Programa de Pós-Graduação Stricto Sensu em Ciências
Genômicas e Biotecnologia
Autor: Daniel Lôpo Polla
Orientador: Dr. Robert Pogue
Coorientador: Dr. Rinaldo Wellerson Pereira
Brasília - DF
2014
SEQUENCIAMENTO DE EXOMA PARCIAL COMO
FERRAMENTA DE DIAGNÓSTICO MOLECULAR DE
DANIEL LÔPO POLLA
SEQUENCIAMENTO DE EXOMA PARCIAL COMO FERRAMENTA DE DIAGNÓSTICO MOLECULAR DE DISPLASIAS ESQUELÉTICAS
Dissertação apresentada ao Programa de Pós-graduação Stricto Sensu em Ciências Genômicas e Biotecnologia da Universidade Católica de Brasília, como requisito parcial para a obtenção do Título de Mestre em Ciências Genômicas e Biotecnologia.
Orientador: Dr. Robert Pogue
Coorientador: Dr. Rinaldo Wellerson Pereira
Ficha elaborada pela Biblioteca Pós-Graduação da UCB
P771s Polla, Daniel Lôpo.
Sequenciamento de exoma parcial como ferramenta de diagnóstico molecular de displasias esqueléticas. / Daniel Lôpo Polla – 2014.
133 f.; il : 30 cm
Dissertação (mestrado) – Universidade Católica de Brasília, 2014.
Orientação: Prof. Dr. Robert Pogue
Coorientação: Prof. Dr. Rinaldo Wellerson Pereira
1. Biotecnologia. 2. Diagnóstico molecular. 3. Displasia esquelética. I. Pogue, Robert, orient. II. Pereira, Rinaldo Wllerson, coorient. III. Título.
Dissertação de autoria de Daniel Lôpo Polla, intitulada “SEQUENCIAMENTO DE EXOMA PARCIAL COMO FERRAMENTA DE DIAGNÓSTICO MOLECULAR DE DISPLASIAS ESQUELÉTICAS”, apresentada como requisito parcial para a obtenção de grau de Mestre em Ciências Genômicas e Biotecnologia da Universidade Católica de Brasília, em 12 de março de 2014, defendida e aprovada pela banca examinadora abaixo assinada:
AGRADECIMENTOS
Agradeço em primeiro lugar a Deus que iluminou o meu caminho durante mais esta caminhada, dando-me força e paciência para superar os desafios.
Aos meus pais, Airton Polla e Marley de Sousa Lôpo Polla, e irmã Rafaela, que me apoiaram, deram forças, me ajudaram a trilhar o meu caminho e confiaram no meu potencial.
À minha tia e madrinha Marly de Sousa Lôpo, pois a realização de mais essa etapa da minha vida não seria possível sem o seu incentivo, afeto e confiança.
A toda minha família, que direta ou indiretamente, me deram forças e me apoiaram a concluir mais uma etapa dessa carreira que eu amo.
Aos professores que me deram força para chegar até aqui, em especial à Prof. Dra. Cristine C Barreto, que me apresentou à pesquisa científica e influenciou diretamente a minha escolha para seguir essa carreira, à Prof. Dra. Juliana Mazzeu, que abriu portas para a continuidade da minha carreira científica com a minha indicação para o doutorado nos Países Baixos, e ao Prof. Dr. Robert Pogue, por todos os ensinamentos, paciência e por ter acreditado em mim e no meu trabalho, sendo peça fundamental para a concretização dessa etapa acadêmica.
À Camila Fernandes, da FCAV-UNESP de Jaboticabal-SP, por ter me auxiliado durante uma etapa crucial do meu trabalho e me ajudou na fragmentação das minhas amostras.
À Dra. Alessandra Reis e ao Adevilton, pela ajuda durante o sequenciamento no MiSeq.
Às Dras. Cristina e Rose, por toda a ajuda nas reuniões para análise dos dados e diagnóstico dos pacientes.
Aos amigos e colegas da UCB (principalmente Lana, Ohana, Tainá, Débora, Daiva e Michelle) que sempre me incentivaram e se prontificaram a me ajudar, além de todos os momentos de desconcentração. Agradeço também a todos do grupo de pesquisa do Prof. Robert, especialmente à Isabela, pela ajuda no sequenciamento das mutações na população e à Mayara que me incentivou e me ajudou sempre que precisei principalmente nas confirmações das mutações nos pacientes por sequenciamento de Sanger e toda a ajuda durante a análise dos dados.
RESUMO
POLLA, Daniel Lôpo. Sequenciamento de exoma parcial como ferramenta de diagnóstico molecular de displasias esqueléticas. 2014. 133p. Dissertação (Mestrado em Ciências Genômicas e Biotecnologia) - Universidade Católica de Brasília, Brasília, 2014.
As displasias esqueléticas compreendem um grande grupo de mais de 450 doenças clinicamente distintas e geneticamente heterogêneas associadas a mutações em mais de 300 genes. Avaliações clínicas e achados radiológicos são utilizados para o diagnóstico dessas doenças. No entanto, devido à heterogeneidade genética dos fenótipos, seu diagnóstico definitivo é complexo. Com o desenvolvimento de técnicas moleculares e a elucidação das bases moleculares dessas doenças, os testes moleculares se tornaram úteis no auxílio do diagnóstico dessas displasias. Uma vez que diversos genes têm sido associados a estas doenças, abordagens padrão para seu diagnóstico molecular, utilizando o sequenciamento de Sanger, podem ser demoradas e ter um alto custo. Para superar essas limitações, foi utilizado o sequenciamento de exoma parcial e construído um painel de 1,4Mb para a captura de todos os éxons de 309 genes envolvidos em displasias esqueléticas, além de 28 regiões genômicas que sofrem deleções associadas com essas doenças. O objetivo principal foi o de fornecer uma ferramenta que possa analisar simultaneamente e com alta confiança todos os genes conhecidos relacionados às displasias esqueléticas, produzindo dados que possam ser cruzados com informações clínicas para chegar a um diagnóstico definitivo. Amostras de DNA de 93 indivíduos, com diagnóstico clínico prévio de displasia esquelética, foram sequenciados multiplexados em oito corridas usando a plataforma de sequenciamento de DNA Illumina® MiSeq®. A reprodutibilidade da metodologia foi testada repetindo todo o processo em três amostras. O sequenciamento de alto desempenho dos éxons capturados resultou em uma cobertura média de 140X. Mutações causais foram caracterizadas em 15 pacientes até o momento, incluindo 7 mutações não reportadas anteriormente. A confirmação de 9 mutações foi realizada por meio do sequenciamento de Sanger e todas foram confirmadas. O painel de genes construído nesse trabalho fornece uma ferramenta de diagnóstico molecular rápida, precisa e de baixo custo e pode auxiliar no diagnóstico definitivo de doenças esqueléticas.
ABSTRACT
POLLA, Daniel Lôpo. Targeted exome sequencing as a molecular diagnostic tool for skeletal dysplasias. 2014. 133p. Dissertation for a diploma (Master’s Degree in
Genomic Sciences and Biotechnology) – Catholic University of Brasília, Brasília, Brazil, 2014.
Skeletal dysplasias comprise a large group of more than 450 clinically distinct and genetically heterogeneous diseases associated with mutations in more than 300 genes. Clinical and radiological findings are used to diagnose these diseases. However, due to the genetic heterogeneity of these disorders, the diagnostic process is complex. With advances in molecular genetics and the elucidation of the molecular basis of many of these diseases, molecular tests have become useful in the diagnosis of skeletal dysplasias. Since many different genes have been associated with these disorders, standard diagnostic approaches using Sanger sequencing can be expensive and time consuming. To overcome these limitations, we used targeted exome sequencing and designed a 1.4Mb panel for simultaneous testing of more than 4800 exons in 309 genes involved in skeletal dysplasias, as well as 28 genomic regions which when deleted are associated with these diseases. The main objective was to provide a tool which can analyze all of the known skeletal dysplasia genes simultaneously with high confidence, producing data which can be crossed with clinical information to reach a definitive diagnosis. DNA samples from 93 individuals with previous clinical diagnosis of skeletal dysplasia were sequenced in 8 multiplexed runs using an Illumina MiSeq sequencer. Reproducibility was tested by repeating the entire procedure for three samples. NGS of the captured exons resulted in an average coverage of 140X. Causative mutations were characterized in 15 patients so far including previously unreported mutations in 7 cases. Analysis of the rest of the data is ongoing. Confirmation with Sanger sequencing was performed for 9 variants and all were confirmed. This NGS panel provides a fast, accurate and cost-effective molecular diagnostic tool and can assist in the diagnosis of skeletal diseases.
LISTA DE ABREVIATURAS
µ micro (10-6)
CCD Charge-coupled device
CDS Coding DNA Sequence
CNV Copy Number Variation
dbNSFP Database for nonsynonymous SNPs' functional predictions dbSNP Single Nucleotide Polimorphism Database
ddNTP Didesoxinucleotídeo dNTP Desoxinucleotídeo dsDNA Double-stranded DNA
FCAV Faculdade de Ciências Agrárias e Veterinárias
g Gramas
Gb Gigabase (109)
HGMD Human Genome Mutation Database indels Inserções/deleções
k Kilo
Kb Kilobase (103)
LOH Loss of heterozigosity m mili (10-3)
M Molar (mol/L)
Mb Megabase (106)
n nano (10-9)
NCBI National Center for Biotechnology Information NGS Next-Generation Sequencing
Nugen Serviço de Genética Clínica da Secretaria de Saúde do DF OMIM Online Mendelian Inheritance in Man
p Pico (10-12)
pb Pares de base
PCR Reação da polimerase em cadeia
PF Passing filter
qPCR PCR quantitativa ROI Regions of interest rpm Rotações por minuto
SNP Single Nucleotide Polimorphism SNV Single Nucleotide Variation UCB Universidade Católica de Brasília UCSC University of California Santa Cruz UnB Universidade de Brasília
SUMÁRIO
1 INTRODUÇÃO ... 10
2 REVISÃO DE LITERATURA ... 13
2.1 TECIDO ESQUELÉTICO ... 13
2.1.1 Dismorfologia ... 14
2.1.2 Displasias esqueléticas ... 16
2.1.2.1 Heterogeneidade genotípica e fenotípica e o desafio no diagnóstico ... 18
2.2 TECNOLOGIAS GENÉTICAS ... 19
2.3 SEQUENCIAMENTO DE EXOMA E EXOMA PARCIAL... 24
3 OBJETIVOS ... 29
3.1 OBJETIVO GERAL ... 29
3.2 OBJETIVOS ESPECÍFICOS ... 29
4 JUSTIFICATIVA ... 30
5 METODOLOGIA ... 31
5.1 CASUÍSTICA ... 31
5.2 COLETA DE AMOSTRAS ... 31
5.3 EXTRAÇÃO DE DNA DAS AMOSTRAS ... 32
5.4 PAINEL DE GENES E SONDAS CUSTOMIZADAS ... 33
5.5 PREPARAÇÃO DAS BIBLIOTECAS PARA SEQUENCIAMENTO NA PLATAFORMA DE ALTO DESEMPENHO ... 34
5.5.1 Fragmentação do DNA genômico ... 35
5.5.2 Preparação da biblioteca de DNA ... 36
5.5.3 Captura e enriquecimento dos fragmentos ... 37
5.5.4 Sequenciamento de DNA na plataforma de alto desempenho ... 39
5.6 ANÁLISE DE DADOS ... 40
5.7 CONFIRMAÇÃO DAS MUTAÇÕES ... 43
6 RESULTADOS ... 45
6.1 PACIENTES ... 45
6.2 PAINEL DE GENES E SONDAS CUSTOMIZADAS ... 45
6.3 PREPARAÇÃO DAS BIBLIOTECAS PARA SEQUENCIAMENTO NA PLATAFORMA DE ALTO DESEMPENHO ... 46
6.3.1 Fragmentação do DNA genômico ... 46
6.3.2 Preparação da biblioteca de DNA ... 47
6.3.3 Captura e enriquecimento dos fragmentos ... 47
6.3.4 Sequenciamento de DNA na plataforma de alto desempenho ... 50
6.4 ANÁLISE DE DADOS ... 53
6.4.1.1 Rede 6 - Síndrome de Sinostose Espondilocarpotársica ... 65
6.4.1.2 Rede 34 - Síndrome de Neurofibromatose-Noonan ... 67
6.4.1.3 Rede 43 - Displasia Geleofísica 2 ... 69
6.4.1.4 Rede 47 - Síndrome EEC3 ... 70
6.4.1.5 Rede 82 - Síndrome de Apert ... 71
6.4.1.6 Rede 85 - Síndrome de Unha-Patela ... 72
6.4.1.7 Rede 99 - Displasia Cleidocraniana ... 74
6.4.1.8 Rede 121 - Síndrome de Weaver ... 76
6.4.1.9 Rede 122 - Síndrome de Townes-Brocks ... 78
6.4.1.10 Rede 150 – Hipocondroplasia ... 79
6.4.1.11 Rede 156 - Síndrome de Wolcott-Rallison ... 80
6.4.1.12 Rede 163 - Displasia Espondiloepifisária Congênita ... 82
6.4.1.13 Rede 174 - Síndrome Tricorinofalangeana Tipo 1 ... 83
7 DISCUSSÃO ... 85
8 CONCLUSÃO ... 93
REFERÊNCIAS BIBLIOGRÁFICAS ... 95
APÊNDICE ... 105
1 INTRODUÇÃO
As displasias esqueléticas são anomalias do desenvolvimento do tecido condro-ósseo, causadas por defeitos na expressão de genes durante o desenvolvimento embrionário ou na vida pós-natal. São divididas em osteodisplasias e condrodisplasias, a primeira está associada às alterações na densidade e mineralização dos ossos e a segunda afeta o desenvolvimento da cartilagem levando a problemas no crescimento (MORTIER, 2001; RIMOIN et al., 2007).
Essas doenças afetam um grande número de fenótipos do esqueleto apresentando grande heterogeneidade genética e fenotípica. (RIMOIN et al., 2007; MAKITIE, 2011; WARMAN et al., 2011). A maioria das displasias esqueléticas é de herança autossômica dominante, no entanto também podem ser herdadas de forma autossômica recessiva, recessiva ligada ao X ou dominante ligada ao X, além de deleções ou duplicações cromossômicas, mosaicismo germinativo e dissomia uniparental (BROOK; DE VRIES, 1998; KRAKOW; RIMOIN, 2010; MAKITIE, 2011). Por isso, baseando-se em dados clínicos, diagnósticos radiológicos e moleculares, a classificação das displasias esqueléticas é complexa (RIMOIN et al., 2007; ALANAY; LACHMAN, 2011; WARMAN et al., 2011).
Atualmente, já existem mais de 450 displasias esqueléticas caracterizadas, das quais a grande maioria está correlacionada com mutações em um ou mais genes dentre mais de 300 genes que agem no desenvolvimento e crescimento dos ossos e codificam proteínas com diversas funções, como fatores de crescimento e transcrição, proteínas estruturais, enzimas, supressores de tumor, entre outras (RIMOIN et al., 2007; ALANAY; LACHMAN, 2011; WARMAN et al., 2011).
Como cada doença tem suas próprias características e complicações, o diagnóstico molecular pode ser utilizado para confirmar o diagnóstico clínico e radiográfico, prever estado de portador para uma doença recessiva nessas famílias em risco, aconselhamento genético e permitir o diagnóstico pré-natal de fetos de alto risco (KRAKOW; RIMOIN, 2010; MAKITIE, 2011).
sequenciamento de DNA são as mais utilizadas, a metodologia de sequenciamento de Sanger por eletroforese capilar e, mais recentemente, as tecnologias de sequenciamento de alto desempenho ou de próxima geração (Next-Generation Sequencing - NGS). Apesar de ser a mais utilizada, a metodologia de Sanger possui limitações de rendimento, escalabilidade e rapidez, tornando-se inviável para utilização mais robusta. Já as plataformas de sequenciamento de alto desempenho foram desenvolvidas para diminuir os custos, aumentar drasticamente a obtenção de dados sequenciados e facilidade de uso, aumentando as possibilidades de aplicações e análises genéticas (SLATKO et al., 2001; METZKER, 2005; SU et al., 2011).
Uma das aplicações para os sequenciadores de alto desempenho é em estudos que envolvam múltiplos genes candidatos para uma doença, sendo possível a análise eficiente de vários loci ao mesmo tempo, como no sequenciamento do exoma, porção genômica formada pelos éxons e que incluem as informações para codificar proteínas (NG et al., 2009; BIESECKER, 2010; TEER; MULLIKIN, 2010; BAMSHAD et al., 2011; BECKER et al., 2011; KU et al., 2012; LEMKE et al., 2012).
A estratégia para a utilização do sequenciamento do exoma é baseada no fato que em doenças mendelianas, a grande maioria das mutações ocorre nas sequências codificantes de genes, seguido por áreas de splicing e as UTRs (untranslated region, região não traduzida) (HODGES et al., 2007; NG et al., 2009; STENSON et al., 2009; TEER; MULLIKIN, 2010; BAMSHAD et al., 2011).
Diversas doenças, como muitas displasias esqueléticas, já possuem seus genes causativos identificados (Anexo B), sendo possível o uso do sequenciamento do exoma parcial com a criação de painéis de genes específicos para triagem de mutações relevantes no diagnóstico e tratamento de doenças, focando o poder de sequenciamento de alto desempenho em uma região específica do DNA (NIGRO; PILUSO, 2012; RIZZO; BUCK, 2012).
2 REVISÃO DE LITERATURA
2.1 TECIDO ESQUELÉTICO
O tecido esquelético é formado por cartilagens e ossos. A primeira é um tecido conectivo avascular e o segundo é um tecido conectivo calcificado, vascularizado e que compõe a maior parte do esqueleto (DÂNGELO, 2002; DRAKE et al., 2010).
As principais funções da cartilagem são suporte para tecidos moles, proporcionar uma superfície adequada para articulações e juntas e proporcionar o desenvolvimento e crescimento de ossos longos. As cartilagens são divididas em três tipos (Figura 1), cartilagem hialina, a mais abundante e fornece sustentação, flexibilidade e resistência; a cartilagem elástica, semelhante à hialina, mas contém fibras elásticas; e a fibrocartilagem, mais resistente à pressão e ao desgaste. Devido à sua avascularização, o tecido cartilaginoso é nutrido por difusão, além de não possuir vasos linfáticos ou nervos (DÂNGELO, 2002; MARIEB, 2006; DRAKE et al., 2010).
O esqueleto pode ser dividido em dois subgrupos (Figura 1), o esqueleto axial, que consiste dos ossos do crânio, coluna vertebral, costelas e esterno; e o esqueleto apendicular, que consiste nos ossos dos membros superiores e inferiores (DÂNGELO, 2002; MARIEB, 2006; DRAKE et al., 2010). Os ossos têm como principais funções a proteção de órgãos vitais, apoio estrutural para o corpo, auxílio nos movimentos, reservatório de minerais como cálcio e fósforo além de abrigar a medula óssea, responsável pela hematopoiese. Ao contrário do tecido cartilaginoso, os ossos são vascularizados e inervados (DRAKE et al., 2010).
Figura 1: Ilustração da composição do tecido esquelético.
A cartilagem hialina pode ser encontrada nas superfícies articulares dos ossos, as fibrocartilagens podem ser encontradas nos discos intervertebrais e as cartilagens elásticas podem ser encontradas na orelha, laringe e epiglote.
Fonte: Adaptado de MARIEB, 2006.
2.1.1 Dismorfologia
A dismorfologia estuda os defeitos congênitos e anomalias do corpo humano, originados antes do nascimento, de acordo com a natureza do defeito estrutural (QUEIßER-LUFT; SPRANGER, 2006) e pode ser classificada em malformações, defeitos primários durante a formação do tecido ou do órgão; disrupções, defeitos nas estruturas em desenvolvimento por eventos intrauterinos;
Legenda:
Ossos do esqueleto axial
Ossos do esqueleto apendicular
Cartilagens hialinas
Fibrocartilagens
deformações, uma estrutura normal é deformada por forças mecânicas internas ou externas; e displasias, em que existe uma desorganização da estrutura celular ou do arranjo celular num tecido específico (STEVENSON; HALL, 2006).
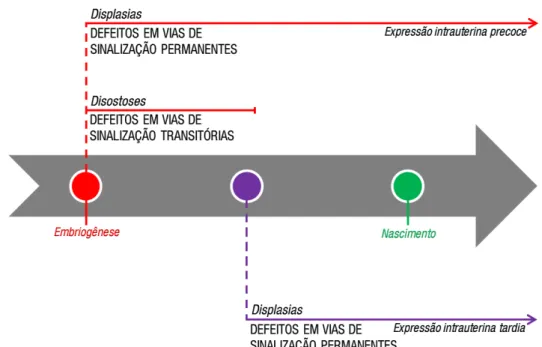
Estes conceitos gerais de dismorfologia foram usados para diferenciar os erros do desenvolvimento ósseo em três subgrupos, disostoses, disrupções ou displasias esqueléticas (Figura 2) (SPRANGER, BRILL; POZNANSKI, 2002).
Figura 2: Esquema ilustrando o conceito de defeito na expressão gênica durante um determinado
período e as anomalias resultantes.
Fonte: Adaptado de SPRANGER et al., 2002.
As disostoses são defeitos na formação de ossos individuais, isolados ou em combinação devido a defeitos em vias de sinalização que atuam brevemente durante a organogênese do esqueleto, portanto é finita, pois o processo molecular defeituoso atua somente durante um espaço de tempo definido. As lesões disostóticas são distribuídas clinicamente de forma assimétrica (SPRANGER, BRILL; POZNANSKI, 2002; NUSSBAUM et al., 2007).
Displasias esqueléticas são anomalias no desenvolvimento do tecido condro-ósseo, causadas por defeitos na expressão embrionária de genes que continuam sendo expressos na vida pós-natal. Podem ser subdividas em osteodisplasias, associadas às alterações na densidade e mineralização dos ossos, e em condrodisplasias, que afetam o desenvolvimento da cartilagem levando a problemas no crescimento (MORTIER, 2001; RIMOIN et al., 2007). As anomalias podem ser reconhecidas no momento do nascimento ou manifestar-se somente na infância ou até mesmo mais tardiamente. Contudo, todos os defeitos que causam as displasias ósseas continuam a afetar o crescimento e desenvolvimento do esqueleto pós-natal, diferente das disostoses, que tradicionalmente tem sido caracterizada como distúrbios limitados espaço-temporalmente durante o início da embriogênese (MORTIER, 2001; SPRANGER, BRILL; POZNANSKI, 2002; STEVENSON; HALL, 2006; RIMOIN et al., 2007).
No entanto as diferenças entre estes três subgrupos já não são muito evidentes, pois mutações em somente um gene podem associadas às disostoses ou às displasias (RIMOIN et al., 2007; KRAKOW; RIMOIN, 2010).
.
2.1.2 Displasias esqueléticas
A regulação do desenvolvimento do esqueleto é realizada por diversos fatores genéticos os quais ainda não são completamente conhecidos. Qualquer alteração genética que afete essa regulação, seja no próprio gene ou na regulação do mesmo, pode gerar uma série de condições clínicas que afetam o esqueleto (STEVENSON; HALL, 2006; MAKITIE, 2011).
Assim, os fenótipos podem variar de gravidade, desde artropatia precoce em indivíduos com estatura média a nanismo grave com mortalidade perinatal. As displasias esqueléticas também podem estar associadas com complicações ortopédicas, cardíacas, neurológicas, renais, auditivas, pulmonárias, visuais e psicológicas (KRAKOW; RIMOIN, 2010; MAKITIE, 2011). Apesar de esses fenótipos serem individualmente raros, a taxa de prevalência em nascidos vivos com displasias esqueléticas é de pelo menos 2 em 10.000 e indivíduos provindos de consanguinidade parental a taxa é aumentada para 9.4 em 10.000, sendo acondroplasia, displasia tanatofórica e osteogênese imperfeita os tipos mais frequentes de displasias esqueléticas (ORIOLI, CASTILLA; BARBOSA-NETO, 1986; AL-GAZALI et al., 2003; STEVENSON; HALL, 2006; ALANAY; LACHMAN, 2011). Por isso o diagnóstico preciso pode ter impacto significativo no tratamento, acompanhamento do paciente e aconselhamento genético (BROOK; DE VRIES, 1998; RIMOIN et al., 2007).
Diversos sistemas de classificação foram criados para que essas doenças fossem diagnosticadas mais facilmente e facilitassem seu tratamento. Inicialmente eram classificadas de acordo com diferentes critérios, como idade de manifestação da patologia, partes do corpo afetadas, padrão de herança e histologia, por exemplo. Devido ao crescimento do conhecimento sobre as bases moleculares do esqueleto e seus defeitos, essas classificações evoluíram. No entanto, diversos fenótipos não possuem suas bases moleculares completamente elucidadas, portanto torna-se necessário agrupar as doenças em grupos de famílias com características clínicas, radiológicas e genéticas similares, que são úteis para facilitar o diagnóstico do paciente. Para uso clínico opta-se a classificação do Grupo de Trabalho Internacional sobre Distúrbios Constitucionais do Osso, inicialmente proposto em 1970 e atualizado desde então (International Working Group on Constitutional Disorders of Bone) (International nomenclature and classification of the osteochondrodysplasias (1997). International Working Group on Constitutional Diseases of Bone, 1998; MORTIER, 2001; SAVARIRAYAN; RIMOIN, 2004; STEVENSON; HALL, 2006; KRAKOW; RIMOIN, 2010; WARMAN et al., 2011).
456 condições patológicas foram observadas e categorizadas em 40 grupos por critérios moleculares, bioquímicos e/ou radiográficos, dentre estas, 316 condições estão associadas com um ou mais de 226 genes diferentes que codificam fatores de transcrição, proteínas da matriz extracelular, enzimas, supressores tumorais, transportadores celulares, transdutores de sinal (ligantes, receptores e proteínas de canal), chaperonas, proteínas de ligação intracelulares, moléculas de processamento de RNA e proteínas citoplasmáticas, além de uma série de genes com produtos ainda sem função conhecida (SUPERTI-FURGA; UNGER, 2007; KRAKOW; RIMOIN, 2010; WARMAN et al., 2011).
Enquanto a caracterização profunda com novos dados clínicos, radiográficos, morfológicos, bioquímicos, metabólicos e moleculares das displasias não for realizada, levando-se em consideração que alterações em um gene podem gerar diversos fenótipos, ou diversos genes podem produzir um mesmo fenótipo, a categorização de displasias esqueléticas em uma única classificação que atenda tanto o diagnóstico clínico quanto a pesquisa é um desafio (BROOK; DE VRIES, 1998; JAKKULA et al., 2005; RIMOIN et al., 2007; MAKITIE, 2011).
2.1.2.1 Heterogeneidade genotípica e fenotípica e o desafio no diagnóstico
LACHMAN; RIMOIN, 2009). Grandes avanços foram feitos nas descobertas de defeitos genéticos causativos dessas doenças. Assim, os dados moleculares podem auxiliar tanto no diagnóstico precoce do paciente quanto no aconselhamento genético de pacientes afetados e suas famílias (MORTIER, 2001; OFFIAH; HALL, 2003; ALANAY; LACHMAN, 2011).
Essas condições podem ser geneticamente herdadas de forma autossômica recessiva, autossômica dominante, recessiva ligada ao X ou dominante ligado ao X, além de mecanismos genéticos mais raros como deleções ou duplicações cromossômicas, mosaicismo germinativo e dissomia uniparental (BROOK; DE VRIES, 1998; KRAKOW; RIMOIN, 2010; MAKITIE, 2011). A maioria das displasias esqueléticas é de herança genética dominante, portanto o diagnóstico ou características parentais prévias podem ajudar no diagnóstico do neonato, porém uma parte dos fenótipos se origina de mutações de novo ou são de herança genética recessiva, ou seja, os pais são fenotipicamente normais e ainda, existe variabilidade intrafamiliar e interfamiliares em muitos desses transtornos (BROOK; DE VRIES, 1998; KRAKOW; RIMOIN, 2010; MAKITIE, 2011).
Como cada doença tem sua própria característica e complicações, o diagnóstico molecular pode ser utilizado para confirmar o diagnóstico clínico e radiográfico, prever estado de portador para uma doença recessiva nessas famílias em risco e permitir o diagnóstico pré-natal de fetos de alto risco (KRAKOW; RIMOIN, 2010; MAKITIE, 2011).
2.2 TECNOLOGIAS GENÉTICAS
ser feitos por análises focadas em mutações específicas, seja ressequenciando um gene específico em busca de mutações ou analisando múltiplos genes em que uma mutação possa levar a um fenótipo semelhante (KORF; REHM, 2013).
Atualmente, a tecnologia amplamente utilizada em laboratórios para o ressequenciamento de genes causativos de doenças mendelianas é feito pela metodologia de eletroforese de capilar utilizando a metodologia de sequenciamento de Sanger (SANGER, NICKLEN; COULSON, 1977; SANGER; COULSON, 1978; SANGER et al., 1980). Essa tecnologia é baseada na incorporação de desoxinucleotídeos (dNTPs) e de didesoxinucleotídeos (ddNTPs), estes marcados com fluorocromos específicos para cada base nitrogenada, a uma nova cadeia de DNA em crescimento, tendo como molde o DNA de interesse. Os ddNTPs, quando incorporados à sequência, interrompem a extensão da mesma, gerando fragmentos de tamanhos aleatórios, que sofrem separação por eletroforese ao percorrerem um capilar e a fluorescência emitida por cada fragmento é então detectada pelo sequenciador e a sequência é montada por um software (SANGER, NICKLEN; COULSON, 1977; SLATKO et al., 2001).
No entanto, a metodologia de Sanger possui limitações de custo-benefício, como rendimento, escalabilidade e rapidez que podem impedir ou retardar o avanço da genômica. Para superar essas limitações, novas tecnologias de sequenciamento, comumente chamadas de sequenciamento de alto desempenho ou de próxima geração, foram desenvolvidas para diminuir os custos, aumentar drasticamente a obtenção de dados sequenciados e facilidade de uso, aumentando as possibilidades de aplicações e análises genéticas (SLATKO et al., 2001; METZKER, 2005; SU et al., 2011).
(aproximadamente 600 pares de base), no entanto o custo é mais alto e a geração de dados é muito menor (SU et al., 2011).
A Life Technologies® desenvolveu o sequenciador Ion Torrent PGM™
(Personal Genome Machine) e o Ion Proton™ System com uma tecnologia baseada em chips semicondutores capazes de detectar a incorporação de nucleotídeos por meio das pequenas mudanças de pH durante o sequenciamento (PURUSHOTHAMAN, TOUMAZOU; OU, 2006; RODRÍGUEZ-EZPELETA, HACKENBERG; ARANSAY, 2012). Sua grande vantagem é a utilização da tecnologia de semicondutores, a rapidez no sequenciamento e não requer a utilização de enzimas especiais ou nucleotídeos marcados (RODRÍGUEZ-EZPELETA, HACKENBERG; ARANSAY, 2012), no entanto essa metodologia possui uma menor capacidade de geração de dados, maior tempo para preparação das amostras e fluxo de trabalho mais complexo (LI et al., 2013). Essa tecnologia ainda está em desenvolvimento e melhorias no seu desempenho ainda estão sendo feitas (LOMAN et al., 2012; LI et al., 2013).
A metodologia de sequenciamento por síntese a partir de terminadores reversíveis da Illumina® (BENTLEY et al., 2008) foi a tecnologia de sequenciamento de alto desempenho mais bem sucedida e amplamente adotada (METZKER, 2005; BENTLEY et al., 2008; SU et al., 2011). A princípio, o conceito por trás dessa tecnologia é similar à metodologia de Sanger, cujas bases nitrogenadas de fragmentos pequenos são sequencialmente identificadas através de fluorescência emitida durante a ressíntese de um fragmento de DNA a partir de uma fita molde de DNA (ILLUMINA, 2012).
Sua metodologia para sequenciamento de DNA (Figura 3) é baseada na fragmentação aleatória do DNA por ultrassonicação ou nebulização, então as extremidades de cada fragmento são reparadas por exonucleases e polimerases para gerar extremidades cegas e a extremidade 5’ de cada fragmento é fosforilada. Logo após é adicionado um único nucleotídeo de adenosina na extremidade 3’ dos
ligados à flow-cell são amplificados por PCR em ponte (bridge PCR) para geração de clusters, necessários para a detecção suficiente de fluorescência durante o sequenciamento. A amplificação por PCR em ponte milhares de clusters de alta densidade, cada cluster contém aproximadamente 1000 cópias da mesma fita de DNA molde, que serão sequenciados pela técnica de sequenciamento por síntese com terminadores reversíveis e fluorocromos removíveis (JANITZ, 2008; SU et al., 2011; ILLUMINA, 2012).
Para o sequenciamento (Figura 3), as fitas reversas de DNA de cada cluster são removidas, seus adaptadores na flow-cell são bloqueados e iniciadores (primers) de sequenciamento são então hibridizados nas fitas molde de DNA para o início do sequenciamento. O sequenciamento é baseado na detecção de fluorescência emitida por um nucleotídeo a partir da sua incorporação na síntese de uma nova fita de DNA baseada no fragmento de DNA anteriormente fixado na flow-cell. A cada ciclo de sequenciamento, uma reação contendo uma DNA polimerase e quatro nucleotídeos com terminadores reversíveis bloqueados marcados com fluorescência específica são injetados na flow-cell e durante cada ciclo, somente um nucleotídeo é incorporado à nova sequência. Após a incorporação, a identidade e posição do nucleotídeo específico na flow-cell são determinadas pela fluorescência emitida pelo mesmo através de uma câmera CCD. No próximo ciclo, o terminador do nucleotídeo é desbloqueado e o marcador de fluorescência é removido da base nitrogenada permitindo que um novo nucleotídeo seja incorporado e uma nova base nitrogenada seja detectada usando a mesma estratégia (JANITZ, 2008; SU et al., 2011; ILLUMINA, 2012). Dependendo do kit de sequenciamento utilizado, podem ocorrer 75, 100, 150 ou 250 ciclos, gerando consequentemente fragmentos com tamanhos de 75, 100, 150 ou 250 pares de base, respectivamente. Através do tipo de equipamento e kit usados, o sequenciamento pode durar de 1 a 15 dias gerando entre 2 e 500Gb de dados.
Figura 3: O processo do sequenciamento por síntese.
Para a preparação da biblioteca de fragmentos, o DNA é fragmentado, então suas extremidades são reparadas gerando extremidades cegas. As extremidades 5’ são fosforiladas e as extremidades 3’ são adeniladas para ligação dos adaptadores. Após a ligação dos adaptadores, o DNA é desnaturado e hibridizado na flow-cell para formação de clusters. As fitas simples de DNA são estendidas, formando fitas duplas e então são desnaturadas, formam uma ponte, são estendidas novamente para a formação de uma ponte de fita dupla, desnaturas e hibridizadas para formar novas pontes de fita simples. Esse processo é repetido 35 vezes para aumentar a densidade de cada cluster. As fitas reversas são clivadas, os adaptadores da flow-cell para as extremidades dessas fitas são bloqueados e as fitas remanescentes são hibridizadas aos iniciadores de sequenciamento. Para o sequenciamento, a DNA polimerase incorpora um nucleotídeo com terminador reversível e marcado com fluorocromo, é realizada a captura da imagem da fluorescência emitida pelos clusters e então a fluorescência de cada nucleotídeo incorporado é retirada, seus terminadores são desbloqueados e o próximo ciclo de sequenciamento reinicia.
Uma das aplicações para os sequenciadores de alto desempenho é em estudos que envolvam múltiplos genes candidatos para uma doença, sendo possível a análise eficiente de vários loci ao mesmo tempo, como no sequenciamento do exoma, porção genômica formada pelos éxons e que codificam proteínas. Esse sequenciamento tem sido muito aplicado nos últimos anos utilizando-se a técnica de sequenciamento de alto desempenho para o diagnóstico molecular de doenças genéticas específicas ainda não elucidadas ou com alta heterogeneidade tanto genotípica quanto fenotípica (NG et al., 2009; BIESECKER, 2010; TEER; MULLIKIN, 2010; BAMSHAD et al., 2011; BECKER et al., 2011; KU et al., 2012; LEMKE et al., 2012).
2.3 SEQUENCIAMENTO DE EXOMA E EXOMA PARCIAL
Com o desenvolvimento e difusão das tecnologias de sequenciamento de alto desempenho, o sequenciamento do genoma humano ficou acessível para laboratórios independentes, principalmente no diagnóstico de doenças. Entretanto, o custo e capacidade necessária para processamento de dados ainda são consideráveis, considerando que a função de grande parte do genoma ainda é desconhecida (NG et al., 2009; TEER; MULLIKIN, 2010). No caso de pacientes sem diagnóstico definido, ou em que não existem genes conhecidos, pode-se realizar o sequenciamento do exoma desse paciente. O exoma é parte do genoma composto pelas regiões que codificam proteínas, constitui somente 1-2% de todo o genoma, ou aproximadamente 30-62 Mb em aproximadamente 180.000 éxons (NG et al., 2009; BIESECKER, 2010).
altamente importante do genoma para a busca de variantes que causam doenças (STENSON et al., 2009; BAMSHAD et al., 2011). Além disso, somente as áreas
codificantes dos genes são “capturadas” e sequenciadas, gerando muito menos
dados, porém mais relevantes e com maior cobertura, auxiliando no diagnóstico de diversas doenças sem gene causativo conhecido (NG et al., 2009; GLAZOV et al., 2011; MIN et al., 2011; CARMICHAEL et al., 2012).
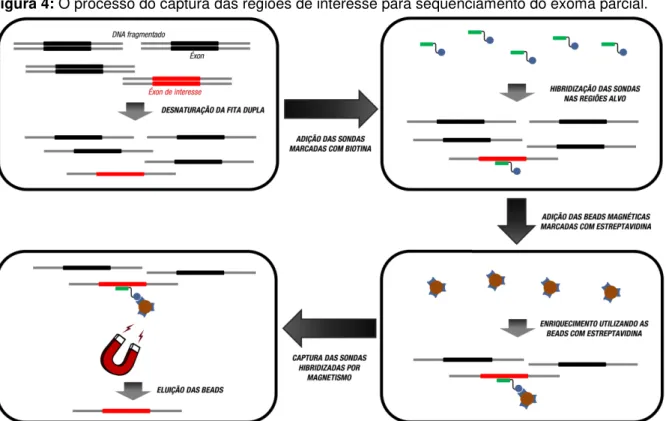
No entanto, diversas doenças, como muitas displasias esqueléticas, já possuem seus genes causativos identificados (Anexo B), sendo possível o uso do sequenciamento do exoma parcial com a criação de painéis de genes específicos para triagem de mutações relevantes no diagnóstico e tratamento de doenças, focando o poder de sequenciamento de alto desempenho em uma região específica do DNA (NIGRO; PILUSO, 2012; RIZZO; BUCK, 2012). Além de possibilitar uma maior taxa de cobertura, o exoma parcial possibilita uma maior acurácia do diagnóstico devido à menor quantidade de material sequenciado, prevenindo a detecção, validação e interpretação de inúmeras variantes em genes não relacionados ao fenótipo do paciente (LEMKE et al., 2012; NIGRO; PILUSO, 2012; RIZZO; BUCK, 2012). Ainda é possível multiplexar os sequenciamentos, ou seja, sequenciar mais de uma amostra ao mesmo tempo. Para o sequenciamento do exoma parcial, as regiões genômicas de interesse são capturadas por hibridização de sondas complementares e enriquecidas antes do sequenciamento para redução da complexidade de uma biblioteca de DNA para sequenciamento (PORRECA et al., 2007; VOELKERDING, DAMES; DURTSCHI, 2009; ILLUMINA, 2012; NIGRO; PILUSO, 2012; RIZZO; BUCK, 2012).
das sondas. Através de separação magnética, os fragmentos que contem as regiões de interesse capturadas (targets, ou alvos) pelas sondas são separados da solução, eluídos das sondas e então amplificados (Figura 4) (PARLA et al., 2011; ILLUMINA, 2012; NIGRO; PILUSO, 2012).
Figura 4: O processo do captura das regiões de interesse para sequenciamento do exoma parcial.
A captura de éxons específicos por sondas customizadas, a partir da desnaturação dos fragmentos da biblioteca de DNA, hibridização das sondas marcadas com biotina e o enriquecimento das sequências utilizando beads marcadas com estreptavidina e no fim a eluição das sequências de interesse.
Fonte: Adaptado de ILLUMINA, 2012.
sequenciamento de Sanger principalmente devido à análise de diversos genes ao mesmo tempo, além de ser mais rápido, menor custo e mais preciso, auxiliando no diagnóstico essas doenças (LEMKE et al., 2012; NIGRO; PILUSO, 2012; VALENCIA et al., 2012; CHEN et al., 2013; CRONA et al., 2013; KOSHIMIZU et al., 2013; SIVAKUMARAN et al., 2013; SULE et al., 2013).
Apesar de essa abordagem ser muito efetiva, ainda possui algumas limitações. A análise dos dados moleculares é limitada aos genes incluídos no painel. Além disso, devido à limitações da metodologia de captura, as regiões de interesse nunca são totalmente capturadas e regiões com sequências de alta similaridade às algumas regiões de interesse podem ser capturadas e sequenciadas (regiões off target). Também, em consequência das sequencias de baixa complexidade no genoma humano, algumas regiões podem não ser sequenciadas e algumas mutações não serão identificadas (JOHANSSON et al., 2011; VALENCIA et al., 2012).
Ainda há a necessidade da confirmação de novos SNVs descobertos utilizando essas plataformas de sequenciamento pela metodologia de Sanger, pois ocorre uma perda de acurácia devido à utilização da DNA polimerase durante a PCR na construção da biblioteca de fragmentos, enriquecimento e durante o sequenciamento, elevando a taxa de erro da mesma. Enquanto o sequenciamento por Sanger possui uma taxa de erro de 1 a cada 10 000 bases sequenciadas, as plataformas de alto desempenho possuam taxas mais elevadas de 1 em 1000 bases sequenciadas em uma cobertura abaixo de 20X (CAMPBELL et al., 2008; HARISMENDY et al., 2009; GULLAPALLI et al., 2012).
Entretanto, a acurácia de detecção de variantes como na metodologia de Sanger tem sido alcançada com cobertura acima de 20X por base sequenciada, indicando que essas plataformas mais recentes de sequenciamento podem ser usadas independentemente para descoberta de variantes sob essas condições (CAMPBELL et al., 2008; GULLAPALLI et al., 2012).
3 OBJETIVOS
3.1 OBJETIVO GERAL
Utilizar uma ferramenta de análise genética eficiente para auxiliar no diagnóstico de pacientes com displasias esqueléticas por meio do sequenciamento de exoma parcial em plataforma de sequenciamento de alto desempenho.
3.2 OBJETIVOS ESPECÍFICOS
Construção de um painel de genes e regiões genômicas associados com doenças esqueléticas;
4 JUSTIFICATIVA
Como as displasias esqueléticas são clinicamente difíceis de serem diagnosticadas devido à heterogeneidade genética e fenotípica e com parte das mutações em seus genes causativos já estão elucidadas, a análise molecular mostrou-se eficaz no auxílio do diagnóstico dessas doenças, juntamente como os dados clínicos e achados radiológicos.
Atualmente, para a análise molecular rotineira de displasias esqueléticas utiliza-se o sequenciamento de Sanger. No entanto, essa metodologia não é eficaz para examinar um grande número de pacientes e genes.
Com o desenvolvimento de novas tecnologias de sequenciamento genético, as quais geraram uma grande quantidade de dados, o sequenciamento de todos os genes conhecidos das displasias esqueléticas torna-se muito mais viável e com um melhor custo benefício que o sequenciamento individual ou de pequenos grupos de genes.
5 METODOLOGIA
Este projeto de pesquisa faz parte do projeto "Uso de Tecnologias Modernas de Análise Genética e Genômica para Caracterizar Doenças Raras do Esqueleto” e foi aprovado pelo Comitê de Ética e Pesquisa da Universidade Católica de Brasília conforme as diretrizes descritas no Ofício de nº 186/2011 e com parecer consubstanciado Nº. CEP/UCB 056/2011 (Anexo A).
5.1 CASUÍSTICA
O grupo de pesquisa do consórcio Rede Centro-Oeste coordenado pelo Prof. Dr. Robert Pogue possui um banco de amostras de pacientes com displasias esqueléticas para o desenvolvimento de estudos genéticos realizados na Universidade Católica de Brasília.
Desse banco de amostras, foram selecionadas noventa e três pacientes adultos e crianças (Apêndice A), com diagnóstico médico que envolva malformações ou anomalias do crescimento do esqueleto, o qual foi requerido que fosse assinado, pelos mesmos ou seus responsáveis, um termo de consentimento livre e esclarecido (Anexo B).
5.2 COLETA DE AMOSTRAS
(NUGEN, Brasília - DF), Hospital Infantil Albert Sabin (Fortaleza - Ceará), Hospital de Reabilitação de Anomalias Craniofaciais da Universidade de São Paulo (Bauru - SP), PUC-Goiás (Goiânia - GO) e Universidade Federal de Alagoas (Maceió –
Alagoas).
5.3 EXTRAÇÃO DE DNA DAS AMOSTRAS
O isolamento do DNA genômico a partir de leucócitos do sangue periférico foi realizada de duas formas, a primeira pelo kit QIAamp DNA Blood Mini Kit 50 (Qiagen®, Hilden, Alemanha) seguindo as especificações do fabricante, para obter-se uma alíquota principal de material genômico utilizando um menor volume de sangue periférico. A segunda metodologia utilizada foi a de salting out, para processamento do restante do sangue periférica e obtenção de amostras de DNA reserva.
Para a extração de DNA pela metodologia de salting out, adicionou-se 750µl de sangue total e 250µl de água destilada em um tubo eppendorf de 2mL, e usou-se um agitador de tubos por 15 segundos para homogeneizar os líquidos. Logo após centrifugou-se por 15 minutos a 5000rpm em centrifuga Eppendorf® Centrifuge 5415D (Eppendorf®, Hamburg, Alemanha). Descartou-se o sobrenadante e adicionou-se novamente água destilada para completar 1mL, homogeneizou-se novamente no agitador de tubos e centrifugou-se por mais 15 minutos a 5000rpm.
Descartou-se novamente o sobrenadante e ressuspendeu-se o pellet formado com 700µl de tampão A (0,32 M Sacarose, 10 mM de Tris HCl pH 7,6, 5 mM MgCl2, 1% de Triton X 100) e centrifugou-se a 5000rpm por 5 minutos.
Em seguida adicionou-se 200µl de NaCl 6M, agitou-se por 15 segundos e centrifugou-se a 3000rpm por 15 minutos. Transferiu-se o sobrenadante para um novo tubo eppendorf de 1,5mL e adicionou-se etanol 100% a temperatura ambiente até completar a capacidade do tubo. Inverteu-se o tubo até a completa precipitação do DNA e centrifugou-se o tubo a 5000rpm por 5 minutos. Descartou-se o sobrenadante e após a completa evaporação do etanol ressuspendeu-se o DNA em 100µl de água Milli-Q (Millipore®) autoclavada e incubou-se a 37oC por 1 hora.
O DNA extraído por ambas as metodologias foi estocado a -80oC.
5.4 PAINEL DE GENES E SONDAS CUSTOMIZADAS
Foram selecionados 309 genes descritos na literatura relacionados com displasias esqueléticas quando mutados (Apêndice B), além de 28 regiões genômicas que sofrem deleções associadas com displasias esqueléticas, sendo as deleções na região reguladora dos genes SOX9 e SHOX, deleções relacionadas com a Síndrome de Kabuki, síndrome de Wolf-Hirschhorn e síndrome de Beckwith-Wiedemann. Para essas regiões foram escolhidos SNPs (single-nucleotide polymorphism, polimorfismo de nucleotídeo único) com alto grau de heterozigose para que essas deleções possam ser indicadas por perda de heterozigose.
Figura 5: Ilustração da plataforma de desenho de exoma parcial DesignStudio com alguns dos genes escolhidos, o número de éxons a serem sequenciados e dados técnicos sobre a eficiência prevista de sequenciamento.
5.5 PREPARAÇÃO DAS BIBLIOTECAS PARA SEQUENCIAMENTO NA PLATAFORMA DE ALTO DESEMPENHO
5.5.1 Fragmentação do DNA genômico
Inicialmente o DNA genômico de cada amostra foi fragmentado para iniciar o preparo de uma biblioteca de pequenos fragmentos gel-free para ser sequenciado uniformemente, seguindo as instruções do fabricante de acordo com o manual Illumina® TruSeq® DNA Sample Preparation V2 Guide low-throughput Rev A. Para a fragmentação das amostras foi utilizado duas metodologias devido à disponibilidade de equipamentos, nebulização e ultrassonicação. Utilizou-se o equipamento Qubit® com o Kit dsDNA BR Assay (Invitrogen™, Carlsbad, CA, EUA) para quantificação das amostras de DNA genômico extraído anteriormente.
A fragmentação por nebulização foi utilizada separadamente no primeiro grupo de doze amostras. Utilizou-se um nebulizador descartável Illumina® Nebulizer Kit e alta pressão para fragmentar o DNA. Para cada amostra, adicionou-se 2µg de DNA em um volume total de 50µl de tampão TE no nebulizador e 700µl de tampão de nebulização. Conectou-se a fonte de ar comprimido ao nebulizador, neste caso nitrogênio líquido, e aplicou-se uma pressão de 43 psi por 6 minutos. Logo após centrifugou-se o nebulizador a 450 xg por 2 minutos. Para purificação dos fragmentos utilizou-se o kit QIAquick PCR Purification Kit (Qiagen®, Hilden, Alemanha), eluiu-se os fragmentos em 50µl de tampão EB do kit e transferiu-se 50µl para um poço de uma placa de PCR de 96 tubos de 0,3mL Thermo-Fast® 96 Non-Skirted (Thermo Fisher Scientific®, Waltham, MA, EUA).
Frequency sweeping e Temperatura 5,5º a 6,0ºC. Após a fragmentação, 52,5µl do DNA fragmentado foram transferidos do microtubo para um poço de uma placa de PCR de 0,3mL de 96 tubos Thermo-Fast® 96 Non-Skirted.
5.5.2 Preparação da biblioteca de DNA
A preparação das bibliotecas de DNA foi realizada seguindo as instruções do fabricante de acordo com o manual Illumina® TruSeq® DNA Sample Preparation V2 Guide low-throughput Rev A. Após a fragmentação do DNA foi feito a reparação das extremidades coesivas dos fragmentos de DNA, causadas pela fragmentação, utilizando éxonucleases 5’ e 3’. Então foi realizada uma etapa de adição de um único nucleotídeo de adenosina na extremidade 3’ dos fragmentos para prevenir a ligação
entre eles na próxima etapa onde são ligados os adaptadores. Nesta etapa foram adicionados múltiplos indexes nas extremidades dos fragmentos, pequenas sequências específicas de DNA que identificam cada amostra como um código de barras, que preparam os fragmentos para a hibridização na flow-cell do sequenciador. Por último foi realizado um processo de enriquecimento dos fragmentos através de uma PCR (reação de polimerase em cadeia) que seletivamente amplifica somente os fragmentos que possuem adaptadores ligados em suas extremidades.
5.5.3 Captura e enriquecimento dos fragmentos
A captura das sequências de interesse foi feita de acordo com as instruções do fabricante de acordo com o manual Illumina ® TruSeq® Enrichment Guide Ver J (Illumina®, San Diego, CA, EUA). A partir da biblioteca de DNA realizada anteriormente, foi preparada a biblioteca para sequenciamento de DNA das regiões genômicas de interesse.
As bibliotecas de DNA foram agrupadas em oito grupos (pools), totalizando doze bibliotecas por grupo (Apêndice C). Para cada grupo, foram adicionados, no máximo, 500ng de DNA de cada uma das doze bibliotecas genômicas (totalizando o
máximo de 6μg de DNA) em um poço de uma placa de PCR de 96 tubos de 0,3mL Thermo-Fast® 96 Non-Skirted, formando uma biblioteca de sequenciamento.
Cada biblioteca de sequenciamento foi misturada com as sondas de captura de regiões específicas selecionadas na construção do painel de gene e sondas customizadas. Foi feita a primeira hibridação que assegura que as regiões específicas se liguem às sondas de captura completamente. A seguir foi feito a primeira lavagem a qual beads de estreptavidina são usadas para capturar as sondas hibridizadas às regiões específicas de interesse. Então foi feito uma segunda hibridização para assegurar que as regiões de interesse sejam enriquecidas e foi feito uma segunda lavagem com beads de estreptavidina. Por último foi feito uma PCR de enriquecimento para amplificar os fragmentos da biblioteca de sequenciamento para a realização do sequenciamento.
Logo após foi feita a validação das bibliotecas de sequenciamento através da quantificação e verificação do tamanho dos fragmentos enriquecidos.
A verificação do tamanho dos fragmentos enriquecidos de todas as bibliotecas foi realizada no equipamento Bioanalyzer® com o chip High Sensitivity (Agilent Technologies®, Santa Clara, CA, EUA).
EUA) na primeira biblioteca e qPCR (PCR quantitativa) nas sete bibliotecas subsequentes devido à disponibilidade dos reagentes.
A quantificação pela metodologia de qPCR foi feita com o kit KAPA Library Quantification Kit for Illumina sequencing platforms (KAPABIOSYSTEMS®, Wilmington, MA, EUA). Esse kit inclui iniciadores específicos para bibliotecas preparadas com os adaptadores da Illumina® além de seis amostras de DNA padrão com suas concentrações conhecidas que são usadas para gerar uma curva padrão a qual é comparada às bibliotecas quantificadas.
Seguindo as especificações do fabricante, preparou-se o Master Mix, logo após foi feita a diluição das bibliotecas de sequenciamento em um tampão de diluição (10mM Tris-HCl, pH 8,0; Tween 20, 0,05%) nas diluições 1:1000, 1:2000, 1:4000 e 1:8000. Então foi preparada a placa para as reações usando uma placa de 96 poços de 0,1mL MicroAmp® Fast Optical 96-Well Reaction with Barcode 0,1mL (Applied Biosystems®, Foster City, CA, EUA), utilizando 10μl de volume final (Master Mix, 6μL; Diluição da biblioteca e DNA padrão 1-6, 4μL) e as alíquotas de DNA padrão e cada diluição das bibliotecas em triplicata. A placa com as reações foi centrifugada por 2 minutos a 280 xg e então colocada no termociclador
StepOnePlus™ Real-Time PCR System (Applied Biosystems®, Foster City, CA, EUA) e programada com o protocolo indicado pela fabricante do kit (Tabela 1).
Tabela 1: Programação do equipamento de qPCR para quantificação das bibliotecas de
sequenciamento.
Temperatura Tempo Ciclos
Ativação inicial/Desnaturação 95ºC 5 minutos 1
Desnaturação 95ºC 30 segundos 35
Anelamento/Extensão/Obtenção
de dados 60ºC 45 segundos
5.5.4 Sequenciamento de DNA na plataforma de alto desempenho
A preparação das bibliotecas para sequenciamento no MiSeq® foi realizada de acordo com as instruções do fabricante de acordo com o manual Illumina® Preparing DNA Libraries for Sequencing on the MiSeq® Rev B (Illumina®, San Diego, CA, EUA). Cada biblioteca com os fragmentos de DNA enriquecidos foram desnaturados com NaOH, diluídos a uma concentração final ideal e otimizada para a formação de clusters (Figura 6) e adicionados no reservatório indicado “Load Samples” do cartucho de reagentes do MiSeq®.
Figura 6: Concentração final das bibliotecas para sequenciamento
.
Cada biblioteca para sequenciamento foi diluída a partir dos dados da quantificação realizada na validação das bibliotecas.
A sample sheet, um arquivo que armazena as informações necessárias para a configuração, desempenho e análise da corrida do sequenciamento, além de indicar qual amostra possui um respectivo index, a configuração do sequenciador e o início da corrida foram realizados de acordo com as instruções do fabricante de acordo com o manual Illumina® MiSeq® System User Guide Rev F (Illumina®, San Diego, CA, EUA) para o kit TruSeq® Custom Enrichment 2x150pb.
8 9 10 11 12
5.6 ANÁLISE DE DADOS
Foi utilizado o software NextGENe® (Softgenetics®, State College, PA, EUA) para alinhamento, detecção de SNP/Indel, cobertura e anotação de variações encontradas nos bancos de dados dbSNP e dbNSFP, este último compila predições de seis algoritmos (SIFT, Polyphen2, LRT, MutationTaster, MutationAssessor e FATHMM), três algoritmos de conservação (PhyloP, GERP++ e SiPhy) além de informações que incluem frequência alélica observado nos dados dos projetos 1000 Genomes Project phase 1 e NHLBI Exome Sequencing Project (LIU, JIAN; BOERWINKLE, 2011; 2013).
usando modelos de alinhamento de sequencias homólogas e domínios conservados de proteínas (SHIHAB et al., 2013).
O PhyloP é uma ferramenta que utiliza o p-value no cálculo da conservação evolucionária de sequências através de árvores filogenéticas (SIEPEL, POLLARD; HAUSSLER, 2006; LIU, JIAN; BOERWINKLE, 2011). O algoritmo GERP++ utiliza sítios conservados pela evolução que mostram menos substituições que o esperado e então agrega esses sítios em sequencias maiores e potencialmente funcionais (DAVYDOV et al., 2010). E a ferramenta SiPhy implementa rigorosos testes estatísticos para detectar padrões de substituições improváveis (GARBER et al., 2009).
Para o alinhamento das sequencias no genoma referência, o software utiliza uma modificação da metodologia Burrows-Wheeler transform (BWT), um algoritmo utilizado no alinhamento de grandes volumes de sequências. O alinhamento foi realizado utilizando a versão hg 19, Genome Reference Consortium GRCh37.p10 do genoma de referência humano e a anotação de variações utilizando a versão 135 do dbSNP e a versão 2.0 do dbNSFP.
Primeiramente foi feita a análise dos dados do alinhamento das sequências ao genoma referência, a filtragem do alinhamento para as regiões de interesse (ROI, regions of interest) do painel de éxons construído, a porcentagem de sequências ‘on target’ e a cobertura média de todas as amostras.
A próxima parte da análise envolveu a procura de mutações (SNP/Indel) nos pacientes (Figura 7). Foram selecionados os bancos de dados dbSNP e dbNSFP para identificar as alterações encontradas no alinhamento das sequências, utilizou-se o filtro para exibir somente mutações de utilizou-sentido trocado e utilizou-sem utilizou-sentido, além de inserções e deleções. Então, de acordo com as predições dos algoritmos, em uma primeira filtragem, foram retiradas todas as variações em que todos os algoritmos as classificassem em benignas, toleradas, polimorfismo ou neutras além das mutações em comum entre os pacientes com diagnósticos distintos.
resultados negativos, todas as variações filtradas foram analisadas no HGMD e OMIM para comparação fenotípica.
Então os resultados foram discutidos com um corpo médico e o diagnóstico molecular foi definido nos pacientes em que as mutações candidatas coincidiram com sua patologia. Nos casos em que não foi possível finalizar o diagnóstico molecular, os pacientes deverão ser reavaliados e os dados moleculares com as mutações candidatas serão reanalisados.
Figura 7: Fluxograma de análise dos dados moleculares obtidos pelo sequenciamento de exoma
5.7 CONFIRMAÇÃO DAS MUTAÇÕES
As mutações encontradas no sequenciamento de exoma parcial pela plataforma de sequenciamento de alto desempenho foram confirmadas através da metodologia de sequenciamento de Sanger por eletroforese de capilar. Além de utilização do DNA das amostras, foram utilizadas as amostras de seus respectivos familiares para análise de herança genética e DNA de uma amostra controle negativo para mutações esqueléticas. Para as mutações ainda não reportadas na literatura, foi feita a análise da frequência alélica em um grupo de 160 cromossomos provindos de 80 indivíduos normais da população Brasileira.
Foi feita uma PCR que amplificou a região genômica que se apresentou mutada no sequenciamento de alto desempenho utilizando iniciadores fabricados pela IDT® (Integrated DNA Technologies®, Coralville, IA, EUA). Foi utilizando 20ng no DNA genômico, a enzima DNA polimerase Platinum® Taq DNA Polymerase (Invitrogen™, Carlsbad, CA, EUA) e as concentrações iniciais e finais de cada reagente para reação de PCR estão listadas na Tabela 2. Para a amplificação, foram utilizados termocicladores Applied Biosystems® Veriti™ Thermal Cycler (Applied Biosystems®, Foster City, CA, EUA). As temperaturas de anelamento de cada iniciador foram ajustadas de acordo com cada par de iniciadores e as reações tiveram um total de 35 ciclos (Figura 8).
Tabela 2: Reagentes para PCR, concentrações e quantidade por reação.
Reagentes Concentração Inicial Concentração Final Quantidade*
Platinum® Taq DNA Polymerase 5 U/μL 1,5 U/μL 0,3 μL
PCR Buffer 10X 1x 1 μL
Desoxinucleotídeos (dNTPs) 2,5 mM 0,2 mM 0,8 μL
Iniciador Direto 10 μM 0,25 μM 0,25 μL
Iniciador Reverso 10 μM 0,25 μM 0,25 μL
MgCl2 50mM 1,5 mM 0,3 μL
DNA - - 20ng
Água Milli-Q (Millipore®)
autoclavada - - q.s.p. 10 μL
Figura 8: Programação do termociclador para as PCRs.
Em vermelho, a temperatura de desnaturação do DNA (95ºC), em verde está representada as variações das temperaturas de anelamento que foram testadas (entre 49-62ºC) e em azul a temperatura que ocorre a extensão (72ºC). Em amarelo estão indicados os tempos de cada estágio. Após as PCRs, as amostras foram estocadas a 10ºC.
Após a amplificação das regiões genômicas foi utilizada a eletroforese de DNA em gel de agarose (0,8%) para visualização das mesmas (SAMBROOK; RUSSELL, 2001). O produto da PCR após analisado por eletroforese foi purificado para evitar qualquer interferência durante o sequenciamento de Sanger, através para remoção de iniciadores e dNTPs não incorporados aos fragmentos durante a PCR. Foi utilizado o kit enzimático Illustra™ ExoStar™ 1-Step (GE® Healthcare, Fairfield, CT, EUA), o qual se utilizou 2,5μL do produto da PCR e 1μL de Illustra™ ExoStar™ para cada amostra, a reação foi incubada a 37°C por 60 minutos e depois a 80°C por 20 minutos para inativação das enzimas.
Após a purificação utilizou-se 1 μL do produto de PCR purificado, 1 μL de
iniciador a 3,3 µM, 3 μL de BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems®, Foster City, CA, EUA) e 5 μL de água Milli-Q (Millipore®, Billerica, MA, EUA) para sequenciamento pelo método de Sanger utilizando-se o sequenciador automático de DNA ABI PRISM® 3130 Genetic Analyzer (Applied Biosystems®, Foster City, CA, EUA).
6 RESULTADOS
6.1 PACIENTES
Após a seleção das amostras, obteve-se um grupo heterogêneo com diferentes diagnósticos (Apêndice A), incluindo hipóteses diagnósticas precisas e inespecíficas.
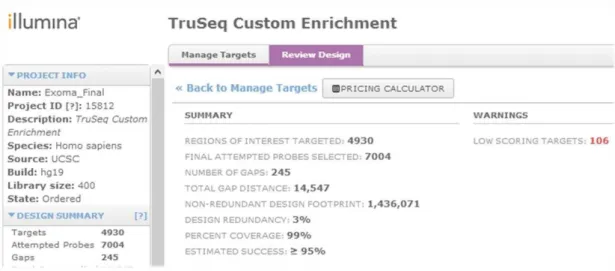
6.2 PAINEL DE GENES E SONDAS CUSTOMIZADAS
Após a seleção dos genes e regiões de interesse foram gerados na plataforma DesignStudio da Illumina® 7004 sondas de captura em 4930 das regiões genômicas de interesse (éxons e regiões de SNPs), com uma porcentagem de
cobertura de 99% dessas regiões e estimativa de sucesso ≥ 95% e com um tamanho de exoma parcial de aproximadamente 1,4Mb (Figura 9).
6.3 PREPARAÇÃO DAS BIBLIOTECAS PARA SEQUENCIAMENTO NA PLATAFORMA DE ALTO DESEMPENHO
6.3.1 Fragmentação do DNA genômico
A fragmentação foi realizada com sucesso pelas metodologias de nebulização e ultrassonicação, que resultaram na variação desejada dos tamanhos dos fragmentos entre 300 e 1000pb. No entanto, a primeira mostrou-se menos eficiente devido à grande perda de amostra (entre 50-90%) durante o procedimento, sendo necessária uma quantidade maior de DNA genômico para obter-se a quantidade necessária de DNA fragmentado para a preparação das bibliotecas
(1μg).
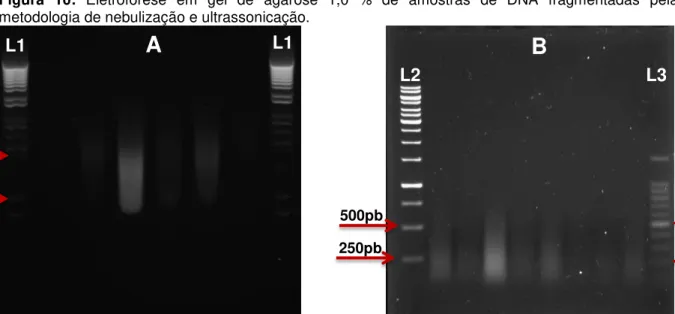
Já a metodologia de ultrassonicação mostrou-se mais eficiente, além do procedimento mais simples e rápido que a nebulização, não há perda de amostra durante o procedimento, além de obter-se uma variação menor no tamanho dos fragmentos (Figura 10).
Figura 10: Eletroforese em gel de agarose 1,0 % de amostras de DNA fragmentadas pela
metodologia de nebulização e ultrassonicação.
L1 – Marcador ladder 1 kb plus (Invitrogen®), L2 – Marcador ladder 1 kb (Invitrogen®), L3- Marcador 100pb (Invitrogen®). (A) Teste de nebulização das 12 primeiras bibliotecas com fragmentos com tamanhos entre 200pb e 1000pb e (B) teste de ultrassonicação das 84 últimas bibliotecas com fragmentos com tamanhos entre 200pb e 600pb.
L1 L1
L2 L3
200pb
250pb 300pb
500pb
500pb 500pb
6.3.2 Preparação da biblioteca de DNA
Após a fragmentação do DNA das amostras, foi realizada a preparação da biblioteca de fragmentos. Bibliotecas de três amostras, Rede 85, Rede 99 e Rede 131, foram feitas em duplicata para validação por replicabilidade da metodologia.
Foi adicionado aos fragmentos de cada amostra uma sequência identificadora específica (index), necessária para a identificação de cada amostra durante o sequenciamento, que é numerada pela fabricante e foi escolhida aleatoriamente para cada amostra. Após a preparação das bibliotecas, estas foram quantificadas (Apêndice C) para a realização da captura e enriquecimento.
Após a quantificação, observou-se que 28 bibliotecas tiveram uma concentração menor que 20ng/µl, abaixo do recomendado pela fabricante para a etapa de captura e enriquecimento dos fragmentos. Isto ocorreu devido a impurezas e/ou baixa qualidade do DNA genômico dessas amostras. Portanto a captura foi feita com o máximo de material genômico obtido após a preparação das bibliotecas.
6.3.3 Captura e enriquecimento dos fragmentos
Para cada grupo de amostras, as instruções da fabricante indicam que sejam adicionados 500ng de DNA de cada uma das doze bibliotecas genômicas
(totalizando 6μg de DNA) em cada grupo de amostras, formando uma biblioteca de sequenciamento.
amostram não são garantidas. Definiu-se que essas amostras seriam sequenciadas e analisadas se possível.
Foram formados os oito grupos de doze amostras, a captura e enriquecimento dos fragmentos foram realizados com sucesso e todas as bibliotecas de sequenciamento foram validadas.
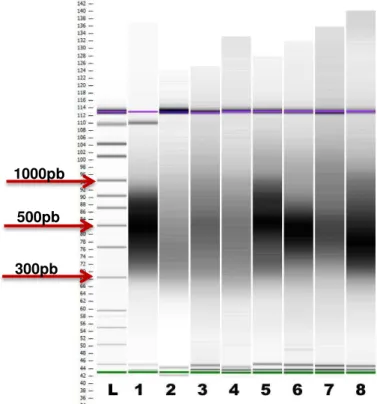
A variação de tamanho dos fragmentos de cada biblioteca de sequenciamento foi satisfatória, entre 300 e 1000pb (Figuras 11 e 12).
Figura 11: Variação do tamanho dos fragmentos observado no gel do Bioanalyser.
As oito bibliotecas de sequenciamento possuem uma variação de tamanhos de fragmentos entre 300 e 1000pb, indicados pelas setas.
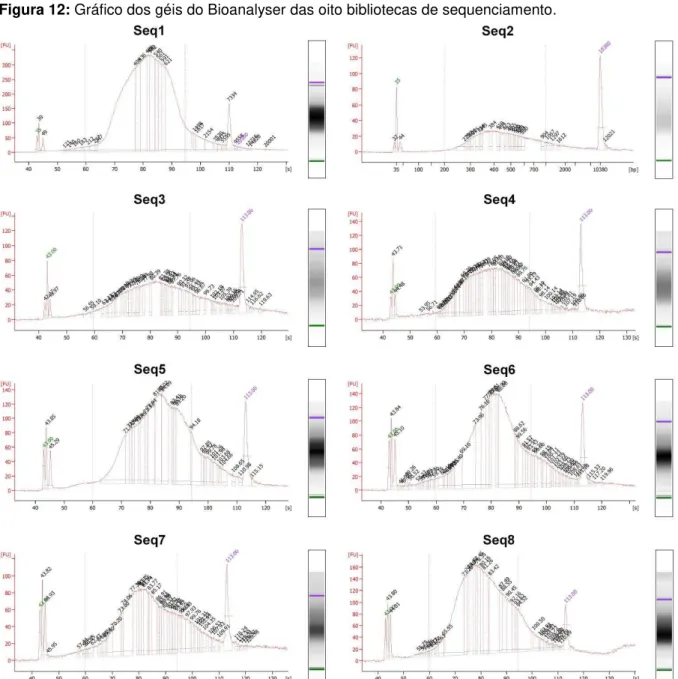
Para a quantificação das bibliotecas de sequenciamento (Tabela 3), utilizaram-se os dados do tamanho médio dos fragmentos de cada biblioteca obtidos pelo Bioanalyser (Figura 12).
Figura 12: Gráfico dos géis do Bioanalyser das oito bibliotecas de sequenciamento.
O pico central indica o tamanho médio dos fragmentos: Seq1, 500pb; Seq2, 384pb; Seq3, 485pb; Seq4, 470pb; Seq5, 535pb; Seq6, 490pb; Seq7, 488pb; e Seq8, 412pb.
Tabela 3: Quantificação das bibliotecas de sequenciamento e tamanho médio dos fragmentos.
Grupo de amostras Quantificação das amostras (nM) Tamanho médio dos fragmentos (pb)
Seq1 35,69 500
Seq2 19,52 384
Seq3 5,71 485
Seq4 13,99 470
Seq5 8,98 535
Seq6 6,41 490
Seq7 4,90 488
Seq8 15,38 412
A metodologia de fluorometria utilizada pelo Qubit, por ser menos sensível, superestima o resultado da quantificação, pois mesmo fragmentos sem adaptadores são quantificados, podendo gerar menos clusters na flow-cell durante o sequenciamento.
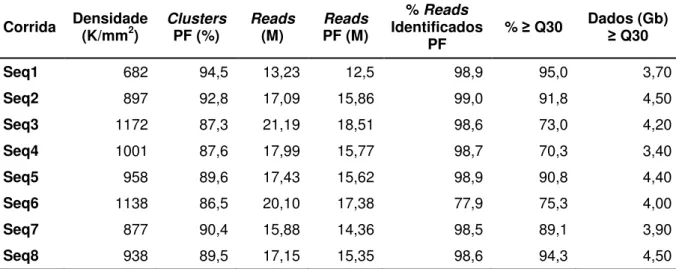
6.3.4 Sequenciamento de DNA na plataforma de alto desempenho
O desempenho ideal esperado do sequenciamento com o kit de 2x150pb (v2) nessa plataforma é de aproximadamente 4,4Gb, 15 milhões de reads gerados e 1 milhão de clusters por mm2 na flow-cell. Para alcançar esse desempenho, utiliza-se a concentração final da biblioteca de sequenciamento indicada pela fabricante de 10 a 12pM.