www.bjorl.org
Brazilian
Journal
of
OTORHINOLARYNGOLOGY
ORIGINAL
ARTICLE
Molecular
approach
of
auditory
neuropathy
夽
Magali
Aparecida
Orate
Menezes
da
Silva
a,
Vânia
Belintani
Piatto
b,∗,
Jose
Victor
Maniglia
aaDepartmentofOtorhinolaryngologyandHeadandNeckSurgery,FaculdadedeMedicinadeSãoJosédoRioPreto(FAMERP),
SãoJosédoRioPreto,SP,Brazil
bDepartmentofAnatomy,FaculdadedeMedicinadeSãoJosédoRioPreto(FAMERP),SãoJosédoRioPreto,SP,Brazil
Received4July2014;accepted12August2014 Availableonline30March2015
KEYWORDS Deafness; Genes;
Molecularbiology; Mutation
Abstract
Introduction:Mutationsintheotoferlingeneareresponsibleforauditoryneuropathy. Objective: Toinvestigatetheprevalenceofmutationsinthemutationsintheotoferlingenein patientswithandwithoutauditoryneuropathy.
Methods:Thisoriginalcross-sectionalcasestudyevaluated16indexcaseswithauditory neu-ropathy,13patientswithsensorineuralhearingloss,and20normal-hearingsubjects.DNAwas extractedfromperipheralbloodleukocytes,andthemutationsintheotoferlingenesiteswere amplifiedbypolymerasechainreaction/restrictionfragmentlengthpolymorphism.
Results:The16indexcasesincludednine(56%)femalesandseven(44%)males.The13deaf patientscomprisedseven(54%)malesandsix(46%)females.Amongthe20normal-hearing sub-jects, 13 (65%) weremales andseven were (35%) females. Thirteen(81%) index cases had wild-type genotype (AA) and three (19%) hadthe heterozygous AG genotype for IVS8-2A-G (intron8)mutation.The5473C-G(exon44)mutationwasfoundinaheterozygousstate(CG) inseven (44%)indexcases andnine(56%) hadthewild-typeallele(CC). Ofthesemutants, two(25%)werecompoundheterozygotesforthemutationsfoundinintron8andexon44.All patientswithsensorineuralhearinglossandnormal-hearingindividualsdidnothavemutations (100%).
Conclusion: Therearedifferencesatthemolecularlevelinpatientswithandwithoutauditory neuropathy.
© 2015Associac¸ãoBrasileira de Otorrinolaringologiae CirurgiaCérvico-Facial. Publishedby ElsevierEditoraLtda.Allrightsreserved.
夽
Pleasecitethisarticleas:daSilvaMA,PiattoVB,ManigliaJV.Molecularapproachofauditoryneuropathy.BrazJOtorhinolaryngol. 2015;81:321---8.
∗Correspondingauthor.
E-mails:[email protected],[email protected](V.B.Piatto).
http://dx.doi.org/10.1016/j.bjorl.2015.03.005
PALAVRAS-CHAVE Surdez;
Genes;
Biologiamolecular; Mutac¸ão
Abordagemmoleculardaneuropatiaauditiva
Resumo
Introduc¸ão:Mutac¸õesnogenedaotoferlina(OTOF)sãoresponsáveispelaneuropatiaauditiva. Objetivo:Investigaraprevalênciademutac¸õesnogeneOTOFempacientescomesem neu-ropatiaauditiva.
Método: Estudodecasosemcortetransversalsendoavaliados16casosíndicecomneuropatia auditiva,13pacientescomdeficiênciaauditivasensorioneural(DASN)e20indivíduosouvintes. DNAfoiextraídodeleucócitosdosangueperiféricoeregiõesdogeneOTOFforamanalisadas pelatécnicaPCR-RFLP.
Resultados: Dos16casosíndice,9(56%)sãodogênerofemininoe7(44%)domasculino.Dos 13pacientescomDASN,7(54%)sãomasculinose6(46%)femininos.Dos20ouvintes,13(65%) sãomasculinose7(35%)femininos.Treze(81%)casosíndiceapresentamogenótiposelvagem (AA)e3(19%) ogenótipoheterozigoto AGparaamutac¸ãoIVS8-2A-G(intron8).A mutac¸ão 5473C-G(exon44)foiencontradaemheterozigose(CG)em7(44%)doscasosíndicee9(56%) apresentamogenótiposelvagem(CC).Destesmutantes,dois(25%)sãoheterozigotoscompostos paraasmutac¸õesencontradasnointron8eexon44.OspacientescomDASNeosouvintesnão apresentammutac¸ões(100%).
Conclusão:Existemdiferenc¸as,aonívelmolecular,empacientescomesemneuropatia audi-tiva.
©2015Associac¸ãoBrasileira deOtorrinolaringologiaeCirurgiaCérvico-Facial.Publicadopor ElsevierEditoraLtda.Todososdireitosreservados.
Introduction
AfterthemutationsinGJB2andGJB6gene,whichaccount forapproximately80%ofautosomal-recessivenonsyndromic hearing impairment (ARNSHI), mutations in the otoferlin gene(OTOF)arethemostfrequentgeneticcauseofdeafness inchildren.This typeofhearingloss (HL)is more compli-cated,consideringthatatanearlystageitmaypresentas auditoryneuropathy(AN)thatisnotdetectedbyneonatal screeningbasedonotoacousticemissiontesting.1
AuditoryneuropathyisauniquetypeofHLinwhich
tym-panogramsarenormal,theacousticreflexandthebrainstem
auditory evoked potentials (BAEP) are absent or severely
altered,andtheouterhaircellfunctionisnormal,asshown
bythe presenceof otoacoustic emissions (OAEs). Patients
withthisdisorder canshowvarieddegreesof HL,in
addi-tiontoseverespeechcomprehensionimpairment,whichis
disproportionaltothedegreeofhearingloss.1
In 2003, the term ‘‘autosomal-recessive nonsyndromic
auditory neuropathy’’ was defined, due not only to the
audiological characteristics, but also to genetic
discover-ies related to mutations in the OTOF gene, which were
associatedwiththistypeofhearingloss.2The OTOFgene,
located on chromosome 2 (2p23-p22), contains 48 exons
and encodes the protein otoferlin, which is expressed in
cochlear inner hair cells and typeI vestibularhair cells.3
Since then, molecular studies have been published that
allowedtheidentificationofmutationsintheOTOFgeneand
associatedthem tothe autosomal-recessivenonsyndromic
AN.2---12
Therefore,thisstudyaimedtoinvestigatetheprevalence
ofmutationsintheOTOFgene:2416T-A(Y730X)inexon18,3
IVS8-2-A-Ginintron8/exon9,62485C-T(Q829X)inexon22,5
5473C-G(Pro1825Ala)inexon44,5and3032T-C(Leu1011Pro)
inexon26,9 inpatientswithANandinpatientswith
non-syndromicHL.
Methods
This cross-sectional study was carried out from April 1,
2010 until July 30, 2011; of 1230 cases of nonsyndromic
sensorineural hearing loss (NSHL) treated at the Hearing
LossOutpatient Clinicofthe institution,16patients were
selected(NPTgroup---neuropathy),ofbothgenders,withAN.
These weredeemedindex cases,accordingtothe
follow-ing inclusion criteria: normal otoscopy,diagnosis of NSHL
by pure tone audiometry, presence of otoacoustic
emis-sionsand/orcochlearmicrophonicinBAEP;absenceofBAEP
wavesorseverealterationintheirmorphology;normalMRI;
absenceof riskfactors (maternal---fetalinfections,
menin-gitis,ototoxicdrugs)orperipheralneuropathies.Exclusion
criteriaincludedadiagnosisofconductiveormixedhearing
loss,thepresenceofriskfactorsorperipheralneuropathies,
andanage>65years.
Atotalof13patientsofbothgenders(NSHLgroup)were
also selected, whohad received a diagnosis of NSHL and
whoseaudiologicaltestsdidnotmatchtheclinicalpicture
of ANandhad nomutationsin theconnexin 26and GJB6
genes.
FortheControlgroup(CGgroup),20individualsofboth
genders were selected, with no hearing complaints, with
normalotoscopyandwhowereunrelatedtothepatientsin
theNPTandNSHLgroups.
Theindexcases,patients,andcontrolswereofthesame
ethnicity,age ≤65years,withnoconsanguinity, andfrom
ThestudywasapprovedbytheEthicsCommitteeofthe
institution(EdictNo.44/2010).GenomicDNAwasextracted
fromperipheralbloodsamplesusingtheGEIllustra
extrac-tionkit--- BloodGenomicprepMiniSpinKitTM(GEHealthcare
UKLtd)accordingtothemanufacturer’sprotocol.
AllfivemutationsanalyzedintheOTOFgeneare
identi-fiedbytheirpositionsintheNCBIGeneBankRefSeqGene:
NG009937.1. Polymerase chain reaction/restriction
frag-mentlengthpolymorphism(PCR/RFLP)testswereusedfor
themolecularanalysis.
Each ofthefivePCRreactionswasprocessedina
tem-perature cycler (Bioer Technology®, Model TC-XPG) using
25uL final reaction volume,containingthe following: 1
---DNA(200---300ng);2---10pmolofeachprimer(forwardand
reverse); 3 --- Reagent Kit FideliTaqTM PCR MasterMix (2×)
(GE HEALTHCARE®)using protocol accordingtothe
manu-facturer’sinstructions.
The followingstandard cycleforthePCR reactionswas
used,differingonlyintheprimerannealingtime,whichis
specificforeachpairused:initialdenaturationat94◦Cfor
3min,35repeatedcyclesof60sat94◦ Cfordenaturation,
60satX◦Cforprimerannealing(forthe2416T-Amutation:
56◦C;IVS8-2A-G:51◦C;2485C-T: 58◦C;5473C-Gmutation:
59◦C;3032T-Cmutation:52◦C),andextensionof2minutes
at72◦
Cand10minat72◦
Cforthefinalextension.The
frag-mentsamplifiedbyPCRwererespectivelysubmittedtoRFLP
analysis,using5Uofthefollowingenzymes:DdeIenzyme,
BGlII,BfaI,FauIandMspI(NewEnglandBiolabs®).
The products of the reactions (PCR/RFLP) were added
tobromophenol blueand submittedtoelectrophoresis on
2% agarose gel in Tris---borate---EDTA1× buffer containing
ethidium bromide(0.5ug/mL), after which the gels were
illuminatedwithUVlightingandphotographed.
Statisticalanalysis
Theresultswerepreviouslysubmittedtodescriptive
statis-ticstodeterminenormality.TheChi-squaredtestwasused
tocompareindependentvariableswithnormaldistribution
and the Kruskal---Wallis test for samples with non-normal
distribution, in addition to odds ratio with a confidence
intervalof95%(95%CI).Thelevelofsignificancewassetat
5%.StatisticaltestswereperformedusingGraphPadInStat
softwarerelease3.0(GraphPadSoftwareInc---SanDiego,
CA, United States). Consistency of genotype distribution
withtheHardy-Weinbergequilibriumwasassessedbyexact
tests.13
Results
Ofatotalof16indexcasesfromtheNPTgroup,nine(56%)
werefemalesandseven(44%)weremales.Agerangedfrom
8to65yearsforfemales(meanM24.2years,SD±20.1)and
formalesbetween1and38years(M14.1years,SD±15.7),
withnostatisticallysignificantdifference(p=0.29).
Ofthe13patientsintheNSHLgroup,seven(54%)were
malesandsix(46%)werefemales.Agevariedfrom7to37
yearsfor males(M22.7 years,SD±12.6)andbetween 12
and45yearsforfemales(M33.3years,SD±12.3),withno
significantdifference(p=0.15).
Regarding the 20 individuals in the CG, 13 (65%) were
malesandseven(35%) werefemales.Age rangedfrom19
to44yearsformales(M28.5years,SD±7.2)andbetween
20---35yearsfor females (M26.3 years,SD±6.3), withno
significantdifference(p=0.49).
Considering the demographicvariables among patients
inthethreegroups,therewasaprevalenceof femalesin
allthree,althoughitwasnotsignificant(p=0.49).Boththe
NSHLandCGgroups hadahigherprevalence intheyoung
adultagerange,totaling55%and95%,respectively.TheNPT
grouphadhigherprevalenceintheinfantgroup(38%).The
statisticalanalysisforthisdemographicvariablewasnot
sig-nificantbetweengroups(p=0.37).Similarly,themeanages
amongthethreegroups showednosignificant differences
(p=0.091;Table1).
Table 2 shows the distribution, in percentages, of the
genotypesfound in thethreegroups assessed for thefive
mutationsintheOTOFgene.IntheNPTgroup,13(81%)index
caseshadthewild genotype(AA)andthree(19%)hadthe
heterozygousgenotype AG for IVS8-2A-G mutation(intron
8).The5473C-Gmutation(exon44)wasfoundin
heterozy-gousform(CG)inseven(44%)oftheindexcasesandnine
(56%)hadthewildallele(CC).IntheNSHLandCGgroups,
100%ofpatientshadnoneoftheanalyzedmutationsinthe
OTOFgene.
Thus, in the NPT group, therewere eight (50%) index
caseswithmutationsin theOTOFgene,one(12.5%;F1.3)
heterozygousforthemutationIVS8-2A-G,five(62.5%;F1.2,
F4.1, FBT1, FBT3.2, and FBT4.2) compound heterozygous
for the mutation 5473C-G, and two (25%; F1.1 and F5GI)
compound heterozygous for the mutations found
respec-tivelyinintron8andexon44.Ofthenormal-hearingfamily
members,themotherofFamily1(F1.M)isacompound
het-erozygousforthemutationsanalyzedinintron8andexon44
andthemothersofFamiliesFBT3andFBT4areheterozygous
onlyforthe5473C-Gmutation(Table3).
The NPT group was assessed for the Hardy---Weinberg
equilibriuminrelation tomutationsfound inintron 8and
exon44. According to the calculations made, allelic
fre-quenciesA(wild-typeallele)andG(mutant allele)ofthe
IVS8-2A-Gmutation,fortheindexcasesoftheNPTgroup,
were A=0.91 and G=0.09. The observed genotypic
fre-quencies of the wild homozygous form (AA-13), mutant
heterozygous (AG-3), and mutant homozygous (GG-0) of
these index cases are consistent with the expected
fre-quencies for the Hardy-Weinberg equilibrium (2=0.171;
p=0.68).Regardingthe5473C-Gmutation,theallelic
fre-quenciesC(wild-typeallele)andG(mutantallele)forthe
indexcasesoftheNPTgroupwereC=0.78andG=0.22.For
this mutation,the genotypic frequencies observed in the
wildhomozygousform(CC-9),mutantheterozygous(GC-7),
and mutant homozygous (GG-0) of these index cases are
alsoin agreement withthe expected frequencies for the
Hardy---Weinbergequilibrium(2=0.125;p=0.26).
Regardingthedegree ofhearinglossfoundintheindex
casesoftheNPTgroup,three(19%)hadmild,five(31%)
mod-erate,five(31%)severe,andthree(19%)profoundhearing
loss.IntheNSHLgroup,four(31%)hadmoderate,two(15%)
severe,andseven(54%)profoundhearingloss.Therewere
nomildcasesinthisgroup.Statisticalanalysisforthe
vari-abledegreeofhearinglossshowednosignificantdifference
Table1 DistributionofdemographicvariablesofindexcasesintheNPTgroup,patientsfromtheNSHLgroup,andindividuals fromtheControlgroup.
Variables NPTgroup(n=16) NSHLgroup(n=13) Controlgroup(n=20) p
Gender n(%) n(%) n(%)
Male 7(44) 6(46) 7(35) 0.49a
Female 9(56) 7(54) 13(65)
Ageclassification n(%) n(%) n(%) 0.37a
Infant 1(06) 0(00) 0(00)
Child 6(38) 2(15) 0(00)
Adolescent 3(19) 2(15) 0(00)
Youngadult 4(25) 7(55) 19(95)
Adult 2(12) 2(15) 1(05)
Elderly 0(00) 0(00) 0(00)
Age M±SD M±SD M±SD p
Years 19.8±18.5 27.6±13.1 19.±18.5 0.091b
M±SD,mean±standarddeviation.
aChi-squaredtest. b Kruskal---Wallistest.
Duetomutationidentificationonlyinintron8andexon
44intheOTOFgene,Table4showsthedegreeofhearingloss
intheNPTgroupindexcasesandgenotypesfoundinthese
cases,throughthemolecularanalysisoftheseregions.
Among the index cases with mutation, those withthe
severe(four; 25%)and profound (three; 19%) hearingloss
weremoreprevalentthanthosewithoutmutation(onlyone
severeHL,6%)ofthesame group,whichshowedahigher
prevalenceof mild(three; 19%) andmoderate(four;25%)
HL,withasignificantdifference(p=0.022).
Discussion
ANisahearingdisordercharacterizedbyseveredistortion
orabsenceofBAEPwithpreservedOAEs,thusindicatingthe
presenceofouterhaircellintegrity.14
AmongthepreviouslyidentifiedcausesofANare
hyper-bilirubinemia, neonatal hypoxia, or neurodegenerative
diseases. However,therehavebeen reportsof‘‘isolated’’
AN, i.e., without any risk factors, and no
physiopatho-logical mechanism has been proposed to explain these
Table2 Distribution,inpercentages,ofthegenotypesfoundinthethreegroupsassessedforthefivemutationsintheOTOF gene.
Genotype NPTgroup(n=16)n(%) NSHLgroup(n=13)n(%) Controlgroup(n=20)n(%)
2416T-AExon18
TT 16(100) 13(100) 20(100)
TA 0 0 0
AA 0 0 0
IVS8-2A-GIntron8
AA 13(81) 13(100) 20(100)
AG 3(19) 0 0
GG 0 0 0
2485C-TExon22
CC 16(100) 13(100) 20(100)
CT 0 0 0
TT 0 0 0
5473C-GExon44
CC 9(56) 13(100) 20(100)
CG 7(44) 0 0
GG 0 0 0
3032T-CExon26
TT 16(100) 13(100) 20(100)
TC 0 0 0
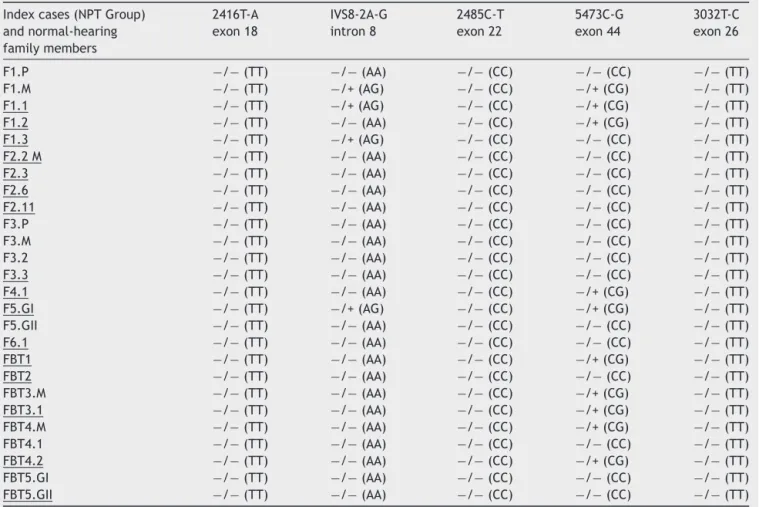
Table3 GenotypesfoundintheNPTgroupindexcasesamongthefivemutationsassessedintheOTOFgene.
Indexcases(NPTGroup) andnormal-hearing familymembers
2416T-A exon18
IVS8-2A-G intron8
2485C-T exon22
5473C-G exon44
3032T-C exon26
F1.P −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F1.M −/−(TT) −/+(AG) −/−(CC) −/+(CG) −/−(TT) F1.1 −/−(TT) −/+(AG) −/−(CC) −/+(CG) −/−(TT) F1.2 −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) F1.3 −/−(TT) −/+(AG) −/−(CC) −/−(CC) −/−(TT) F2.2M −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F2.3 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F2.6 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F2.11 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F3.P −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F3.M −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F3.2 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F3.3 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F4.1 −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) F5.GI −/−(TT) −/+(AG) −/−(CC) −/+(CG) −/−(TT) F5.GII −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) F6.1 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) FBT1 −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) FBT2 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) FBT3.M −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) FBT3.1 −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) FBT4.M −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) FBT4.1 −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) FBT4.2 −/−(TT) −/−(AA) −/−(CC) −/+(CG) −/−(TT) FBT5.GI −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT) FBT5.GII −/−(TT) −/−(AA) −/−(CC) −/−(CC) −/−(TT)
Underlined,indexcaseswithauditoryneuropathy(NPT).F,family;P,fatheroftheindexcase;M,motheroftheindexcase;GI,twin;I GII,twinII;−/−,wildhomozygousgenotypefortheanalyzedmutation;−/+,heterozygousgenotypefortheanalyzedmutation;−/+, heterozygousgenotypeforthemutationanalyzedinnormal-hearingfamilymember.
Table4 DegreeofhearinglossandthegenotypesfoundinIntron8andExon44oftheOTOFgene,inindexcasesofNPTgroup.
GroupNPT Degreeofhearingloss IVS8-2A-Gintron8 5473C-Gexon44
F1.1 Profound −/+(AG) −/+(CG)
F1.2 Severe −/−(AA) −/+(CG)
F1.3 Severe −/+(AG) −/−(CC)
F2.2M Mild −/−(AA) −/−(CC)
F2.3 Moderate −/−(AA) −/−(CC)
F2.6 Mild −/−(AA) −/−(CC)
F2.11 Moderate −/−(AA) −/−(CC)
F3.3 Moderate −/−(AA) −/−(CC)
F4.1 Severe −/−(AA) −/+(CG)
F5.GI Profound −/+(AG) −/+(CG)
F6.1 Severe −/−(AA) −/−(CC)
FBT1 Moderate −/−(AA) −/+(CG)
FBT2 Moderate −/−(AA) −/−(CC)
FBT3.1 Severe −/−(AA) −/+(CG)
FBT4.2 Profound −/−(AA) −/+(CG)
cases. Recent advances in genetic research has allowed a new approach to elucidate the physiopathology of this disease.15,16
Given the above, this study aimed to investigate the
prevalenceoffivemutationsin theOTOFgeneinpatients
withANdiagnosis,innormal-hearingsubjectsandinpatients
with NSHL. These were included in order to determine
whethermutationsintheOTOFgenecouldbeidentifiedin
normal-hearingsubjectsandinpatientswithNSHLwithout
mutationsinothergenesandwithoutAN.
Regardingthedemographicvariables,therewasa
preva-lenceoffemalesinthethreestudygroups,althoughitwas
notstatisticallysignificant.TheNSHLandCGgroups hada
higherprevalenceintheyoungadultagerangeandtheNPT
grouphadahigherprevalenceintheinfantagerange,but
withoutasignificantdifference.Thesedataareconsistent
withthefewpublishedreportsontheprevalenceofgender
andagerangerelatedtotheOTOFgene.17,18
Mostreports of mutations identifiedin the OTOF gene
have been described in homozygous form and in
con-sanguineous families.3,4,6,9,18---22 Among the five mutations
analyzed in the OTOF gene, only the IVS8-2A-G mutation
inintron 8and 5473C-Gmutationin exon44 werefound,
affecting 50% of the study index cases. In contrast to
the literature,all index cases were the offspring of
non-consanguineousmarriages,demonstratingthatmutationsin
theOTOF gene that areassociated withAN mayoccur in
thesecases.
Heterozygous cases in patients with AN have been
described,withorwithoutconsanguinity.2,5,8,10---12,23,24Inthe
present study, the IVS8-2A-G mutation was found in
het-erozygousstate(onemutantallele)in12.5%ofindexcases,
withthesameoccurringwiththe5473C-Gmutation,which
wasfoundin62.5%ofcases.In25%ofcases,thetwo
muta-tions identified occurred at the same time in the same
individual,which,therefore,arecalledcompound
heterozy-gotes. These two mutations, as well as the other three
analyzed, were not found in the NSHL group (100%) nor
in the CG group. These data are in agreement with the
literature.2,5,8,10---12,23,24
In 2000, the IVS8-2A-G mutationwasfirst described in
itshomozygousstate inthreebrothersfromIndia born to
consanguineous parents and in its heterozygous state in
theparentsandunaffectedsiblings,andwasnotidentified
in109normal-hearingindividuals fromthecontrolgroup.6
Therehavebeen nofurtherreportsonthismutation.The
5473C-Gmutationwasdescribedincompound
heterozygos-ityinaSpanishfamily,withthe2485C-Tmutationinexon
22,identifiedinanotherallele.5
Similarly,therehavebeennofurtherreportsofthe
afore-mentionedmutationinexon44.
The IVS8-2A-G mutation,by creatinga prematurestop
codon,andthe5473C-Gmutation,byalteringthebinding
capacitywith Ca+2 in the wild-type gene, are considered
pathogenicwhenintheirhomozygousstate.3,5Inthisstudy,
bothwerefound incompound heterozygousstates onlyin
indexcaseswithAN(F1.1andF5.GI),whichcouldexplain
themolecularcauseofhearinglossintheseindividuals,as
theANphenotypehasbeenobserved inpatientswithtwo
mutationsaffectingallisoformsofotoferlinoronemutation
affectingthelong isoformandanother,theshortisoform.
Thismeans that anycombination of biallelic mutationsin
theOTOFgenemayresultinAN,astheyarepartiallyrelated
tothenature(truncatedor non-truncated)orthelocation
(longorshortisoform)ofthesemutations.23
However, the same mutations were also found in
compound heterozygosis in the normal-hearing mother
(F1.M)ofthecompoundheterozygousindexcase(F1.1).As
themotherhasnormalhearing,thecauseofHLintheindex
casemaybeduetootherexistingmutationsinotherexons
or other splicesites, ormore complexmutations,suchas
large deletions or other sequence rearrangements, which
requireamorespecificmolecularapproachthanusedhere.
Similarly,thesehypothesesmayalsobeappliedtoexplain
theothersixHLindexcases,astheyarecarriersofoneof
themutationsidentifiedinonlyoneofthealleles.
Brazilian references have not reported the mutations
identified in this study,10---12 and only one of them
identi-fiedthe2485C-Tmutationinexon22withheterozygosityin
0.5%ofcasesofpatientswithnon-syndromicsensorineural
HL(1/200).10
Estimates of the prevalence of AN in deaf individuals
rangefrom1%to19%.25---28InBrazil,theprevalenceofAN
amongallcasesofdeafness is1.2%.29 This rangeof
varia-tionmaybeduetothefactthatdifferentpopulationswere
studied ordue todifferencesinthecriteriausedto
iden-tifypatients withthis disease.The prevalence of ANin a
population withoutriskfactorsis notwellestablished.9 In
thepresentstudy,allthecriteriaforinclusionandexclusion
werestrictlyfollowed,thusdeterminingtheprevalenceof
1.3% of cases withAN in a population with sensorineural
hearinglossduringthestudyperiod.
Moreover,theBrazilianestimateofthemagnitudeofHL
in patients with ANis 29.6% mild, 55.5% moderate, 7.4%
severe, and7.5% profound HL.29 Inthe present study 19%
hadmild,31%moderate,31%severe,and19%profoundHL.
Among the indexcaseswithmutations in theOTOF gene,
wefoundastatisticallysignificantprevalenceofsevereand
profound HL compared to the mild and moderate HL in
indexcaseswithoutmutation.Theintensityofhearingloss
causedbyANcanrangefrommildtoprofound,witha
dis-proportionate decreaseinunderstandingspeechthatdoes
notcorrespondtothepure-toneaudiogram.
Thereissomehomogeneityinthedescriptionofthe
phe-notype ofpatients withmutations intheOTOF gene,that
ischaracterizedbyacongenitalorpre-lingualprofoundHL
notassociatedwithotherabnormalities,astheindexcases
studied.Therefore,ANistheonlyphenotypicexpressionof
mutationsintheOTOFgene,withhearinglossidentifiedby
therelativepreservationofouterhaircellfunction.9,30
ThepresenceofOAEsinthefirstmonthsoflifehas
impor-tantimplicationsforneonatalscreeningprograms,suchas
initialscreening.InthecaseofAN,thereisariskthatthis
losswillgoundiagnosed,for althoughthesepatientshave
severe toprofound degree ofhearing loss,theywould be
includedamongthecasesclassifiedas‘‘noalterationor
nor-mal’’ becauseOAEsreflect theauditory function inouter
haircellsandtheirpresencedoesnotexcludealterationsof
innerhaircells,insynapses,orintheauditorynerve.28
InthecontextofHLinvestigation,itisimportanttotake
into accountthe typeof AN loss,which is often
misdiag-nosed. Therefore, when assessing a child or patient with
deafness in the presence of OAEs, the hypothesis of AN
OTOFgeneresearchshouldbeperformed,astheywere
per-formedinthisstudy,allowinganaccurateetiologicdiagnosis
toprovide appropriate audiological treatment in atimely
manner.28
Potentiallimitationsregardingthestudyfindingsdeserve
consideration;despite thepositiveresults,theyshouldbe
interpreted with caution and should be corroborated by
independent, multicenter studies to determine the true
prevalenceofmutationsintheOTOFgeneandthe
associa-tionwithANintheBrazilianpopulation.
Conclusion
TheevidenceofassociationbetweenmutationsintheOTOF
gene and AN indicates that there are differences at the
molecular level in patients with andwithout this typeof
hearingloss;intheabsenceofriskfactors,thecombination
ofseveredistortionorabsenceofBAEPinthepresenceof
OAEsshoulddirectthediagnosisofANandthescreeningfor
mutationsin the OTOFgene. Moleculartests arevaluable
diagnosticcomplements,notonlyindividually,butalsoasa
screeningmethodforaneonatalorpopulationstudy.
Funding
ThisstudywassupportedbyResearchGrant(BAP-FAMERP).
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgements
Tothepatientsandtheirparents/guardianswho,by
agree-ingtoparticipateinthisresearch,willcontributetoabetter
futureforthechildrenofBrazil.Totheinstitutioninwhich
thisresearchwasperformed, byencouragingresearchand
teachingqualification.
References
1.IdanN, Brownstein Z,ShivatzkiS, Avraham KB.Advances in geneticdiagnostics for hereditary hearing loss. J Basic Clin PhysiolPharmacol.2013;24:165---70.
2.VargaR,KelleyPM,KeatsBJ.Non-syndromicrecessiveauditory neuropathyistheresultofmutationsintheotoferlin(OTOF) gene.JMedGenet.2003;40:45---50.
3.YasunagaS,GratiM,Cohen-SalmonM,El-AmraouiA,Mustapha M,SalemN,etal.AmutationinOTOF,encodingotoferlin,a FER-1-likeprotein,causesDFNB9,anonsyndromicformofdeafness. NatGenet.1999;21:363---9.
4.AdatoA,RaskinL,PetitC.DeafnessheterogeneityinaDruze isolatefromtheMiddleEast:novelOTOFandPDSmutations, lowprevalenceofGJB235delGmutationandindicationfora newlocus.EurJHumGenet.2000;8:437---42.
5.MigliosiV,Modamio-HøybiørS,Moreno-PelayoMA, Rodriguez-BallesterosM, Villamar M, TelleriaD, et al. Q829X, a novel mutationinthegeneencodingotoferlin(OTOF),isfrequently foundinSpanishpatientswithprelingualnon-syndromichearing loss.JMedGenet.2002;39:502---6.
6.YasunagaS,GratiM,ChardenouxS,SmithTN,FriedmanTB, Lal-waniAK,etal.OTOFencodesmultiplelongandshortisoforms:
geneticevidencethatthelongonesunderlierecessivedeafness DFNB9.AmJHumGenet.2000;67:591---600.
7.MirghomizadehF,PfisterM,ApaydinF,PetitC,KupkaS,Pusch CM,etal.SubstitutionsintheconservedC2Cdomainof otorfe-lincauseDFNB9,aformofnonsyndromicautosomalrecessive deafness.NeurobiolDis.2002;10:157---64.
8.Rodriguez-Ballesteros M, del Castillo FJ, Martín Y, Moreno-PelayoMA,MoreraC,PrietoF,etal. Auditoryneuropathyin patientscarryingmutations intheotoferlingeneOTOF.Hum Mutat.2003;22:451---6.
9.TekinM,AkcayozD,IncesuluA.Anovelmissensemutationin aC2 domainofOTOFresultsinautosomalrecessiveauditory neuropathy.AmJMedGenet.2005;138:6---10.
10.FáveroML,RomanosJ,Mingroni-Neto-R.C.Neuropatiaauditiva decorrente de mutac¸ão no geneOTOF. Arq Otorrinolaringol. 2005;9:325---30.
11.Oliveira CA,Alexandrino F,Christiani TV, Steiner CE, Cunha JLR, Guerra ATM, et al. Molecular genetics study of deaf-ness in Brazil: 8-year experience. Am J Med Genet Part A. 2007;143:1574---9.
12.Romanos J, Kimura L, Fávero ML, Izarra FA, de Melo Auric-chio MT, Batissoco AC, et al. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J Hum Genet. 2009;54:382---5.
13.RodriguezS,GauntTR.Day INM.Hardy-Weinbergequilibrium testingofbiologicalascertainmentformendelian randomiza-tionstudies.AmJEpidemiol.2009;169:505-514.Availablefrom: http://www.oege.org/software/hwe-mr-calc.shtml
14.LoundonN,MarcollaA,RouxI,RouillonI,DenoyelleF,Feldmann D,etal.Auditoryneuropathyorendocochlearhearingloss?Otol Neurotol.2005;26:748---54.
15.ManchaiahVK,ZhaoF,DaneshAA.Thegeneticbasisofauditory neuropathyspectrumdisorder(ANSD).IntJPediatr Otorhino-laryngol.2011;75:151---8.
16.Cooper DN, Ball EV, Stenson PD. The Human GeneMutation Database; 2014. Available from:http://www.hgmd.cf.ac.uk/ ac/index.php
17.SheykholeslamiK, Kaga K,Kaga M. Anisolatedand sporadic auditory neuropathy(auditory nerve disease): reportof five patients.JLaryngolOtol.2001;115:530---4.
18.RouillonI,MarcollaA,RouxI,MarlinSD,FeldmannD,CoudecR, etal.Resultsofcochlearintwochildrenwithmutationsinthe OTOFgene.IntJPediatrOtorhinolaryngol.2006;70:689---96. 19.HousemanMJ,JacksonAP,AlGazaliLI.Anovelmutationina
familywithnon-syndromicsensorineuralhearinglossthat dis-ruptsthenewlycharacterisedOTOFlongisoforms.JMedGenet. 2001;38:e25.
20.Gallo-TeránJ,Megía LópezR, Morales-AnguloC,delCastillo I, Moreno-Pelayo MA, Mazón Gutiérrez A, et al. Evalua-tion of a family with sensorineural hearing loss due to the Q829XmutationintheOTOFgene.ActaOtorrinolaringolEsp. 2004;55:120---5.
21.Gallo-Terán J, Morales-Angulo C, Rodríguez-Ballesteros M. Prevalenceofthe35delGmutationintheGJB2gene,del (GJB6-D13S1830) in the GJB6 gene, Q829X in the OTOF gene and A1555Gin themitochondrial12S rRNAgeneinsubjects with non-syndromic sensorineural hearing impairment of congeni-tal/childhoodonset.ActaOtorrinolaringolEsp.2005;56:463---8. 22.ChoiBY,AhmedZM,RiazuddinS,BhinderMA,ShahzadM, Hus-nainT, et al.Identitiesand frequenciesofmutations ofthe otoferlingene(OTOF)causingDFNB9deafnessinPakistan.Clin Genet.2009;75:237---43.
23.SantarelliR, Del CastilloI,Rodríguez-BallesterosM,Scimemi P,CamaE,ArslanE,etal.Abnormalcochlearpotentialsfrom deafpatientswithmutationsintheotoferlingene.JAssocRes Otolaryngol.2009;10:545---56.
withanotoferlinmutation:deafeningfever!BiochemBiophys ResCommun.2010;394:737---42.
25.Psarommatis IM, Tsakanikos MD, Komtrogianni AD. Profound hearinglossandpresenceofclick-evokedotoacousticemissions intheneonate:areportoftwocases.IntJPediatr Otorhino-laryngol.1997;39:237---43.
26.RanceG.Auditoryneuropathy/dys-synchronyanditsperceptual consequences.TrendAmplif.2005;9:1---43.
27.FoerstA,BeutnerD,Lang-RothR.Prevalenceofauditory neu-ropathy/synaptopathyinapopulationofchildrenwithprofound hearingloss.IntJPediatrOtorhinolaryngol.2006;70:1415---22.
28.Maris M, Venstermans C, Boudewyns AN. Auditory neuropa-thy/dyssynchrony as a cause of failed neonatal hearing screening.IntJPediatrOtorhinolaryngol.2011;75:973---5. 29.PenidoRC,IsaacML.Prevalenceofauditoryneuropathy
spec-trum disorder in an auditory health care service. Braz J Otorhinolaryngol.2013;79:429---33.