdysplasia
14 Male,cranialasymmetry,cleftpalate(soft),ocular hypertelorism,esophagealatresiatypeIIIB, camptodactylyofthe3rd,4th,and5thfingers, clinodactylyofthe5thfinger,clubfeet 15 Male,analatresia,clubfeet
16 Female,analatresia,hypoplasticgenitalia,CHD 17 Female,arthrogryposismultiplexcongenital
(amyoplasia)
19 Female,non-syndromicunilateralcleftlip(left)and cleftpalate
22a Female,microcephaly,cerebellarhypoplasia,
olygohydramnios,pulmonaryhypoplasia,renal dysplasia,genitalhypoplasia,frontalmicrogyria, occipitalencephalocoele,cerebellarhypoplasia
23 Female,omphalocele,microcephaly
24 Male,micrognathia,singleuppermedianincisor 25 Female,non-syndromiccleftlipandpalate
(bilateral)
29 Male,CHD(tetralogyofFallot)
30 Male,non-syndromiccleftlipandpalate(bilateral) 31a Male,arthrogryposismultiplexcongenita
(amyoplasia),CHD(tetralogyofFallot),hipdysplasia 32a Female,bilateralmulticysticdysplastickidney,
oligohydramnios
33 Male,cleftpalate,clubfeet
34 Male,cleftlip(left)andcleftpalate,widow’speak, widelyspacednipples,genitalhypoplasia,
hypospadias
35 Female,HPE,oligohydramnios,microcephaly, unilateralchoanalatresia
37 Male,non-syndromiccleftlipandpalate(bilateral) 38 Male,esophagealatresiatypeIIIB
40 Male,intrauterinegrowthretardation
41 Female,gastrosquisis
42 Female,CHD(tetralogyofFallot) 43 Male,non-syndromiccleftpalate
44 Male,microgyria,incompletelissencephaly, micrognathia
46 Male,meningocele,clubfoot(left)
47 Female,gastroschisis
48a Male,bilateralmulticysticdysplastickidney,
oligohydramnios,bilateralpulmonaryhypoplasia
49 Female,gastroschisis
50 Male,arthrogryposismultiplexcongenita, micrognathia
CDH,congenitaldiaphragmatichernia; CHD,congenitalheart defect;HPE,holoprosencephaly.
a Patientdied.
available from neonates with congenital anomalies of unknown etiology. In addition, this study evaluated the contribution of array-CGH as a first-line diagnostic tool in neonates with congenital anomalies in a birth defects monitoring program at a public maternity hospital in SouthernBrazil.
Todate,thelargestnewbornscreening(in20,126 unse-lected cases) using array-CGH analysis as a first-line test revealedthat87/20,126(0.43%)oftheneonatalcaseshad chromosomalimbalances(53casesofaneuploidies,23 dele-tions,and11duplications).4
Reddyetal.14reportedtheresultsofapopulation-based
studyof532 stillbirths.Inthissample, array-CGHanalysis yieldedmoreresultsthandidkaryotypeanalysis(87.4%vs. 70.5%),providedbetterdetectionofgeneticabnormalities (aneuploidyor pathogenic CNVs, 8.3% vs. 5.8%), andalso identified more genomic imbalances among 67 stillbirths withcongenitalanomalies(29.9%vs.19.4%).
When selective screening is performed, the use of array-based technologies demonstrates the ability to detectpathogenicimbalancesinapproximately14%-18%of postnatalcaseswithdevelopmentaldelays,intellectual dis-abilities,andMCAsreferredforanalysis.2,15---18 Thepresent
study verifiedgenomic imbalances in 4/35 (14.3%) of the cases that could be defined as pathogenic and causally relatedtotheabnormalphenotypes.Althoughthisstudywas performedinarelativelysmallcohort,therateofpositive findingsdetectedthrougharray-CGHisintherangereported inseveralpostnatalseries.
AlthoughaclearassociationexistsbetweenCNVsinboth syndromic and non-syndromic congenital anomalies, only fewlargecohortstudieshavespecificallyperformed whole-genome array-CGH analysis in samples of neonates with birthdefects.Luetal.19reportedthefrequencyofgenomic
imbalances identified in 638 neonates with various birth defectsreferredforchromosomalmicroarrayanalysis.They usedthreedifferentarrayplatformswithincreasingly exten-sivegenomiccoverageandcomparedtheresultsobtained. Overall,17.1%ofpatientswereidentifiedwithclinically sig-nificantabnormalities,withdetectionratesof13.7%,16.6%, and19.9%,dependingonthearrayplatformused.
In the present study, a previous karyotype analysis wasavailable in 32 cases andshowed that the frequency of chromosomal imbalances detected was 1/32 (3.1%). Thedetection yieldof genomic imbalances notpreviously detectedbykaryotypeanalysisincreasedto9/32cases(28%) withtheuseofarray-CGH,whichwasinagreementwiththe expectedincreaseddetectionyield.In4/35cases(11.4%), CNVscouldbedefinedaspathogenicandcausallyrelatedto theabnormalphenotypes.Ratedifferencesbetween differ-entstudiesmaybeduetothecohortsize,differencesinthe resolutionofthearrayplatformused,thecriteriaforpatient selection,andtheinterpretationoftheclinicalrelevanceof theCNVs.
Among the 4/35 pathogenic cases, in two cases (31 and48), the identified abnormalities found had not been previously associated with well-recognized syndromes. In the two other cases (1 and 14), causative alterations hadalreadybeen associated withwell-defined microdele-tion syndromes20 (Wolf-Hirschhorn Syndrome [WHS] and
Table2 Detailsofthearray-CGHfrom12sampleswithchromosomalimbalances.
Case Del/Dup Chromosome bandlocation
Size(Mb) Genomic coordinates (hg19)
Classification Mainclinicalassociated features
1a del 4p16.3-p15.33 12.90 71,552-12,976,346 Pathogenic Female,CDH,microtia,
hypertelorism,CHD
(dextropositionoftheheart), analatresia
14 del 17p13.3-p13.1 7.22 87,309-7,306,339 Pathogenic Male,cranialasymmetry,cleft
palate(soft),ocular hypertelorism,esophageal atresiatypeIIIB,
camptodactyly,clinodactylyof thefifthfinger,clubfeet
16 del 8p11.2 0.13 39,258,894-39,386,158 Benign Female,analatresia,clubfeet
17 del 11p14.2-p14.1 0.22 27,006,061-27,225,374 VOUS Female,distalarthrogryposis,
clubfeet
22a dup 9q31.3-q32 1.53 113,919,284-115,449,137 VOUS Female,microcephaly,
cerebellarhypoplasia, olygohydramnios,pulmonary hypoplasia,renaldysplasia, genitalhypoplasia,frontal microgyria,occipital encephalocele,cerebellar hypoplasia
25 dup 9p13.3-p13.2 1.89 36,163,040-38,050,778 VOUS Female,non-syndromiccleft
lipandpalate(bilateral)
30 del 15q11.1-.q11.2 1.91 20,575,646-22,486,999 Benign Male,non-syndromiccleftlip
andpalate(bilateral)
31a del 1q44 1.52 247,695,693-249,212,668 VOUS Male,arthrogryposismultiplex
congenita(amyoplasia),CHD (TetralogyofFallot),hip dysplasia
dup 6q22.31-q27 49.75 118,718,417-168,473,515 Pathogenic
34 dup 22q11.23 0.23 25,664,618-25,892,253 Benign Male,cleftlip(left)andcleft
palate,widow’speak,widely spacednipples,genital hypoplasia,hypospadias
37 dup 3q29 0.19 197,574,293-197,766,791 VOUS Male,non-syndromiccleftlip
andpalate(bilateral)
38 dup 2p22.3 0.64 32,654,837-33,294,782 Benign Male,esophagealatresiatype
IIIB
48a del 10q26.3 2.37 132,720,766-135,089,504 Pathogenic Male,bilateralmulticystic
dysplastickidney, oligohydramnios,bilateral pulmonaryhypoplasia
CDH,congenitaldiaphragmatichernia;CHD,congenitalheartdefect;VOUS,variantofuncertainsignificance;array-CGH, microarray-basedcomparativegenomichybridization;del,deletion;dup,duplication.
aPatientdied.
imbalancescouldhavebeenpreviouslydiagnosedby karyo-typeanalysisorbyFISHanalysisalone(usinglocus-specific probes for the critical chromosome region)if the clinical findings at the time of referral were indicative of a par-ticular microdeletion syndrome that could inform exactly whichregion(s)and/orchromosome(s)toinvestigate. How-ever, both samples were from subjects in whom neither
beof differentsizes, leadingtoabroad phenotypic spec-trum.
One of the two cases with arthrogryposis multiplex congenita (case 31) showed a large duplication of the long arm of chromosome 6 at bands q22.31-q27 and a smallerdeletionofthelongarmofchromosome1atband q44. The retrieval of laboratory records showed that a chromosomalabnormality of unknown origin involving the long arm of chromosome 6 was previously recorded, but no chromosomal imbalance involving chromosome 1 was identified. At that time, there was an expectation that parental karyotypeswould be performed tobetter define the type and origin (de novo or familial) of the extra materialonchromosome6.Array-CGHanalysisallowedfor additional genomic information regarding the previously identifiedduplicationat chromosome6 andthe detection of an additionalgenomic imbalance (deletionat chromo-some1)thatwasnotpreviouslyreported.Frequently,more than one CNV is identified in an individual. It is evident alreadyfromthekaryotype analysisthatthe chromosome duplications must involve many genes and be causally related to the congenital anomalies, as assumed in case 31. However, it has been recognized that the presence of another CNV could reduce or aggravate the clinical phenotype.21,22
From the twosamples withsyndromic cleft lip and/or cleft palate (cases 14 and 34) and the three with non-syndromic cleft lip and cleft palate (cases 25, 30, and 37),onecase (14)hadaclinicallysignificant7.2Mb dele-tionatchromosome17p13.3-p13.1thatcoincideswiththe known MDS microdeletion syndrome. In the other four cases, benign CNVs (30 and 34) or VOUS were identi-fied (cases 25 and 37). Approximately 30% of cleft lip and palate cases and 50% of cleft palate cases are rec-ognized as components of MCA syndromes.23 However,
both genetic and environmental factors are known to contributetotheoccurrenceofcleftlip andpalate, com-plicating the elucidation of the causative mechanisms. Considerableeffortshavebeenmadeinseekingcandidate gene(s)fornon-syndromiccleftsthrougharray-CGH, show-ing that it is an effective method for isolating candidate loci.24,25
The clinical relevance of 5/13 (36.7%) CNVs among the 12 cases withgenomic imbalances remains uncertain at present, as there is insufficient evidence to conclude whethertheCNVswereeitherpathogenicorbenign.When CNVs are detected that have no strong track record for clinicalimportance,theinterpretationofwhethertheyare causalforthebirthdefectcanbechallenging.Itshouldalso be considered that the CNV is potentially inherited from ahealthy parentand,in thiscase, couldbe apathogenic variant with incomplete penetrance or a benign familiar variation.Thehighlyvariablenatureofthegenomemeans thatcaremustbetakeninassigningpathogenicitytoCNVs detectedbyarray-CGH.FromtheCNVsclassifiedasVOUSin thisstudy,itmightbeexpectedthatparentalstudieswould beperformedtoallowabetterinterpretationandtoprovide valuableinformationforgeneticcounselingpriortoafuture pregnancy.Indeed,itisimportanttoreportdataon chromo-someimbalanceswithunclearclinicalsignificance,because someofthedatamayrepresentrecurrentCNVsthatcouldbe
associatedwithnovel syndromes.Reportsofpatients with similargenomicimbalancesandclinicalfindingscanleadto theidentificationofnewlyrecognizedgenomicdisordersor candidategenesassociatedwithisolatedcongenital anoma-lies.
Infourcases(16,30,34,and38),normalvariants clas-sified as benign were detected. It is recognized that all humansdifferintheirchromosomes atthesubmicroscopic leveland thateventhe genomesof normal,healthy indi-viduals have a high number of copy number changes.26
WhenseveralindividualswerescreenedforCNVs,atotalof 1,447copynumbervariableregionscovering360Mb(12%of genome)wereidentified.27CNVsareoftenrelativelysmall,
canbeinheritedfromaphenotypicallynormalparent,occur inmoregene-sparsechromosomalregions,andcontainmore repetitiveDNAsequences.ThedetectionofbenignCNVswas reportedinthisstudyfromgenomicregionsthatconsistently harborbenignvariants;thismightreducetheneedto per-formparentalstudiesin neonates inwhomproven benign CNVswereidentified.
A limitation of this study was the inability to distin-guish de novo from inherited genomic imbalances due to the unavailability of parental DNA. De novo CNVs in clinically significant gene regions are more likely to be causative.However,inheritedpathogenic CNVsshouldnot beexcludedasacauseofcongenitalanomaliesbecauseof theirvariableexpressivityandincompletepenetrance.28,29
PathogenicCNVsmaybeinheritedfromanapparently nor-mal parentand contribute to the abnormalphenotype in thechild. These types of CNVs arethought of as suscep-tibility loci, in that they increase the chance of a child developingcongenitalanomalies butmaynotbesufficient tocauseaphenotypebythemselves.Parentalstudiesshould be recommended for individuals for whom clinically sig-nificantfindingswerereported,todeterminewhetherthe CNVfindingsrepresentdenovoorfamiliarevents.Incases of a de novo chromosome imbalance, it is also recom-mendedtoobtaintheparentalkaryotypeinordertoexclude a balanced translocation in one of the parents. Although several common strategies have been proposed to help interpretthefindingsofgenomicimbalances,29,30thereare
no universal criteria thus far. It is essential to have the most accurate and up-to-date information on the clini-calsignificance of detected genomic imbalances, as well asCNVs at different positions in the genome, pathogenic mutations or polymorphisms in other individual genes, or nongenetic causes that might be required for a congen-ital anomaly to be expressed. Caution must be taken in the clinical interpretation of the array-CGH results. Fur-therconsultationsatgeneticsclinicsandextendedanalysis infamily members maybe necessary to provide accurate counselingtothefamiliesandtocalculate therecurrence risks.
inwhomthepresenceofchromosomalimbalanceswas sus-pected.
This study demonstrated the feasibilityand usefulness of array-CGH to identify deletions and duplications in stored DNA samples. It was shown that a proportion of neonateswithcongenitalanomaliesofunknowncausehad chromosomalimbalancesassociatedwiththeirphenotypes. Furthermore, this study demonstrated the detection of chromosomal abnormalities consistent with genetic syn-dromes at an early age, when often, only a few clinical findingsareclear.
Inconclusion,retrospectiveorprospectivearray-CGHas a first-line diagnostic tool would benefit families by pro-viding a more accurate diagnosis and impact the overall management in a significant number of cases from birth defectsmonitoringprograms.
Funding
CNPq/Brazil,grant402012/2010-0.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgments
Theauthorswould liketothanktheConselho Nacionalde DesenvolvimentoCientíficoeTecnológico(CNPq)for finan-cialsupport(grant402012/2010-0).
References
1.SchinzelA.Catalogueofunbalancedchromosomeaberrations inman.2nded.Berlin:WalterdeGruyter;2001.
2.HochstenbachR,Buizer-VoskampJE,VorstmanJA,OphoffRA. Genomearraysforthedetectionofcopynumbervariationsin idiopathicmentalretardation,idiopathicgeneralizedepilepsy and neuropsychiatric disorders: lessonsfor diagnostic work-flow and research. Cytogenet Genome Res. 2011;135:174---202.
3.Wellesley D, Dolk H, Boyd PA, Greenlees R, Haeusler M, Nelen V, et al. Rare chromosome abnormalities,prevalence andprenataldiagnosisratesfrompopulation-basedcongenital anomalyregistersinEurope.EurJHumGenet. 2012;20:521---6.
4.ParkSJ,JungEH,RyuRS,KangHW,ChungHD,KangHY.The clinicalapplicationofarrayCGHforthedetectionof chromo-somaldefectsin20,126unselectednewborns.MolCytogenet. 2013;6:21.
5.FernandesMS,Ashton-ProllaP,MatteU,MeurerL,OsvaldtA, BittelbrunnAC,etal.The Hospitalde ClinicasdePorto Ale-gre normative for the storage and use of human biological materialsandtheirassociatedinformationinresearch:an inter-disciplinaryapproach.RevistaHCPA.2010;30:169---79.
6.VermeeschJR, MelotteC,Froyen G, VanVoorenS, DuttaB, MaasN,etal.Molecularkaryotyping:arrayCGHquality crite-riaforconstitutionalgeneticdiagnosis.JHistochemCytochem. 2005;53:413---22.
7.DatabaseofChromosomalImbalanceandPhenotypeinHumans Using Ensemble Resources (Decipher). [cited 26 Jan 2014]. Availablefrom:http://decipher.sanger.ac.uk/
8.MacDonald JR, Ziman R, YuenRK, Feuk L, Scherer SW. The DatabaseofGenomicVariants:acurated collectionof struc-tural variation in the human genome. Nucleic Acids Res. 2014;42:D986---92.
9.European Cytogeneticists Association Register of Unbal-anced Chromosome Aberrations (ECARUCA). [cited 23 Feb 2014]. Available from: http://umcecaruca01.extern.umcn. nl:8080/ecaruca/ecaruca.jsp
10.EnsemblGenomeBrowser.[cited23Feb2014].Availablefrom: http://www.ensembl.org/index.html
11.The International Standards for Cytogenomic Arrays (ISCA). [cited 23 Feb 2014]. Available from: https://www. iscaconsortium.org/index.php
12.NationalCenterforBiotechnologyInformation(NCBI).[cited23 Feb2014].Availablefrom:http://www.ncbi.nlm.nih.gov/ 13.UniversityCaliforniaSantaCruz(UCSC)GenomeBrowser.[cited
23Feb2014].Availablefrom:http://genome.ucsc.edu/ 14.Reddy UM, Page GP, Saade GR, Silver RM, Thorsten VR,
Parker CB, et al. Karyotype versus microarray testing for geneticabnormalitiesafterstillbirth.NEnglJMed.2012;367: 2185---93.
15.IourovIY,VorsanovaSG,KurinnaiaOS,ZelenovaMA,Silvanovich AP,YurovYB.MolecularkaryotypingbyarrayCGHinaRussian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 2012;5: 46.
16.Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175---84.
17.Ahn JW, Bint S, Bergbaum A, Mann K, Hall RP, Ogilvie CM. ArrayCGHasafirstlinediagnostictestinplaceof karyotyp-ingfor postnatal referrals - resultsfrom four years’clinical application for over 8,700 patients. Mol Cytogenet. 2013;6: 16.
18.HillmanSC,McMullanDJ,HallG,TogneriFS,JamesN,Maher EJ,etal.Useofprenatalchromosomalmicroarray:prospective cohortstudyand systematicreviewandmeta-analysis. Ultra-soundObstetGynecol.2013;41:610---20.
19.LuXY,PhungMT, Shaw CA,PhamK, NeilSE,Patel A, et al. Genomicimbalancesinneonateswithbirthdefects:high detec-tionratesbyusingchromosomalmicroarrayanalysis.Pediatrics. 2008;122:1310---8.
20.VissersLE,StankiewiczP.Microdeletionandmicroduplication syndromes.MethodsMolBiol.2012;838:29---75.
21.Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367:1321---31.
22.GirirajanS.Genomicdisorders:complexityatmultiplelevels. GenomeMed.2013;5:43.
23.StanierP,MooreGE.Geneticsofcleftlipandpalate:syndromic genescontributetotheincidenceofnon-syndromicclefts.Hum MolGenet.2004;13:R73---81.
24.OsoegawaK,VessereGM,UtamiKH,MansillaMA,JohnsonMK, RileyBM,etal.Identificationofnovelcandidategenes associ-atedwithcleftlipandpalateusingarraycomparativegenomic hybridisation.JMedGenet.2008;45:81---6.
25.ShiM,MostowskaA,JugessurA,JohnsonMK,MansillaMA, Chris-tensenK, etal.Identification ofmicrodeletions incandidate genesforcleftlipand/orpalate.BirthDefectsResAClinMol Teratol.2009;85:42---51.
26.IafrateAJ,FeukL,RiveraMN,ListewnikML,DonahoePK,QiY, etal.Detectionoflarge-scalevariationinthehumangenome. NatGenet.2004;36:949---51.
28.Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW,etal.AmericanCollegeofMedicalGenetics recommenda-tionsforthedesignandperformanceexpectationsforclinical genomic copy number microarrays intended for use in the postnatalsettingfordetectionofconstitutionalabnormalities. GenetMed.2011;13:676---9.
29.Vermeesch JR, Brady PD, Sanlaville D, Kok K, Hastings RJ. Genome-widearrays:qualitycriteriaandplatformstobeused inroutinediagnostics.HumMutat.2012;33:906---15.