Article
J. Braz. Chem. Soc., Vol. 26, No. 3, 411-419, 2015. Printed in Brazil - ©2015 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
A
*e-mail: [email protected]
Non-Thermal Plasma Induced Total Mineralization of Glyphosate in Water in the
Presence of Iron II Ions
Moïse Fouodjouo,a,b Samuel Laminsi,a Georges Y. Kamgang,a Michele T. Menguea,c and
Nito A. Debacher*,b
aMineral Chemistry Laboratory, Department of Inorganic Chemistry,
University of Yaoundé 1, P.O. Box 812, Yaoundé, Cameroon
bChemistry Department, Federal University of Santa Catarina (UFSC),
88040-900 Florianopolis-SC, Brazil
cDépartement des sciences du bois, Université Laval (UL) 2405,
rue de la Terrasse, Quebec City, Canada
The present work focused on the mineralization of herbicide glyphosate (N-(phosphonomethyl)
glycine) (C3H8NO5P) in aqueous phase by Glidarc plasma following the orthophosphates released
and the reduction of the total organic carbon (TOC). Furthermore, the effect of initial concentration, pH and the degradation process of glyphosate in the presence of Fe2+ were also examined. The results
showed that the efficiency of degradation increased with the presence of ferrous ions as catalysts in aqueous medium and also the release of orthophosphate is more efficient in acid medium with pH range of 3.5-6.3. The kinetics of the release of orthophosphate and the TOC removal obey the first order rate law. The ninhydrin and orthophosphates tests showed that the degradation of glyphosate in water by Glidarc plasma was mainly by ●OH and NO● attacked with C–N and C–P
bond cleavage leading to complete mineralization.
Keywords: glyphosate, Glidarc discharge, orthophosphate release, mineralization
Introduction
Glyphosate also called N-(phosphonomethyl) glycine
(NPG) of chemical formula C3H8NO5P is a nonselective
systemic broad spectrum herbicide used for weed control. Glyphosate is absorbed by the leaves and carried by the phloem till the extremities of roots and rhizomes, where it inhibits the biosynthesis of aromatic amino acids by bonding to 5-enolpyruvylshikimate-3-phosphate syntase enzyme.1 This herbicide was extensively used in European
and American agricultural exploitations in the early 1970s. Faced with the problem of famine that characterizes most third-world countries, particularly African countries, the use of this herbicide, based on its ability of fertilizing and destroying annual and perennial weeds was intensified and widespread with leitmotiv the food safety for a growing population. High solubility of glyphosate in water1 and
the fact that less than 1% often reaches the target makes that, a component of herbicide spills unto surface waters. It thus becomes a potential contaminant with harmful
effects on the environment.2 In water, glyphosate is rapidly
dissipated through adsorption on suspended matters and sediments, and has a half-life of 19 to 45 days for the whole system.3 Although recognized to be non toxic for mammals,
human beings and birds,4 Bolognesis et al.5 showed that
glyphosate produces positive genotoxic effects in vitro in
human peripheral blood, evidenced by sister chromatids exchange and in vivo in mouse. Similarly in their previous
works, Clements et al.6 reported positive DNA damage
for comet assay in erythrocytes of Rana castesbeiana
tadpoles following exposure to glyphosate. In the same vein, Poletta et al.7 and Manas et al.8 respectively revealed DNA
damage after performing comet assay and micronucleus test on the erythrocytes of broad-snouted Caiman latirostris
and fetal bovin serum exposed to glyphosate. In spite of its hydrolytic decomposition with a greater than 35 days mean half-life at various pH levels and temperatures,9
this herbicide is classified among the organic molecules frequently detected in streams and rivers, and generally exceeds the European standard for drinking (1 µg L−1) and
surface water (1-3 µg L−1).10 Although being very sensitive
in water and soil yields more toxic intermediates,3 i.e.,
aminomethylphosphonic acid (AMPA) and sarcosine, which could further be degraded slowly into water, phosphate and carbon dioxide. Within the purview of the relevance of this ecological problem, the control and elimination of glyphosate and other organic pollutants becomes imperious for environmental safety.
Previous studies reported the glyphosate degradation using processes such as hydrogen peroxide-based advanced oxidation processes (H2O2/UV, H2O2/O3/UV, H2O2/Cl−,
Fenton, photo-Fenton, Fe(III)/Na2C2O4/UV, FeOH/UV,
H2O2/UVC),11-13 titanium dioxide-based advanced oxidation
processes (TiO2/UV)14,15 and manganese oxide in aqueous
suspension.16 Efficiencies obtained with these processes are
relative and the costs of set up often expensive. Non-thermal plasma (NTP), an environmental friendly technique, can be a good alternative for organic pollutants abatement in water. For some time in the past, NTP has shown its proofs to be a promising technology for organic pollutants abatement in gaseous, liquid, and solid phases.17 Environmental
applications of electric discharges are being considered increasingly; they imply the chemical properties of the activated species generated by the discharge.18 There are
many types of non-thermal plasmas,19 but in this work, we
focused on gliding arc discharge commonly called Glidarc. The Glidarc device is an inexpensive source of NTP, which operates at conditions close to atmospheric pressure and room temperature. The analysis with spectroscopy emission has identified ●OH and NO● radicals as responsible of
oxidative properties of Glidarc plasma plume when humid air is used as feeding gas.20 These radicals are precursors
of other active species, which endow the reaction medium a very high enhanced chemical reactivity.19 The chemical
reactivity of Glidarc has been utilized with success in both the degradation of several dissolved organic compounds21,22
and industrial effluents.23
The present work focused on the study of the mechanism of mineralization of glyphosate in aqueous phase by Glidarc plasma. Mineralization via the orthophosphates released and the total organic carbon (TOC) was studied. Furthermore, the effect of initial concentration, initial pH and the degradation process of glyphosate in the presence of Fe2+ were also
examined. The kinetics was studied and a plausible degradation pathway of glyphosate in water by NTP was proposed.
Experimental
Materials and reagents
Glyphosate (Figure 1) 98.5% was purchased from Dr Ehrenstorfer Gmbh (Bgm Schlosser-Str., Augsburg,
Germany). Aqueous solutions of glyphosate were prepared by dissolving 6.0 mg of highly soluble glyphosate salt in distilled water. All other reagents were of analytical grade and were used without purification. Molar solutions of NaOH and H2SO4 were used to adjust the pH values to study
the influence of initial pH on glyphosate degradation by humid air plasma. 4.0 mol L−1 acetic acid-sodium acetate
buffer (HAc-NaAC; pH 5.5) was prepared for ninhydrin method,24 which involves the reaction of alpha-amino acid
and ninhydrin resulting in a colored solution absorbing in 570 nm.
Apparatus
The gliding arc device used for this study was described previously.17 It derived from the original device
described by Czernichowski et al.17 Figure 2 presents
a scheme of the experimental device. When a suitable voltage (5-10 kV) and electric current (160 mA)25 are
applied between two aluminum divergent electrodes, an electric arc forms at the shortest electrode gap (3 mm) and glides along the electrode’s axis pushed on by a gas flow (humid air). The arc length increases on moving and its temperature decreases, so that the arc turns from thermal plasma to quenched plasma on breaking into a plume. A new arc then forms at the narrowest gap and the cycle resumes as a large plasma plume. The plasma plume leaks the liquid target brought to its contact at overall room temperature and pressure, close to atmospheric conditions.17 The electric energy is transferred to gas
molecules, which become excited and are broken into radicals giving highly reactive species such as ●OH and
NO● according to the following equations.18
H2O + e–→H• + OH• + e– (1) O2 + e–→O(3P) + O(1D) + e– (2) N2 + e–→N(4S) + N(2D) + e– (3)
N(2D) + O2→NO• + O (4) H• + O
2→HO•2 (5)
HO•
2 + NO•→NO2 + OH• (6)
The diffusion process in the liquid is improved by convection in the liquid phase due to the airflow and magnetic stirring. Treatments were carried out in a closed batch reactor for different plasma-exposure time. The reactor is equipped with a cooling system to avoid
HO N
O
P O
OH OH H
evaporation and the temperature was kept constant at 24 ± 0.1 °C for all treatments.
Analytical methods
The pH adjustment was followed using a multifunctional digital pH-meter branded HANNA. FeSO4 was added to
the solution as a ferrous ions (Fe2+) catalyst source. The
monitoring of reduction of glyphosate concentration in solution after Glidarc plasma treatment, proceeded as follows: glyphosate solution (1 mL) and ninhydrin solution (1 mL) were put in screw-capped test tubes and heated in a boiling water bath for a pre-determined period of time. After heating, the tubes were immediately cooled in an ice-bath, then; 5 mL of 50% ethanol was added into each tube and thoroughly mixed with a Vortex mixer for 30 s. The residual glyphosate was followed by spectrophotometry at 570 nm. The reduction product obtained from ninhydrin then reacts with NH3 and excess ninhydrin to yield a blue colored
substance.24 Orthophosphates (PO
4-P) formed as result
of degradation of glyphosate were measured according to the standard methods with a spectrophotometer at 700 nm, based on the formation of a blue molybdenum complex and after digestion using peroxodisulfate (K2S2O8), respectively.26 All experiments (manipulations
and analysis) were duplicated. Glyphosate degradation was also evaluated by analyzing the TOC content of the samples using a Shimadzu TOC-VE analyzer equipped with a non selective infrared detector. Calibration was done before,
and destruction of the parent pesticide (glyphosate) was then confirmed by measuring its TOC in both untreated and treated samples by comparison with pure glyphosate standards.
Calculations
The release of orthophosphates in the solution [%(PO4-P)] and the TOC abatement rate were respectively
calculated using equations 7 and 8.
4
[PO – P] – [PO – P]4 t 4 0
%(PO – P) = 100
[PO – P]4 tot
× (7)
0
0
( )
100
t
TOC TOC
TOC
η= − × (8)
%(PO4-P) is the orthophosphates release efficiency, [PO4-P]t in mg L−1 the orthophosphates concentration at
treatment time t, [PO4-P]0 the residual concentration of orthophosphate found before treatment27 and [PO
4-P]tot the
total concentration of orthophosphate releasable calculated considering that each molecule of glyphosate releases one molecule of orthosphosphate. η is the TOC reduction
percentage, TOC0 the initial total organic carbon and TOCt
the TOC at treatment time, t.
Results
The study of the degradation of glyphosate by Glidarc humid air plasma was followed by measuring the orthophosphates released and the TOC values as a function of exposure time together with ninhydrin test to highlight the breaking of C–N bond.
NPG + OH•
→oxidation products (9)
The working parameters fixed were: the distance between the ends of the electrodes and the liquid surface (1.5 cm), the lengths of the electrodes (6.0 cm), the nozzle diameter by which the plasmogen gas flows (1 mm) and the gas flow rate (800.0 L h−1).
For the study, the pH of the pristine solution was 6.3 and the initial concentration of glyphosate 33.0 µmol L−1. The
TOC found was 215.0 mg L−1 and the residual concentration
of orthophosphates 0.097 mg L−1.
Glyphosate degradation
Figure 3 shows the visible spectrum of glyphosate degradation after ninhydrin colorimetric assay. From d
L
E
P
H
D
Magnetic stirrer Humid air
I
Target solution W
A
Figure 2. Experimental setup of Glidarc plasma. (A = air entrance;
this figure, it is clear that, just after 5 min of exposure, the intensity of the signal decreases substantially and disappears after 30 min of treatment.
Release of orthophosphates by cleavage of C–P bond
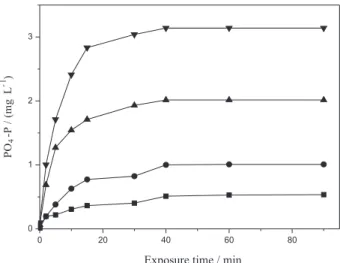
Figure 4 shows that glyphosate exposed to Glidarc plasma released orthophosphates (PO4-P). It also shows the
variations of orthophosphates formed for various exposure times. Orthophosphate release is continuous and increases with exposure time. The curve leveled off after 30 min of treatment.
Kinetics of orthophosphate released
From Figure 4, it can be observed that, the kinetics of release of orthophosphates obey the first-order rate law
with a rate constant of 7.83 × 10−2 min−1.The interval of
time (0 ≤ t < 30 min) is the most significant and relevant because 93.72% of orthophosphates were released within this period. After 30 min, a landing is observed showing the decrease in the degradation rate of glyphosate (Figure 4).
Influence of initial pH on the release of orthophosphates
Figure 5 shows the plasma degradation of glyphosate with formation of orthophosphates in the range of pH 3.5-11.0. Glyphosate can be efficiently transformed into orthophosphates at pH 3.5-6.3. It also shows that the degradation was slow for the higher values of pH (pH > 6). Initial pH values governed the degradation of glyphosate by plasma.
Effect of initial concentrations on the release of orthophosphates
As shown in Figure 6, the efficiency of orthophosphate released increases with the increase in the initial concentrations. For example, when the initial concentration of glyphosate is 33.0 µmol L−1 and pH 3.5, 93.72%
of orthophosphates was produced after 30 min of exposure, whereas under the same conditions with an initial concentration of 5.5 µmol L−1, only 75.78% of
orthophosphates was released. It is observed that as for higher initial concentration, higher degradation occurred.
Effect of Fe2+ catalysis
Figure 7 shows that the formation of PO4-P increased with the presence of ferrous ions acting as catalyst in the aqueous medium. Addition of Fe2+ 0.1 mmol L−1 permitted
to obtain 90.60% of orthophosphate release after 15 min
Figure 3. The visible absorbance spectra of glyphosate after ninhydrin
colorimetric test. (a) 0 min; (b) 5 min; (c) 10 min; (d) 15 min; (e) 30 min. [NPG]0 = 33.0 µmol L−1, pH
0 6.3.
0 20 40 60 80
0 1 2 3 4
Exposure time / min
I II
P
O4
-P
/
(m
g
L
-1 )
Figure 4. Profile of orthophosphates formation against exposure
time during plasma treatment under the following conditions (initial concentration [NPG]0 = 33.0 µmol L−1, pH 6.3). Zone I is characterized by first-order in relation to substrate. Zone II the orthophosphate production rate decreases and tends to zero.
P
O4
-P
/
(m
g
L
-1)
0 20 40 60 80
0 1 2 3
Exposure time / min
Figure 5. Effect of initial pH ( 3.5; 5.0; 6.3; 7.5; 9.0; 11.0)
of plasma exposure compared to 60.61% obtained for the plasma treatment alone. It is also noticed that an increase in the concentration of ferrous ions increased the formation of PO4-P. With an initial concentration
[NPG]0 = 33.0 µmol L−1 and an pH 6.3, the formation
of orthophosphate was fast in the presence of Fe2+ and
equilibrium was attained after 40 min of exposure. However, it is observed that excessive addition of catalyst acts negatively on the glyphosate degradation: the uses of 0.2 mmol L−1 ferrous ions diminished the efficacy of
PO4-P release from 90.60% to 80.44% after 15 min of exposure to Glidarc plasma.
TOC content
Figure 8 shows that glyphosate exposed to plasma can be mineralized into CO2. The TOC concentration decreased
with time and an abatement of 68.8% was reached after 90 min of treatment in the absence of iron II ions.
The degradation was slow between 0-10 min. Figure 8 also shows the plot of ln(TOC) versus exposure time and
suggests that the kinetics of TOC reduction is first-order with a kinetic constant k = 1.39 × 10−2 min−1.
Figure 9 shows the combined effect of plasma and ferrous ions on glyphosate degradation. The mineralization of glyphosate is more efficient when ferrous ions are added to the target solution. Table 1 gives the rate constants for each concentration of ferrous ions added to the medium.
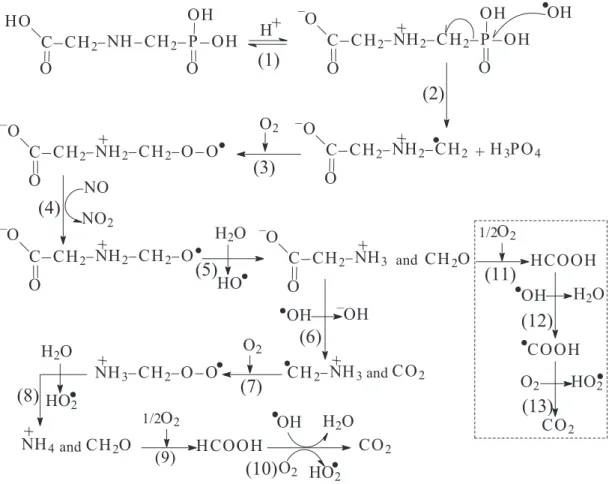
Glyphosate degradation pathway
As mentioned previously, glyphosate can be degraded by Glidarc plasma via C–N and C–P bond cleavages
P
O4
-P
/
(m
g
L
-1 )
0 20 40 60 80
0 1 2 3
Exposure time / min
Figure 6. Effect of initial concentrations of glyphosate: 5.5; 11.0;
22.0; 33.0 µmol L−1 on the release of orthophosphates at initial pH 3.5.
0 20 40 60 80
0 1 2 3
Exposure time / min
P
O4
-P
/
(m
g
L
-1 )
Figure 7. Influence of ferrous ions concentration: 0.0; 0.02;
0.05; 0.1; 0.2 mmol L−1 on the catalysis of PO
4-P released ([NPG]0 = 33.0 µmol L−1, pH 3.5).
0 20 40 60 80 100 50
100 150 200
0 20 40 60 80 100
4,0 4,5 5,0 5,5
ln(T
OC)
Exposure time / min
4.0 4.5 5.0 5.5
T
O
C
/
(m
g
L
-1 )
Exposure time / min
Figure 8. Plot of total organic compound (TOC) against exposure time.
The insert shows a linear correlation between ln(TOC) and exposure time which fit the first order rate law, (k = 1.39 × 10−2 min−1).
0 20 40 60 80
0 20 40 60 80 100
A
b
a
te
m
e
n
t
ra
te
/
%
Exposure time / min
Figure 9. Influence of ferrous ions concentration: 0.0; 0.02;
leading to mineralization (Figure 10). The ninhydrin method based on the formation of the colored Ruhemann purple compound of primary amine showed that glyphosate possibly underwent C–N bonds breakage. The C–P bond cleavage also occurred and was evidenced by the formation of orthophosphates and TOC removal.
Discussion
The above results showed that glyphosate is effectively degraded by humid air plasma. The formation of orthophosphates and decrease in the TOC concentration are proofs of the degradation of this compound and can
be considered as the results of the interaction between the strong oxidizing agents present in humid air plasma and the solute at the plasma-liquid interface. Previous work suggested that ●OH and NO● were the main activated
species present in humid air plasma plume.21●OH is the
most powerful oxidizing agent and, as other radicals, it has a very short half-life (1 ms).28 Therefore, the entire ●OH formed by conventional electron impact dissociation
of water molecules could not, during that short time, react with target molecules. Consequently, part of radicals, which did not react because of the dilution of the target, combine together to form H2O2 in aqueous phase. This was evidenced and measured by previous works.29,30 Since ●OH
is a stronger oxidizing agent after fluorine with a standard potential (●OH/H
2O) = 2.72 V,31 it likely participated in
the plasma-chemical degradation processes of organic compounds. Apart from ●OH and H
2O2, some other
species such as HO2●, ONOO−, O
3 and O,30,32 are formed
and are also thermodynamically able to oxidize organic compounds. Previous investigation on the degradation of glyphosate also showed that the mechanism path involves many radical steps.13
Figure 4 shows that the glyphosate can be attacked by reactive species (●OHand NO●) of Glidarc humid air
Table 1. Rate constants of TOC removal
[Fe2+] / (mmol L−1)
Rate constant k / min−1
Correlation coefficient R2
Standard error on rate constant 0.00 1.39 × 10−2 0.989 9.96 × 10−4 0.02 5.98 × 10−2 0.995 5.22 × 10−4 0.05 6.26 × 10−2 0.970 0.11 × 10−4 0.10 7.14 × 10−2 0.987 9.17 × 10−4 0.20 6.51 × 10−2 0.963 0.14 × 10−4
Figure 10. A proposed pathway of glyphosate degradation by Glidarc plasma.
C
HO
O
C H
2NH CH
2P
OH
OH
O
C
O
O
C H
2NH
2C H
2P
OH
OH
O
OH
–
– –
– –
–
C
O
O
C H
2NH
2C H
2O O
O
2C
O
O
C H
2NH
2C H
2+
H
3PO
4NO
NO
2C
O
O
C H
2NH
2C H
2O
H
2O
HO
C
O
O
CH
2NH
3CH
2NH
3C O
2NH
3CH
2O O
O
2OH
OH
C H
2O
NH
4O
21/2
H COO H
H
2O
OH
O
2HO
2CO
2and
H
2O
HO
2and
H
2O
OH
CO OH
O
2HO
2CO
2H
(1)
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
and
CH
2O
O
21/2
HCOOH
plasma leading to the formation of PO43−. This formation
can be explained by the breakage of C–P and O–H bonds in the phosphonate group of the substrate. The abstraction of hydrogen follows radical mechanism and radical ●H formed
is ionized and contributed to acidify the aqueous medium (Figure 10). First order kinetic model is in agreement with the experimental data of the orthophosphates released. The rate-limiting factors are the concentrations of the plasma active species (depends on the energy provided to the discharge) at the reaction zone and the concentration of the substrate.33,34
The level-off after 30 min corresponds to a drastic decrease in the effective contact between the plasma and the glyphosate, when the volume of organic phase becomes too low to cover the entire liquid surface and thus to maintain an efficient interface. The orthophosphate production rate decreases and tends to zero. Kinetics of formation of orthophosphates in each time interval (Figure 4) showed that the mechanism of their formation by degradation of glyphosate is a multi-step with overall first order.33
The initial pHand the initial concentration of glyphosate also influenced the rate of formation of orthophosphates by degradation by humid air plasma. As shown in Figure 5, the degradation of glyphosate is more efficient for low values of initial pH. Glyphosate is an amphoteric compound; the basic form of glyphosate quickly undergoes degradation by plasma than the acid form (Figure 10). This pH dependence of glyphosate degradation can also be attributed to the speciation of ●OH and NO● radical, since there are more
efficient in acidic medium.29 Oxidising degradation reactions
of organic compounds often involve protons; thus, the kinetic rate very often depends on the acidity of the medium. Furthermore, the pH values govern the relative amount of
●O
2−/HO2●, which is also precursor of H2O2 formation and
leads to more ●OH. Besides previous works using plasma
for degradation of organic compounds,35,36 reported that,
the increasing of initial pH decreases the rate constant. This observation is attributed to the fact that ●OH, HO
2 ● and H
2O2
have higher oxidation effect in acidic medium.37
The initial concentration of target molecules plays an important role in the treatment of waste water by Glidarc discharge since the final application is in the industries where the effluents are relatively concentrated. It comes out from Figure 6 that the orthophosphate release is more efficient for concentrated solutions. This is due to the fact that with concentrated solutions, the probability of collision between primary (●OH, NO●) and secondary
(H2O2, NO2) active species and molecules of glyphosate
at the reaction interface is more important.35 The effective
surface between the plasma and glyphosate is low for dilute solutions33 and the rate of degradation is earlier governs
by diffusion process comparatively to more concentrated
solutions that the reaction rate is for a relative long time controlled by ●OH.38 It could therefore be suggested that
higher rate of degradation occurred for higher initial concentrations contrary to results obtained in previous works.39 However, after 90 min of exposure, almost the
same rate of orthophosphates is produced both for dilute and concentrated solutions of glyphosate.
When glyphosate solution is mixed with ferrous ions and exposed to Glidarc plasma flame, the rate of degradation increases due to H2O2 consumption by Fe2+ to form ●OH
radicals leading to increase the rate of degradation of glyphosate.40 The Gliding arc plasma in humid air/Fenton
mechanism that accompanies the above suggestions is:
H• + O
2→HO•2 (10)
HO•
2 + HO•2→H2O2 + O2 (11)
HO•
2 + NO•→NO2 + OH• (12)
OH• + OH•
→H2O2 (13)
H2O2 + Fe2+→Fe3+ + OH• + OH– (14) Fe2+ + OH•
→Fe3+ + OH– (15)
RH + OH•→R• + H
2O→products (16)
H2O2 + OH•
→H2O + HO•
2 (17)
Fe3+ + HO•
2→Fe2+ + HO2+ (18)
Although Abdelmalek et al.40 in their previous work
found that the catalytic activity of ferrous ions is probably negligible in humid air plasma because of eventual oxidation by nitrites, which parasites their reaction with hydrogen peroxide to form ●OH, our study showed that the catalytic
effect of Fe2+ was not negligible in the case of degradation of
glyphosate. This may be due to the presence in the medium of PO43− ions, which can probably hinder the reaction of
nitrites with ferrous ions by electron acceptor limitation. An excess of catalyst reduces the mass transfer and therefore the kinetics of degradation likewise. Figure 7 showed that the production of PO43− increases with an increase Fe2+
concentration. Its proofs that ferrous ions can sufficiently utilize the residual hydrogen peroxide generated by plasma to produce a larger amount of the reactive, non-selective radicals ●OH(equation 9 to equation 18), which rapidly
mineralize glyphosate and produce orthophosphates.41
However, an excess of Fe2+ scavenged OH radicals and
slowed the degradation of glyphosate as described by nonoxidizing reaction (equation 16).41,42
species at the reaction interface.33 As we can see, the
mineralization is appreciable and goes in straight line with the TOC reduction measured in the case of degradation of other organic compound by gliding arc discharge in humid air.23,32,43,44 The kinetics of TOC reduction was
studied and showed to be first order (Figure 8). The rate of mineralization of glyphosate when exposed to Glidarc in humid air increases with the addition of ferrous ions in the buffer aqueous medium (Figure 9) due to the increase of ●OH coming from decomposition of H
2O245-47 produced
in the discharges and evidenced by Burlica et al.29 The
TOC reduction also increases with the concentration of the catalyst. The kinetic rates of TOC reduction increased with the concentration of ferrous ions to a certain value and decreased (Table 1). This may be due to the fact that the excessive addition of ferrous ions operates negatively on glyphosate degradation. This phenomenon can be explained by the existence of parasitical reaction, which consumes ●OH radicals.46 Similar results were obtained by
Souza et al. who uses the Fenton process.48 The low rate
of mineralization (28% after 120 min of treatment) that they obtained may be due to the fact that they have been working with the commercial formulation.
Figures 3, 4 and 8 show that glyphosate is degraded by Glidarc plasma via C–N and C–P bonds breakage followed by a considerable reduction in TOC as reported in the previous works using advanced oxidized processes (AOPs).13-15,49 The cleavage of C–N and C–P bonds is
attributed to ●OH radicals. Thus, Figure 10 proposes a
probable reaction pathway of glyphosate degradation by Glidarc plasma.
As can be seen in Figure 10, the ●OH radical attacks
glyphosate (step 2), which leads to the formation of a carbon centred radical −OOC-CH
2-NH2+-CH2-O-O● and
orthophosphates.16 The generated radical can react with
molecular oxygen (step 3)50 present in the medium at high
concentration to give a new radical −OOC-CH
2-NH2+-CH2
-O-O●, which reacts directly with NO● (step 4) and then H 2O
(step 5) to form −OOC-CH
2-NH3+, and CH2O.49 Glycine
may undergo decarboxylation due to the presence of ●OH
(step 6) to give CO2 and ●CH2-NH3+ radical. ●CH2-NH3+ then
reacts with O2 and ●OH (step 7 and 8), respectively to yield
formaldehyde and NH4+. The formaldehyde generated in
the process can be directly oxidized into formic acid by the dissolved oxygen and mineralized into CO2 by combined
action of ●OH and once more oxygen.49,51
Conclusions
The aim of this work was to study the degradation of glyphosate by the gliding arc discharge. It was found that
gliding arc discharge can efficiently degrade glyphosate evidenced by the formation of orthophosphates. The efficiency of orthophosphate increases with the increase of initial concentration. The degradation is more efficient in acid medium with the best pH values in the range of 3.5-6.3. The gliding arc discharge mineralizes glyphosate evidenced by a measure of the percentage of TOC reduction. The ferrous ions can catalyze the degradation of glyphosate, thus improving the efficiency of orthophosphate released and the TOC reduction. The kinetics of the release of orthophosphate by gliding arc discharge obeyed first-order rate law with kinetic constant 7.83 × 10−2 min−1. The kinetics
of the TOC reduction was also first order with a kinetic constant 1.39 × 10−2 min−1. The degradation of glyphosate
by gliding arc discharge was mainly done by hydroxyl radical and NO● followed by the C–N and C–P cleavages.
Acknowledgments
The authors of this work thank the Twas-CNPq Doctoral programme and the Laboratory of Interfacial Electrochemistry and Analytical Chemistry (LEICA) of the University of Rouen for the donation of equipments to the Laboratory of Mineral Chemistry of the University of Yaoundé I, which immensely help to the accomplishment of a part of this work. We are also indebted to Dr Luís Otávio de Brito Benoteli for his contribution to the revision of this paper, and the International Foundation of Sciences (IFS) for the grant given to Dr Nzali Serge. The materials provided to him equally helped in some analysis of this work. Also, the authors like to thank Prof Emmanuel Ngameni for his significantly contribution to the critical review of the manuscript during the evaluations of reviewers of the JCBS.
References
1. Moses, M.; Toxicol. Ind. Health1993, 9, 913.
2. Gasnier, C.; Dumont, C.; Benachour, N.; Clair, E.; Chagnon, M. C.; Séralini, G-E.; Toxicology 2009, 262, 184.
3. Vereecken, H.; Pest Manage. Sci. 2005, 61, 1139.
4. US Environmental Protection Agency (US EPA-738-F-93-011);
Office of Prevention of Pesticides and Toxic Substances; Washington DC, 1993.
5. Bolegnesis, C.; Bonatti, S.; Degan, P.; Gallerani, E.; Peluso, M.; Rabboni, R.; Roggieri, P.; Abbondandolo, A.; J. Agric. Food Chem.1997, 45, 1957.
6. Clements, C.; Ralph, S.; Petras, M.; Environ. Mol. Mutagen. 1997, 29, 277.
8. Manas, F.; Peralta, L.; Raviolo, J.; Ovando, H. G.; Weyers, A.; Ugnia, L.; Cid, M. G.; Larripa, I.; Gorla, N.; Environ. Toxicol. Pharmacol. 2009, 28, 37.
9. Kollman, W.; Segawa, R.; Department of Pesticide Regulation;
EPA: Sacramento, 1995.
10. Botta, F.; Lavison, G.; Coututier, G.; Alliot, F.; Moreau-Guigon, E.; Fauchon, N.; Guery, B.; Chevreuil, M.; Blanchoud, H.; Chemosphere2009, 77, 133.
11. Junges, C. M.; Vidal, E. E.; Attademo, A. M.; Mariani, M. L.; Cardell, L.; Negro, A. C.; Cassan, A.; Peltzer, P. M.; Lajmanovich, R. C.; Zalazar, C. S.; J. Environ. Sci. Health, Part B 2013, 48, 163.
12. Ikehata, K.; El-Din, M. G.; J. Environ. Eng. Sci. 2006, 5, 81. 13. Chen, Y.; Wu, F.; Lin, Y.; Deng, N.; Bazhin, N.; Glebov, E.;
J. Hazard. Mater. 2007, 148, 360.
14. Echavia, G. R. M.; Matzusawa, F.; Negishi, N.; Chemosphere 2009, 76, 995.
15. Muneer, M.; Boxall, C.; Int. J. Photoenergy2008, ID 197346,
1.
16. Barret, K. A.; Mcbride, M. B.; Environ. Sci. Technol.2005, 39,
9223.
17. Czernikowski, A.; Pure Appl. Chem. 1994, 66, 1301.
18. Brisset, J-L.; Moussa, D.; Doubla, A.; Hnatiuc, E.; Hnatiuc, B.; Kamgang, G. Y.; Herry, J-M.; Naitali, M.; Bellon-Fontaine, M-N.; Ind. Eng. Chem. Res. 2008, 47, 5761.
19. Ghezzar, M. R.; Abdelmalek, F.; Belhadj, M.; Benderdouche, M.; Addou, A.; Appl. Catal., B2007, 72, 304.
20. Benstaali, B.; Boubert, P.; Cheron, B. G.; Abdou, A.; Brisset, J-L.; Plasma Chem. Plasma Process.2002, 22, 553. 21. Fouodjouo, M.; Laminsi, S.; Djepang, S. A.; Tadom, D.; Brisset,
J-L.; Int. J. Res. Chem. Environ.2013, 3, 316.
22. Laminsi, S.; Acayanka, E.; Teke-Ndifon, P.; Djowe-Tiya, A.; Brisset, J-L.; Environ. Eng. Manage. J. 2012, 11, 1821. 23. Mountapmbeme-Kouotou, P.; Laminsi, S.; Acayanka, E.;
Brisset, J-L.; Environ. Monit. Assess. 2013, 185, 5789. 24. Fisher, J. L.; Bunting, L. S.; Rosenberg, E. L.; Clin. Chem.
1963, 9, 573.
25. Hnatiuc, E.; Procédés Électriques de Mesure et de Traitement des Polluants, Lavoisier, ed., Tec&Doc Lavoisier: Paris, 2002, ch.10.
26. SEPA; Water and Waste Water Monitor and Analysis Methods, 4th ed.; Chinese Environmental Science Press (CESP): Beijing, 2002.
27. Lesueur, C.; Pfeffer, M.; Fuerhacker, M.; Chemosphere2005, 59, 685.
28. Bhattachrjee, S.; Curr. Sci.2005, 89, 1113.
29. Burlica, R.; Kirkpatrick, M. J.; Locke, B.; J. Electrostat. 2006,
64, 35.
30. Chang, J. S.; Non Thermal Plasma Technique for Pollution Control: Part A and Part B; Nato-ASI Series; Penetrante, B. M.;
Schultheis, S. E., eds.; Springer Verlag-Ecological Sciences 34: Berlin, 1993.
31. Antelman, M. S.; The Encyclopaedia of Chemical Electrodes Potentials, 2nd ed., Plenum Press: London, 1982.
32. Brisset, J-L.; Hnatiuc, E.; Plasma Chem. Plasma Process.2012,
32, 655.
33. Moussa, D.; Brisset, J-L.; J. Hazard. Mat. 2003, 102, 189. 34. Brisset, J-L.; Benstaali, B.; Moussa, D.; Fanmoe, J.;
Njoyim-Tamungang, E.; Plasma Sources Sci. Technol.2011, 1, 20. 35. Wang, B.; Sun, B.; Zhu, X.; Yan, Z.; Liu, Y.; Liu, H.; Contrib.
Plasma Phys. 2013, 53, 697.
36. Liu, Y.; Jiang, X.; Plasma Chem. Plasma Process. 2008, 28,
15.
37. Sugiarto, A. T.; Ito, S.; Ohshima, T.; Satoa, M.; Skalny, J. D.;
J. Electrostat. 2003, 58, 135.
38. Benetoli, L. O. D. B.; Cadorin, B. M.; Baldissarelli, V. Z.; Geremias, R.; De Souza, I. G.; Debacher, N. A.; J. Hazard. Mat. 2012, 55, 237.
39. Ni, G.; Zhao, G.; Jiang, Y.; Li, J.; Meng, Y.; Wang, X.; Plasma Processes Polym. 2013, 10, 353.
40. Abdelmalek, F.; Torres, R. A.; Combet, E.; Petrier, C.; Pulgarin, C.; Addou, A.; Sep. Purif. Technol.2008, 63, 30.
41. Hao, X.; Zhou, M.; Xin, Q.; Lei, L.; Chemosphere2007, 66, 2185.
42. Panizza, M.; Cerisola, G.; Water Res. 2009, 43, 339.
43. Tsagou-Sobze, E. B.; Moussa, D.; Doubla, A.; Hnatiuc, E.; Brisset, J.-L.; J. Hazard. Mat. 2008, 152, 446.
44. Marotta, E.; Ceriani, E.; Shapoval, V.; Schiorlin, M.; Ceretta, C.; Rea, M.; Paradisi, C.; Eur. Phys. J.: Appl. Phys. 2011, 55, 13811. 45. Boye, B.; Dieng, M. M.; Brillas, E.; Environ. Sci. Technol. 2002,
36, 3030.
46. Oturan, M. A.; Oturan, N.; Lahitte, C.; Trévin, S.; J. Electroanal. Chem. 2001, 507, 96.
47. Joshi, A. A.; Locke, B.; Arce, P.; Finney, W. C.; J. Hazard. Mat. 1995, 41, 3.
48. Souza, D. R.; Trovó, A. G.; Antoniosi-Filho, N. R.; Silva, M. A. A.; Machado, A. E. H.; J. Braz. Chem. Soc. 2013, 24, 1451. 49. Manassero, A.; Passalia, C.; Negro, A. C.; Cassano, A. E.;
Zalazar, C. S.; Water Res. 2010, 44, 3875.
50. Fanmoe, J.; Kamgang, J. O.; Moussa, D.; Brisset, J-L.; Phys. Chem. News2003, 14, 1.
51. Krẏsa, J.; Waldner, G.; Mestànkovà, H.; Jirkovskẏ, J.; Grabner, G.; Appl. Catal., B2006, 64, 290.
Submitted: June 6, 2014

![Figure 4. Profile of orthophosphates formation against exposure time during plasma treatment under the following conditions (initial concentration [NPG] 0 = 33.0 µmol L −1 , pH 6.3)](https://thumb-eu.123doks.com/thumbv2/123dok_br/18999321.463225/4.892.68.411.655.918/profile-orthophosphates-formation-exposure-treatment-following-conditions-concentration.webp)