rev bras hematol hemoter. 2015;37(3):198–201
w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Case
report
Very
mild
forms
of
Hb
S/beta
+
-thalassemia
in
Brazilian
children
André
Rolim
Belisário
a,b,∗,
Rahyssa
Rodrigues
Sales
b,
Marcos
Borato
Viana
a aUniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,BrazilbFundac¸ãoCentrodeHematologiaeHemoterapiadeMinasGerais(HEMOMINAS),BeloHorizonte,MG,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received17November2014 Accepted19January2015 Availableonline15April2015
Introduction
Theclinical phenotypeof sicklecell/beta+-thalassemia(Hb
S/+-Thal)ishighlyvariable,andseverityisassociatedwith thequantitativedegreeofdecreaseintheproductionofthe betaglobinchains.1 Evidenceshowsthatdifferences inthe productionofhemoglobinA(HbA)andseveritycorrespond todifferentmolecularbeta-thalassemia(-Thal)mutations.2 Apreviousreportproposed aclassificationofHb S/+-Thal phenotypesbasedontherelativeconcentrationofHbA:Type I:1–7%ofHbA;TypeII:7–14%ofHbA;andTypeIII:14–25%ofHb A.2However,some-Thalmutationsleadtolowimpairment of-globinproductionandtheresultingphenotypedoesnot fitthisproposedclassification.Inasinglepatientcasereport, the−92(C>T)mutationwasassociatedwithahighlevelof HbA(45%)inanadultSicilianpatient withHb S/+-Thal.3 Recently, a combination of two sequence variants, IVS-II-839(T>C) andIVS-II-844 (C>A), was associatedwithavery mildphenotypeofsicklecelldisease(SCD).4Becauseofthe markedclinicalvariabilityofHbS/+-Thalpatients,molecular
∗ Correspondingauthorat:RuadasGoiabeiras,779,LagoaSanta,33400-000,Brazil.
E-mailaddress:[email protected](A.R.Belisário).
predictorsofdiseaseseveritywouldbehelpfultoguide treat-ment choicesinchildrenwiththisgenotype. Theobjective ofthisstudywastocharacterizethehematological parame-ters,clinicalfeatures,andmolecularbasisofverymildforms ofHbS/+-Thal inanewborncohortofMinasGeraisstate, Brazil.
Case
series
ThisisacaseseriesinvolvingchildrenfromtheMinasGerais StateSCDnewborncohort,Brazil.Aspartofayetincomplete study,56outof96(58.3%)childrenwithHbS/0-thalorHb S/+-Thalfromthatcohortbornbetween1998and2013had alreadybeensubmittedtoDNAanalysistoidentifythe-thal mutationscausingHbS/-thal.Childrenwereconsideredtobe eligibleforthisreportiftheyhadHbAconcentrationsequalto orabove25%confirmedbyhemoglobinelectrophoresis. Chil-drenhad hemoglobinFSApatternsdiagnosedbyisoelectric focusingandhigh-performanceliquidchromatographyinthe NewbornScreeningProgramandhavebeenfollowedupsince
http://dx.doi.org/10.1016/j.bjhh.2015.03.010
revbrashematolhemoter.2015;37(3):198–201
199
diagnosisintheoutpatientcareunitofFundac¸ãoHemominas, whichissituatedinthestatecapital,BeloHorizonte.
Laboratoryandclinicaldatawereretrievedbychartreview afterapprovalofthelocalinstitutionalreviewboard. Hema-tologicandgeneticstudieswereperformedontheparentsof twochildrentoelucidatetheinheritancepatternofthe-thal mutations.
Completebloodcountwasperformedusinganelectronic cellcounter(modelT-890,BeckmanCoulter,Hialeah,FL,USA orCell-Dynruby,Abbott,IL,USA).Hemoglobin electrophore-siswasconductedinanalkalinemedium(SPIFEkits,Helena Laboratories,Beaumont,TX,USA)andthepercentageofHb Fwasquantified byradialimmunodiffusion(HbFQUIPlate, Helena Laboratories,Beaumont, TX,USA). Thereticulocyte countwasmeasuredbyopticalmicroscopyusingbrilliant cre-sylblue.
GenomicDNAwasisolatedwithaQIAGENkit(QIAamp®
DNABloodMiniKit,Qiagen,Hilden,Germany).TheHBBgene was amplified with specific primers. Sequence data were generatedwith anABI 3130xl capillarysequencer (Applied Biosystems,FosterCity,CA,USA)usingstandardprotocols.A multiplexgappolymerasechainreaction(PCR)assaywasused todetectthemostcommonalpha-thalassemiadeletions.5
Writteninformed consentwasobtainedfrom parentsor guardiansofeverychildinaccordancewiththenormsofthe DeclarationofHelsinkiguidelineswiththechild’sassentbeing obtainedwhenappropriate.
Thestudyincludedfourunrelatedchildrenwith diagno-sisofHbS/+-Thal.Themeanagewas8.0±2.4years(range: 5.5–11.2years)and all weremale. These childrenwere fol-lowedforameanof7.7±2.4years(range:5.2–11years).
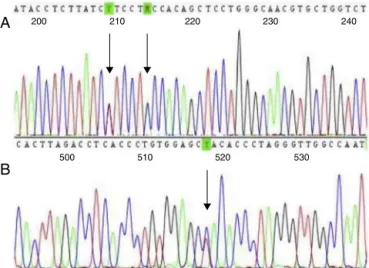
Twochildrenhad a−92(C>T)mutation(HBB:c.−142C>T) andtwohadIVS-II-844(C>A)(HBB:c.316-7C>A)plusIVS-II-839 (T>C)(HBB:c.316-12T>C)mutations(Figure1).Geneticfamily studiesshowedthattheIVS-II-844(C>A)andIVS-II-839(T>C) mutationswere incisinbothchildrenbecauseboth muta-tionswere inheritedfromjustoneoftheparents.Nochild hadco-inheritedalpha-thalassemiadeletions.Themean rel-ativeconcentrationofHbAwas40.4%andthatofHbSwas
200
500
A
B
510 520 530
210 220 230 240
Figure1–Sequencingfour-colorchromatogramshowing (A)IVS-II-844(C>A)andIVS-II-839(T>C)mutationsincis;
and(B)the−92(C>T)mutation.
54%.Genetic,laboratory,andfamilydataaresummarizedin
Table1.
Allchildrenwereclinicallyoligosymptomaticandled nor-mal lives.Except foran obstruction ofthe airways caused byadenoid glands, childnumber I wasasymptomatic dur-ing the follow-up period. Child number II suffered from two infectious episodes: virus infection, for which a five-dayhospitalizationwasrequired,andimpetigo,treatedwith benzathinepenicillin. Hehashad recurrentheadaches,but the neurologic physical examination was normal. Except for an episode of sinusitis, child number III was asymp-tomatic during the follow-up period. Child number IV sufferedfromrecurrentheadaches,buttheneurologic phys-icalexaminationwasnormal.Noneofthechildrenhavehad severe acute clinical manifestationsof SCD such as acute painfulcrises,stroke,acutechestsyndrome,oracutesplenic sequestration.
Discussion
OurstudydescribedagroupofHbS/+-Thalpatientswith nor-malornearlynormalhematologicaldataandtheabsenceof complicationsattributabletoSCD.Tothebestofour knowl-edge,thisisthefirststudytoreportontheoccurrenceof−92 (C>T),andIVS-II-844(C>A)/IVS-II-839(T>C)-thalmutations inBrazil.
Thedegreeof-chainsynthesisinHbS/+-Thalpatients depends primarilyon the -Thal molecular mutation. The IVS-II-844(C>A)/IVS-II-839(T>C)mutationaffectsthe consen-sussplicesiteandinterfereswithprocessingoftheprimary mRNAtranscript. Itmodifiesthe conservedpolypyrimidine tract ofthespliceacceptorsitecausing areductionofbeta globinchainexpressiontoaround60%ofnormal.4,6Carriers ofthemutationsdescribedinthisreportshowedameanHb Alevelof39.7%andwereoligosymptomatic.Thisfindingis consistentwiththefirstreportofthesetwovariantsinfour unrelatedCanadianfamiliesofAfricanancestry.Theprobands wereasymptomaticHbS/+-Thalpatientsandtheadultshad HbAlevelsofabout44%.4Thisisthesecondstudyworldwide toreportthesetwolinkedsequencevariantsincis.The IVS-II-844mutationhasalsobeenidentifiedinbeta-thalassemia patientswithouttheconcomitantIVS-II-839mutationbeing reported.7–9
The−92(C>T)isatranscriptionalmutationaffectingthe -globingenepromoter.6Thisstudyshowedthatthis muta-tionleadstoanearlyasymptomaticphenotypeofSCDwitha meanHbAlevelof41.4%.Thisfindingisinagreementwith thoseofotherinvestigators.3Thismutationwasfirstreported inassociationwiththeSalleleinaSicilianadultwithahigh levelofHbA(45%)whowasclinicallyasymptomatic.3This isthesecondstudytoreportthe−92(C>T)mutationinHb S/+-Thal patients. Heterozygousbeta-thalassemia patients affectedbythismutationalsoshowedasilentformofdisease, whereascompoundheterozygoteswithcodon39orIVS-II-745 mutationsdevelopedbeta-thalassemiaintermedia.10
200
rev
bras
hema
tol
hemoter.
2
0
1
5;
3
7(3)
:198–201
Table1–Genetic,laboratory,andfamilycharacteristicsofchildrenwithverymildformsofS+-thalinMinasGerais,Brazil.
FamilyI FamilyII FamilyIII FamilyIV
Familyrelationship ChildI Mother Father ChildII Mother Father ChildIII ChildIV
Beta-globin genotype
S/ThalIVSII−844/839 A/ThalIVSII−844/839 NA S/ThalIVSII−844/839 S/A A/ThalIVSII−844/839 S/Thal−92C>T S/Thal−92 C>T
Age(years) 7.4 8.0 5.5 11.2
Gender Male Female Male Male Female Male Male Male
Hemoglobin(g/dL) 12.9±0.4 11.2 16.1 12.0±0.8 14.5 16.8 11.4±1.1 11.5±0.6 Hematocrit(%) 40±1.1 35.1 47.1 37.5±2.5 43 50.2 35.4±2.1 35.3±1.4 Redbloodcells
(106/L)
5.5±0.3 4.1 5.5 5.3±0.3 4.5 5.8 4.7±1.1 4.6±0.2
Meancorpuscular volume(fL)
74.5±3.4 85.7 85.3 75.0±1.1 95.5 86.2 78±0.8 78.4±2.5
Meancorpuscular hemoglobin(pg)
24±0.7 27.2 29.1 23.5±0.4 33.7 33.5 25.7 25.2±1.2
Reticulocytecount (%)
1.2±0.7 2.1 0.7 1.1±0.4 1 0.9 0.9±0.7 0.9±0.6
Leukocytes(103/L) 11.3±2.6 8.2 7.8 5.5±0.7 8.5 3.5 9.6±2.5 5.8±1.0
Platelets(103/L) 387.7±50 239.5 233 273.8±26.5 180 169 255.5±14.8 312.1±63.6
HemoglobinA(%) 38.3±1.3 97 51 41.0±2.2 52 95.5 41 41.4±0.9 HemoglobinS(%) 55.2±2.1 0 45 54.2±1.8 44 0 54 53.6±0.5
HemoglobinF(%) 3.5±1.0 1 2 2.0±0.7 2 1.5 2 1.8±0.4
HemoglobinA2(%) 3.0±0 2 2 2.8±0.4 2 3 3 3.2±0.8
Alpha-globin genotype
␣␣/␣␣ ␣␣/␣␣ NA ␣␣/␣␣ ␣␣/␣␣ ␣␣/␣␣ ␣␣/␣␣ ␣␣/␣␣
revbrashematolhemoter.2015;37(3):198–201
201
the Hb S/+-Thal phenotypic classificationas proposed by Serjeantetal.basedontheamountofHbAproduced.2This phenotypicclassificationwasbasedondatafromJamaican patientswithSCDandsoitdoesnotrepresentallHbS/+-Thal patientsworldwide.As the originalclassificationislimited topatientswith1–25%ofHbA,weaccordinglyproposehere afourthHbS/+-Thal phenotype,withanamountofHb A rangingfrom25to45%.Moleculardiagnosisofthesetypesof HbS/+-Thalbynewbornscreeningprogramsconfera‘good’ prognosisandwillsurelyreduceanxietyandstressinfamily caregiversofchildrenrecentlydiagnosedwiththissubtypeof SCD.
Afewlimitationsofourstudywarrantmention.Giventhat thestudiedcaseswerechildren,possiblelong-term compli-cationscannotbepredicted.Owingtothesmall numberof patients,thepresentreportmaynotfullyrepresenttheserare HbS/+-Thalgenotypes.
In summary, −92 (C>T) and IVS-II-844 (C>A)/IVS-II-839 (T>C)mutationsassociatedwiththeS alleleleadtoavery mildformofHb S/+-Thal,whichwe havenamedType IV (25–45%ofHbA).Researchisneededtodeterminewhether othermodifyinggeneticorenvironmentalfactorsmay aggra-vatethephenotypeofaffectedpatients.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgments
Theauthors acknowledgeall subjectsandparentsfortheir cooperationinthestudy.Theresearchwassupportedby Con-selhoNacionaldeDesenvolvimentoCientíficoeTecnológico (CNPq, Grant n◦. 304530/2011-5), Fundac¸ão de Amparo à PesquisadoEstadodeMinasGerais(FAPEMG,Grantn◦. PPM-00266-13),Fundac¸ãoCentrodeHematologiaeHemoterapiade
MinasGerais(Hemominas),andNúcleodeAc¸õesePesquisa emApoioDiagnóstico(Nupad–UFMG).
r
e
f
e
r
e
n
c
e
s
1.SerjeantGR.Thenaturalhistoryofsicklecelldisease.Cold SpringHarborPerspectMed.2013;3(10):a011783.
2.SerjeantGR,SerjeantBE,FraserRA,HambletonIR,HiggsDR, KulozikAE,etal.HbS-beta-thalassemia:molecular, hematologicalandclinicalcomparisons.Hemoglobin. 2011;35(1):1–12.
3.DivokyV,BaysalE,SchiliroG,DibenedettoSP,HuismanTH.A mildtypeofHbS-beta(+)-thalassemia[−92(C→T)]ina Sicilianfamily.AmJHematol.1993;42(2):225–6.
4.WayeJS,EngB,HellensL,HohenadelBA,NakamuraLM, WalkerL.Mildbeta(+)-thalassemiaassociatedwithtwo linkedsequencevariants:IVS-II-839(T>C)andIVS-II-844 (C>A).Hemoglobin.2013;37(4):378–86.
5.TanAS,QuahTC,LowPS,ChongSS.Arapidandreliable 7-deletionmultiplexpolymerasechainreactionassayfor alpha-thalassemia.Blood.2001;98(1):250–1.
6.TheinSL.Themolecularbasisofbeta-thalassemia.Cold SpringHarbPerspectMed.2013;3(5):a011700.
7.BiancoI,LeroneM,FogliettaE,DeiddaG,CappabiancaMP, MorlupiL,etal.Phenotypesofindividualswithabetathal classicalalleleassociatedeitherwithabetathalsilentallele orwithalphaglobingenetriplication.Haematologica. 1997;82(5):513–25.
8.MurruS,LoudianosG,DeianaM,CamaschellaC,Sciarratta GV,AgostiS,etal.Molecularcharacterizationof
beta-thalassemiaintermediainpatientsofItaliandescent andidentificationofthreenovelbeta-thalassemiamutations. Blood.1991;77(6):1342–7.
9.RosatelliMC,PischeddaA,MeloniA,SabaL,PomoA,TraviM, etal.Homozygousbeta-thalassaemiaresultinginthe beta-thalassaemiacarrierstatephenotype.BrJHaematol. 1994;88(3):562–5.
10.RosatelliMC,FaaV,MeloniA,FiorenzaF,GalanelloR, GasperiniD,etal.Apromotermutation,C→Tatposition−92, leadingtosilentbeta-thalassaemia.BrJHaematol.