Capítulo 5
5.1 - INTRODUÇÃO
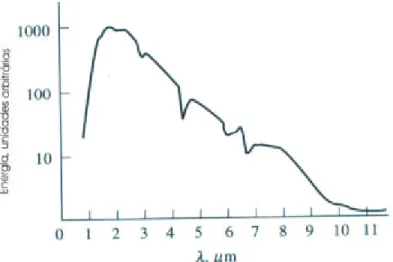
Região do Infravermelho
νν 3,8 x 1014 Hz 1,2 x 1014 Hz 6,0 x 1012 Hz 3,0 x 1011 Hz
λλ 0,78 µm 2,5 µm 50 µm 1000 µm
(780 nm) (5 x 105 nm)
λλ
==
νν
1
14000 cm-1 4000 cm-1 200 cm-1 10 cm-1INFRAVERMELHO PRÓXIMO
INFRAVERMELHO MÉDIO
INFRAVERMELHO AFASTADO
⇓ ⇓
região mais utilizada nos métodos analíticos
Figura 5.1 – Espectro de absorção no IV de filme de poliestireno
5.1.1 - APLICAÇÕES DA ESPECTROSCOPIA NO IV
Ä
Ä Análises quantitativas e qualitativas
Ø
Ø Análises qualitativas ⇒⇒ identificação de compostos orgânicos →→ espectros no IV
• na maior parte dos compostos orgânicos →→ espectros podem ser considerados a sua “impressão digital” →→ identificação qualitativa →→ mais precisa e com aplicação mais ampla que UV/VIS
Ø
Ø Análises quantitativas ⇒⇒ elevada seletividade do método
• determinação da concentração de uma dada espécie presente numa mistura complexa com poucos ou nenhuma etapa preliminar de separação
Ä
Ä Análises por CG/IV ⇒⇒ espectroscopia no IV acoplada à cromatografia gasosa
Ø
Ø Cromatografia gasosa →→ separação dos componentes de uma mistura
Ø
Ø Espectroscopia IV →→ identificação de cada componente
5.2 - ABSORÇÃO DA RADIAÇÃO INFRAVERMELHA
Ä
Ä Radiação IV →→ não é suficientemente energética para provocar transições
eletrônicas observadas com a radiação UV/VIS ↓ ↓
absorção restrita a espécies moleculares para as quais existam diferenças de energia entre os níveis vibracionais e rotacionais
Ä
Ä Absorção radiação IV →→ alteração no momento de dipolo da molécula associado aos
movimentos vibracionais e/ou rotacionais da mesma
Ex:
1) H Cl →→ molécula polar
momento de dipolo →→ diferença de carga dos átomos distância interatômica
Ø
Ø Como a molécula vibra →→ flutuação regular no momento de dipolo
Ø
Ø Se a freqüência da radiação foridêntica à freqüência de vibração da molécula →→
absorção da radiação →→transferência de energia para a molécula →→ alteração da amplitude de vibração molecular →→ alteração no momento de dipolo
-2) O2, N2 ou Cl2 →→ distribuição homogênea de carga ↓
↓
mesmo com vibração e rotação dos átomos ↓
↓
não apresentam dipolo →→ moléculas apolares ↓
↓
não absorvem radiação infravermelha
Ä
Ä Transições Rotacionais
Ø
Ø Envolvem energia muito baixa
- radiação λ > 100 µm ou ν < 100 cm-1 (infravermelho longínquo)
Ø
Ø Níveis rotacionais →→ quantizados
•• Espectros de absorção
- gases →→ região IV longínquo →→ linhas discretas bem definidas
-líquidos/sólidos →→ colisões intermoleculares e interações causam alargamento das linhas →→ espectro contínuo de linhas alargadas
Ä
Ä Transições Vibracionais/Rotacionais
Ø
Ø Níveis de energia vibracional →→ quantizados →→ maior parte das moléculas ⇒⇒
∆
∆Evibracional →→ radiação no IV médio
Ø
Ø Espectros de absorção
• gases →→ série de linhas espaçadas próximas →→ existência de vários níveis rotacionais para cada nível vibracional
Resumindo:
Condições para absorção da radiação IV
F
F Energia da radiação (E = hνν) deve coincidir com a diferença de energia entre os estados vibracionais excitado e fundamental da molécula ⇒⇒ freqüência da radiação é idêntica à freqüência de vibração molecular
F
F A molécula deve apresentar momento de dipolo como conseqüência de seus movimentos vibracionais ou rotacionais
5.3 - VIBRAÇÕES MOLECULARES
Existem dois tipos de vibrações moleculares:
F estiramento ou deformação axial ⇒⇒ variação na distância interatômica ao longo do
eixo da ligação
F deformações angulares ⇒⇒ variação no ângulo entre as ligações
5.3.1 - ESTIRAMENTO
Envolve alterações na distância entre os átomos ligantes ao longo da ligação. Corresponde ao movimento rítmico ao longo do eixo da ligação de modo que a distância interatômica aumente e diminua alternadamente.
Estiramento simétrico Estiramento assimétrico
Figura 5.2 – Tipos de estiramento simétrico e assimétrico
OBS:
5.3.2 - DEFORMAÇÕES ANGULARES
Correspondem a variações nos ângulos de ligação, seja internamente ao grupo de átomos seja deste grupo de átomos em relação à molécula com um todo (movimentos dos átomos fora do eixo de ligação)
Deformação angular simétrica no plano
Os dois átomos ligados a um átomo central aproximam-se e afastam-se um do outro com deformação no ângulo da ligação ⇒⇒ tesoura
Deformação angular assimétrica no plano
A unidade estrutural executa um movimento de vai e vem em relação ao plano de simetria da molécula ⇒⇒
balanço
Deformação angular simétrica fora do plano
A unidade estrutural executa um movimento de vai e vem em relação a um plano perpendicular ao plano de simetria da molécula ⇒⇒ sacudida
Deformação angular assimétrica fora do plano
A unidade estrutural roda num sentido e noutro em torno da ligação que a une ao resto da molécula ⇒⇒ torção
5.4 - MODELO MECÂNICO DAS VIBRAÇÕES DE ESTIRAMENTO
Aproximação:
- modelo mecânico de duas massas conectadas por mola
F = - K y
(1)sendo: F = força restauradora
K = constante de força da mola
•• Energia potencial (E)
dE = - F dy
(2)Substituindo (1) em (2)
dE = K y dy
(3)Integrando entre a posição de equilíbrio (y = 0) e y
∫∫
==
∫∫
E
0
y
0
dy
y
K
dE
∴
∴
K
y
22
1
E
==
(4)O movimento da massa em função do tempo pode ser deduzido da 2a lei de Newton:
F = m.a
(5)2 2
dt
y
d
a
==
(6)Combinando-se as equações (1), (5) e (6)
y
.
K
dt
y
d
.
m
2 2
−−
==
(7)Figura 5.4 – Diagrama de energia potencial de um oscilador harmônico
Solução:
y = A.cos (2
ππνν
0t + b)
(8)y
4
)
t
2
cos(
.
A
4
dt
y
d
2 0 2 0 2 0 2 2 2νν
ππ
πν
πν
νν
ππ
==
−−
−−
==
(9)Da equação (7) tem-se:
y
m
K
dt
y
d
2 2−−
==
(10)Como ⇒⇒ equação (10) = equação (9)
2 0 2
4
m
K
νν
ππ
==
∴
∴
m
K
2
1
0ππ
νν ==
(11)sendo: ν0 = freqüência do oscilador mecânico
A equação (11) pode ser modificada para descrever o comportamento de um sistema com duas massas:
2 1 2 1 0
m
.
m
)
m
m
(
K
2
1
K
2
1
++
==
==
ππ
µµ
ππ
νν
(12))
m
m
(
m
.
m
2 1 2 1++
==
Ä
Ä Tratamento quântico das vibrações
Modelo oscilador harmônico considerando o caráter quantizado das vibrações:
µµ
ππ
νν
K
2
1
.
2
1
v
++
==
(13)sendo: v = número quântico vibracional
Combinando-se as equações (12) e (13), tem-se:
0
2
1
v
νν
νν
++
==
(14) ØØ Energia vibracional
0
.
h
.
2
1
v
h
E
νν
νν
++
==
==
(15)- apenas certos valores discretos de energia podem estar associados à vibração quantizada
Ø
Ø Transição vibracional (∆E)
•• Regras de seleção ⇒⇒ teoria quântica só ocorrem as transições nas quais o número quântico vibracional varia de uma unidade
∆
∆v = ±± 1
- Níveis vibracionais ⇒⇒ igualmente espaçados →→ um único pico de absorção é observado para uma dada vibração molecular
Ex:
Transição ⇒⇒νν = 0 →→ νν = 1 Transição ⇒⇒ νν = 1 →→ νν = 2
0 0 21h
E == νν
∆
∆E = h νν0
0 1 23h
E == νν
0 1 23h
E == νν
∆
∆E = h νν0
0 2 25h
Quaisquer que sejam os níveis vibracionais envolvidos a energia envolvida é a mesma ↓
↓
é também idêntica à energia prevista pelo modelo da mecânica clássica para a energia de vibração de duas massas por uma mola
↓ ↓
µµ
ππ
νν
∆
∆
K
2
h
h
E
==
0==
(16) ouµµ
ππ
νν
νν
K
2
1
0==
==
(17)Em termos do número de onda (ν)
2 1 2 1 12 2 1 2 1
m
.
m
)
m
m
(
K
10
x
3
,
5
m
.
m
)
m
m
(
K
c
2
1
K
c
2
1
++
==
++
==
==
−−ππ
µµ
ππ
νν
(18)sendo: [ν] = cm-1
[K] = N/m ou din/cm [c] = m/s ou cm/s [µ] = kg ou g
OBS:
Ligações simples ⇒⇒ K ≈ 3,0 x 102 – 8,0 x 102 N/m (5,0 x 102 N/m) Ligações duplas ⇒⇒ K ≈ 1,0 x 103 N/m
Ligações triplas ⇒⇒ K ≈ 1,5 x 103 N/m
Exercício:
Cálcular a freqüência aproximada para os seguintes estiramentos: (a) C – H
(b) F – H
2 1 2 1
m
.
m
)
m
m
(
K
c
2
1
++
==
ππ
νν
onde: ν = freqüência vibracional (cm-1) (no de onda)
c = velocidade da luz (cm/s)
K = constante de força da ligação (dina/cm)
m1, m2 = massa dos átomos 1 e 2, respectivamente (g)
(a) estiramento C - H
)
10
x
8
,
19
x
10
x
64
,
1
(
10
x
)
64
,
1
8
,
19
(
10
x
0
,
5
)
10
x
0
,
3
(
2
1
24 24 24 510 −− −−
−−
++
==
ππ
νν
νν = 3,05 x 103 cm-1 = 3050 cm-1 (λλ = 3,28 µµm)
OBS:
Na prática observa-se bandas associadas ao estiramento C – H para grupos metil e metileno na faixa de 2960-2850cm-1 →→ desvios podem ser associados a fenômenos não
considerados na fórmula →→ interações entre os átomos considerados no cálculo e o resto da molécula
2) estiramento F - H mF = 31,2 x 10-24 g
)
10
x
2
,
31
x
10
x
64
,
1
(
10
x
)
64
,
1
2
,
31
(
10
x
0
,
5
)
10
x
3
(
2
1
24 24 24 510 −− −−
−−
++
==
ππ
νν
ν = 3010 cm-1
νF - H < νC - H
na realidade νF - H = 4138 cm-1
νC - H = 3050 cm-1
a constante de força (K) da ligação F - H é maior do que a da C - H ↓
↓
quanto maior a diferença entre a eletronegatividade do par de átomos, maior a constante de força da ligação
24 24 24 10
10
x
2
,
31
x
10
x
64
,
1
10
x
)
64
,
1
2
,
31
(
K
)
10
x
3
(
2
1
4138
−− −−−−
++
==
ππ
Ø
Ø Tratamento mecânica clássica ou mecânica quântica →→ oscilador harmônico (Figura
4) →→ energia potencial do oscilador varia periodicamente com a variação na distância entre as massas
↓ ↓
não descreve bem a vibração molecular:
- quando os dois átomos se aproximam →→ repulsão coulômbica entre os núcleos
→→ força que age na mesma direção da força restauradora →→ E aumenta mais rapidamente que o previsto pelo modelo do oscilador harmônico
- quando a distância interatômica se aproxima daquela onde ocorre a dissociação dos átomos →→ decréscimo da força restauradora →→ decréscimo de E mais rapidamente que o previsto pelo modelo do oscilador harmônico
⇓ ⇓
Oscilador anarmônico
Figura 5.5 – Diagrama de energia potencial do oscilador anarmônico
OBS:
1) Para números quânticos (v) elevados →→ ∆E torna-se menor →→ regra de seleção não é obtida rigorosamente →→ transições com ∆v = ± 2 ou ± 3 são observadas →→ sobretons.
3) Os espectros vibracionais podem se complicar devido à possibilidade de interações entre as diferentes vibrações da molécula →→ picos a freqüências iguais à soma ou à diferença entre as freqüências das vibrações fundamentais →→ aparecem quando um fóton excita dois modos vibracionais simultaneamente
5.5 - ESTIMATIVA DO NÚMERO DE VIBRAÇÕES FUNDAMENTAIS
Ä
Ä Graus de liberdade de uma molécula = Σ graus de liberdade de seus átomos
individualmente
•• Cada átomo ⇒⇒ 3 graus de liberdade ⇒⇒ x, y, z •• Molécula ⇒⇒ 3 N graus de liberdade
Ä
Ä Movimentos da molécula:
•• translação ⇒⇒ perda de três graus de liberdade
•• rotação ⇒⇒ perda de até 3 graus de liberdade
•• vibrações dos átomos uns em relação aos outros
Ä
Ä Número de vibrações fundamentais da molécula⇒⇒ 3 N - 6
5.5.1 - ESPECTROS DE ABSORÇÃO NO IV DE MOLÉCULAS POLIATÔMICAS
Ä
Ä Aumento do número de bandas de absorção no espectro no IV das moléculas com o
aumento do número de átomos
••Número de vibrações fundamentais ⇒⇒ 3 N – 6 (valor teórico nem sempre é observado)
Ä
Ä Fatores que tendem a reduzir o número de vibrações fundamentais
••simetria da molécula é tal que uma dada vibração não altera o seu momento de dipolo
••as energias de duas ou mais vibrações são idênticas ou quase idênticas
••intensidade de absorção é muito baixa para ser detectada
Ä
Ä Acoplamento Vibracional
Ø
Ø Interações entre as vibrações alterando a freqüência de vibração teórica:
••forte acoplamento entre vibrações de estiramento →→ existe átomo comum às duas vibrações
••acoplamento entre vibrações de deformação →→ existência de ligação comum entre os átomos que vibram
••acoplamento entre estiramento e deformação →→ ligação que vibra forma um dos lados do ângulo que se deforma
••a interação é grande quando os grupos acoplados têm energias individuais aproximadamente iguais
••pouca ou nenhuma interação é observada entre grupos separados por duas ou mais ligações
••acoplamento requer que as vibrações sejam da mesma classe de simetria
Ex:
1) CO2⇒⇒ molécula linear com três átomos
3 N graus de liberdade rotação: - 2 G. L. translação: - 3 G. L.
número de vibrações fundamentais = 3 N –5
(1) estiramento simétrico (2) estiramento assimétrico (νν = 2350 cm-1)
(3) deformação angular simétrica no plano (νν = 667 cm-1)
Na prática ⇒⇒ 2 bandas de absorção ⇒⇒ 2350 cm-1 e 667 cm-1
- (1) é inativa no IV →→ não altera o momento de dipolo da molécula
- (3) e (4) são idênticas →→ movimentos de deformação possíveis nos planos em torno do eixo de ligação
2) H2O⇒⇒ molécula angular com 3 átomos
3 N graus de liberdade rotação – 3 G.L.
translação – 3 G. L.
número de vibrações fundamentais = 3N – 6
(1) estiramento simétrico (νν = 3650 cm-1) (2) estiramento assimétrico (νν=3760cm-1)
(3) deformação angular no plano (δδ = 1596 cm-1)
OBS:
1) Estiramentos ocorrem a ν maiores do que as deformações.
2) Estiramentos simétricos e assimétricos geralmente ocorrem a ν próximos.
Ex: estiramento C - O
- metanol CH3 - O - H ν = 1034 cm-1
- etanol CH3 – CH2 - O - H ν = 1053 cm-1
- butanol-2 CH3 - CH2 - CH - CH3 ν = 1105 cm-1
OH
Estas variações resultam do acoplamento do estiramento C - O com o estiramento C - C vizinho ou com as vibrações C – H
5.6-INSTRUMENTOS PARA MEDIDAS DE ABSORÇÃO MOLECULAR NO INFRAVERMELHO
Ä
Ä Componentes básicos:
FONTE AMOSTRA
DISP. SELEÇÃO
λλ
DETETOR LEITURA /
REGISTRO
5.6.1 - FONTES:
Ø
Ø Características:
∗∗ estabilidade
∗∗ fornecer espectro contínuo na região de trabalho
∗∗ potência suficiente para ser detectada
5.6.1.1 –Fontes de IV
Figura 5.6 – Curvas de radiação do corpo negro
Ø
Øsólidos inertes aquecidos
5.6.1.1.1 - Filamento de Nernst (IV médio)
•• Óxidos de terras raras (Th, Ce) moldados em um cilindro de φ = 1 a 2 mm e L = 20 mm
•• Máximo de energia = 1,4 µm (1400 nm)
•• Fonte incandescente (1200 K- 2200 K)
•• Fragilidade mecânica
•• Necessita de aquecimento prévio para se tornar condutor
•• Resistência decresce com aumento da temperatura →→ dispositivo para limitar a corrente
Figura 5.7 – Distribuição espectral da energia de um filamento de Nernst operando a 2200K
5.6.1.1.2 - Globar (IV médio e longínquo)
•• Fonte incandescente (1300 K – 1500 K)
•• Bastão de carbeto de silício φ = 5 mm e L = 50 mm
•• Distribuição espectral de energia semelhante à do filamento de Nernst
•• Máxima intensidade de energia 1,8 –2,0 µm
•• Refrigeração contínua com água corrente →→ evitar arco voltaico
•• Coeficiente de resistência positivo
5.6.1.1.3 - Hélice de Ni-Cr
•• Fio espiral de Ni-Cr aquecido por corrente elétrica
•• Fonte incandescente (1100 K)
•• Intensidade menor que Globar ou filamento de Nernst, mas é mais durável
5.6.1.1.4 - Lâmpada de arco de mercúrio (IV longínquo λλ > 50 µµm)
•• Tubo de quartzo contendo vapor de Hg a p > 1 atm
•• Passagem de corrente elétrica através do vapor forma uma fonte interna de plasma radiação contínua na região IV longínquo
5.6.1.1.5 - Lâmpada de filamento de W (IV próximo)
•• Fonte adequada para IV próximo⇒⇒ 4000 – 12800 cm-1 (2,5 a 0,78 µm)
5.6.1.1.6 - Laser de CO2 sintonizável
•• Fonte para monitoramento de poluentes no ar atmosférico e para determinação de espécies em soluções aquosas
•• Espectro de banda entre 900 e 1100 cm-1 constituído por linhas (≈ 100) muito próximas
•• Útil para determinação quantitativa de butadieno, amoníaco, benzeno, etanol, dióxido de nitrogênio e tricloroetileno
•• Potência em cada linha é muito maior que a das fontes incandescentes
5.6.2 - DETECTORES
Ä
Ä Radiação IV ⇒⇒ detectores ⇒⇒ três tipos:
•• térmicos →→ espectrofotômetros dispersivos e fotômetros
•• piroelétricos →→ espectrofotômetros dispersivos, fotômetros e espectrômetros interferométricos
5.6.2.1 - Térmicos
Ø
Ø Radiação IV é absorvida por um pequeno corpo negro e a variação de temperatura
resultante é medida
Ø
Ø Capacidade térmica do material deve ser a menor possível para detectar a variação de
temperatura (≈ 10-3 K)
Ø
Ø Características:
•• resposta depende do efeito de aquecimento da radiação
•• usados em ampla faixa de λ
•• detectores encapsulados e abrigados de radiação térmica produzida por objetos na vizinhança ⇒⇒ evitar ruídos térmicos
•• a radiação é modulada para minimizar sinais estranhos
5.6.2.1.1 - Termopar
Ø
Ø Par bimetálico cujas extremidades são soldadas (ex: Bi e Sb) →→ junção dos dois
metais →→ geração de mV proporcional a ∆T entre eles
Ø
Ø Junção é enegrecida (melhorar a capacidade de absorver calor) e selada numa câmara
sob vácuo com janela transparente à radiação IV
Ø
Ø Bom detector →→ responder a diferenças de temperaturas na faixa de 10-6 K
Ø
Ø Detector mais utilizado no IV médio
Ø
Ø Tempo de resposta : 40 ms
5.6.2.1.2 - Bolômetro
Ø
Ø Tipo de termômetro de resistência construído por tiras de metal (Pt ou Ni) ou por
material semicondutor (ex: Si ou Ge) →→ apresentam grande variação na resistência com a temperatura
Ø
Ø Menos utilizados que termopares
Ø
5.6.2.2 - Detector piroelétrico
Ø
Ø Pastilha de material piroelétrico, que é um isolante (dielétrico) com propriedades
térmicas e elétricas especiais (sulfato de triglicina - (NH2CH2COOH)3.H2SO4,
usualmente deuterado)
↓↓
Cristal de sulfato de triglicina montado entre dois eletrodos paralelos, um deles transparente à radiação IV →→ capacitor dependente de T
↓ ↓
radiação IV incide no cristal ↓
↓
alteração na distribuição de cargas no cristal ↓
↓
detectada como corrente no circuito externo
Ø
Ø Magnitude da corrente é proporcional à área do cristal e à taxa de polarização com a
temperatura
Ø
ØTempo de resposta muito rápido →→ uso nos espectrofotômetros interferométricos
5.6.2.3 - Fotocondutores
Ø
Ø Consistem num filme de material semicondutor (PbS, PbTe, PbSe, InSb) com
espessura de 0,1 µm depositado numa placa de vidro não condutora e selado sob vácuo num invólucro para proteção do semicondutor
Ø
Ø Baseiam-se no efeito fotocondutor interno em conseqüência da transição de um elétron
de uma banda de valência (não-condutora) para uma banda de condução →→ decréscimo da resistência elétrica do semicondutor
Ø
Ø Montagem →→ detector em série com fonte de alimentação e resistência →→ queda
de voltagem na resistência →→ medir a potência do feixe de IV
Ø
Ø IV próximo (1 a 3 µm) →→ PbS (operado a T ambiente)
Ø
Ø IV médio/afastado →→ HgTe/CdTe (resfriados com N2 líquido (minimizar ruído térmico)
Ø
Ø Mais rápidos (tempo de resposta < 10 µs) e mais sensíveis que detectores térmicos e
5.7 - INSTRUMENTOS PARA MEDIDA DE ABSORÇÃO DE RADIAÇÃO IV
Três tipos de instrumentos para medidas de absorção de radiação IV encontram-se disponíveis comercialmente:
•• espectrofotômetros dispersivos à base de redes de difração - uso em análises qualitativas
•• espectrômetros multiplex (interferométricos - uso de transformada de Fourrier)
- uso em análises quantitativas e qualitativas
•• fotômetros não dispersivos
- determinação quantitativa de espécies orgânicas presentes na atmosfera
5.7.1 - ESPECTROFOTÔMETROS DISPERSIVOS
5.7.1.1 - Características básicas:
Ø
Ø feixe duplo:
- redução/eliminação de problemas devidos à variação na tensã o de alimentação da fonte
- eliminação dos sinais de fundo devido à presença do CO2 e da H2O na atmosfera
Ø
Ø monocromador reticular ⇒⇒ elemento de dispersão →→ rede de difração
Ø
Ø registrador acoplado →→ obtenção dos espectros de absorção
Figura 5.9 – Representação esquemática de espectrofotômetro dispersivo de feixe duplo para medidas de absorção no IV
Ø
Ø Comentários:
•• Chopper de baixa freqüência (5 a 13 cps)
- alternar envio do sinal da referência e da amostra ao detector
- como estes sinais tem freqüência definida é possível ao detector discriminá-los dos sinais de radiações estranhas provenientes das vizinhanças do detector
•• Monocromador →→ arranjo ótico semelhante ao dos espectrofotômetros UV/VIS
•• Amostra →→ entre a fonte e o monocromador
- RIV não possui energia suficiente para causar decomposição fotoquímica da amostra
•• Atenuador →→ atuar de modo que a potência do feixe de referência fique semelhante à do feixe transmitido pela amostra
- movimento sincronizado com registrador ↓
↓
posição do registrador →→ transmitância da amostra
- sistema de atenuação do feixe de referência →→ limitações na performance do equipamento:
∗∗ resposta do atenuador →→ retardo em relação às variações de transmitância, particularmente quando o sinal de transmitância varia rapidamente
∗∗ acionamento mecânico da pena e atenuador →→ posições da pena ultrapassam os valores reais de transmitância
∗∗ T → 0% →→ praticamente não chega radiação proveniente da amostra ao detetor →→ difícil atingir o ponto nulo
5.7.2 - ESPECTRÔMETROS POR TRANSFORMADA DE FOURRIER
Ø
Ø inicialmente caros ( > U$ 100000)
volumosos
ajustes mecânicos freqüentes
uso limitado
Ø
Ø atualmente mais baratos ( U$ 15000 – 20000 )
compactos manutenção fácil
substituindo gradativamente os instrumentos dispersivos
5.7.2.1 - Instrumentos analíticos multiplex
Ø
Ø Dispositivos de canal único no qual todos os elementos do sinal são observados
simultaneamente
Ø
Ø Não possuem dispositivo para seleção de λ (filtros ou monocromadores)
Ø
Ø Fazem uso da transformada de Fourrier para decodificar o sinal ⇒⇒
INSTRUMENTOS POR TRANSFORMADA DE FOURRIER
5.7.2.1.1 - Vantagens da espectroscopia por transformada de Fourrier
Œ equipamentos têm menos componentes óticos e não possuem fendas para atenuar a
radiação →→ potência da radiação que alcança o detetor é maior que nos instrumentos dispersivos assim como a relação sinal/ruído ⇒⇒Vantagem Jacquinot
• elevada exatidão e precisão na determinação dos λ (ou ν)
- possibilidade de calcular a média dos sinais
- melhora a relação sinal/ruído
Ž todas as freqüências da fonte alcançam o detetor simultaneamente →→ redução do
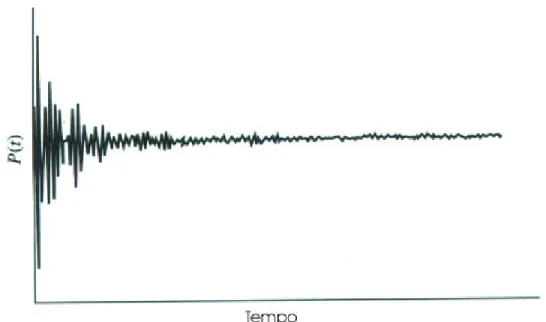
5.7.2.1.2 - Espectroscopia no domínio tempo X domínio de freqüências
Figura 5.11 – Representação de espectros nos domínios tempo (a, b) e freqüência (c, d, e)
Domínio tempo e domínio freqüência ↓
↓
mesmas informações ↓
↓
convertidos um no outro:
P(t) = k [cos(2
ππνν
1t) + cos (2
ππνν
2t)]
k = constante; t = tempo
5.7.2.1.3 - Obtenção de espectros no domínio tempo ( P = f(t) )
Ø
Ø Espectroscopia ótica →→ radiações com alta freqüência (1011 – 1015 Hz) →→ não existem transdutores que respondam a variações de potência de radiações com freqüências tão elevadas
↓ ↓
Necessidade de “modular” o sinal:
sinal de alta freqüência sinal de freqüência mensurável sem
distorcer a dependência original de P = f(t)
Interferômetro de Michelson
Ø
Ø Movimentação de um dos espelhos →→ flutuação da potência que alcança o detetor:
- posição = 0 (M = F) →→ recombinação dos feixes “em fase” →→ interferência construtiva máxima →→ P é máxima
- posição =
4
λλ
−−
==
2
F
M
λλ
→→ interferência destrutiva máxima →→ P=0- posição =
2
λλ
(M – F =
λλ
)
→→ interferência construtiva máxima →→ P émáxima
Definindo: δδ = retardação = 2 (M-F) ⇒⇒ diferença entre o percurso dos feixes divididos ↓
↓
P(t) vs δδ ⇒⇒ interferograma
( freqüência(δ) << freqüência(ν) )
sendo: vM = velocidade de movimentação do espelho (cm/s)
τ = tempo para espelho percorrer
2
λλ
cm (1 ciclo) (s)f = freqüência de oscilação do sinal no detetor (s-1)
2
x
v
Mττ ==
λλ
(19)λλ
ττ
Mv
2
1
f
==
==
(20)ν
ν
M
v
2
f
==
(21)Ø
Ø Relação entre a freqüência da fonte (ν) e a do interferograma (f)
λλ
M
v
2
f
==
∴
∴
νν
λλ
==
c
c
v
2
f
==
Mνν
(22)Ex: vM = 1,5 cm/s
νν
10
10
x
3
5
,
1
x
2
f
==
∴
∴
f = 10
-10νν
f <<<
νν
Ø
Ø Transformação dos interferogramas em espectros
- radiação monocromática
P(
δδ
) = B(
νν
) cos 2
ππ
f t
(23)sendo: P(δ) = potência no domínio tempo
B(ν) = termo proporcional à potência no domínio ν
Substituindo (21) em (23)
P(
δδ
) = B(
νν
) cos 4
ππ
v
Mνν
t
(24)Mas:
δδ
= 2 v
Mt
(25)Então:
P(
δδ
) = B(
νν
) cos 2
ππ
δδ
νν
(26)Se no interferograma →→ 2 ν diferentes
P(
δδ
) = B(
νν
1) cos (2
ππ
δδ
νν
1) + B(
νν
2) cos (2
ππ
δδ
νν
2)
(27)Para um espectro contínuo
νν
πδν
πδν
νν
δδ
)
B
(
)
cos(2
)
.
d
(
P
==
∫∫
+∞ +∞
∞ ∞ −−
(28)
↓ ↓
Figura 5.14 – Interferogramas e espectros óticos correspondentes
Espectroscopia por Transformada de Fourrier
obtenção do espectro P(δδ) vs δδ
↓ ↓
interferograma
cálculos matemáticos complexos
↓ ↓
computadores
Figura 5.15 a – Transformada de Fourrier
5.7.2.1.4 - Componentes dos espectrômetros por transformada de Fourrier
Ø
Ø Interferômetro de Michelson
•• mecanismo para movimentação do espelho
∗∗ velocidade de movimentação do espelho ⇒⇒ constante
∗∗ posição do espelho conhecida exatamente a cada instante
- IV longínquo parafuso micrométrico acionado por motor
- IV médio/próximo mecanismo mais sofisticado
•• modelos com três interferômetros
∗∗ fonte de IV
∗∗ luz branca
- determinação exata do ponto zero de retardação
∗∗ laser (He/Ne)
- obtenção dos interferogramas a intervalos de aquisição precisamente espaçados
•• interferômetro triplo
∗∗ elevada precisão na determinação das freqüências espectrais ⇒⇒ importante quando se calcula a média das várias varreduras espectrais
Figura 5.17 – Sinais no domínio tempo para os três interferômetros presentes num espectrômetro FTIR
•• modelos com interferômetro único
∗ feixe laser paralelo ao feixe IV
∗ δ = 0 ⇒⇒ calculado pelo interferograma do feixe IV
Ø
Ø Divisor de feixe
•• dividir o feixe 50% transmitido 50% refletido
∗ IV afastado →→ filme de Mylar entre duas placas de sólido com baixo índice de refração
∗ IV médio →→ filme de Si ou Ge depositado sobre CsI, CsBr, NaCl ou KBr
∗ IV próximo →→ filme de Fe2O3 depositado sobre CaF2
Ø
Ø Fontes e transdutores
•• Fontes →→ as mesmas dos instrumentos dispersivos
•• Detectores
∗∗ Detector piroelétrico →→ sulfato de triglicina →→ IV médio
∗∗ Detector fotocondutor
- maior sensibilidade
- resposta mais rápida
u Cd/Hg Te ou InSb refrigerados com N2 líquido
5.7.2.1.5 - Características dos espectrômetros por transformada de Fourrier
Ø
Ø Feixe simples
Ø
Ø Operação:
Œ obtenção do interferograma do material de referência (branco) →→ média de 20 a
30 aquisições →→ tratamento de dados armazenados como espectro de referência
• obtenção do interferograma da amostra →→ média de 20 a 30 aquisições →→
relação entre os dados espectrais da amostra e da referência ⇒⇒ espectro de absorção
OBS:
Ø
Ø Preço:
v
v equipamentos simples ≈ U$ 16000
7800 – 350 cm-1
v
vequipamentos sofisticados ≈ U$ 150000
25000 – 10 cm-1 IV/VIS
fontes, detectores e divisores de feixe intercambiáveis
Ø
Ø Resolução instrumentos comerciais ⇒⇒ 8 – 0,01 cm-1
8.2.1.6 - Vantagens sobre os instrumentos dispersivos
Ø
Ø IV médio ⇒⇒ FTIR
•• melhor relação sinal ruído
•• rapidez na aquisição dos espectros
•• resolução elevada ( < 0,1 cm-1)
•• determinação dos ν com maior precisão e exatidão
•• sistema ótico →→ quantidade de energia que atravessa o instrumento 10 a 100 x maior
- parcialmente perdida →→ detector menos sensível pois responde mais rápido
•• não tem problema de radiação estranha
8.2.1.7 - Aplicações do instrumento
Ø
Ø Trabalhos em que elevada resolução é necessária (misturas gasosas)
Ø
Ø Amostras com absorbâncias elevadas
Ø
Ø Substâncias com bandas de absorção fracas
•• espécies quimicamente adsorvidas em catalisadores
Ø
Ø Análises que requerem rápida determinação do espectro
•• estudos cinéticos, detecção de efluentes cromatográficos
Ø
Ø Obtenção de espectros de reflexão
Ø
5.7.3 - INSTRUMENTOS NÃO DISPERSIVOS
5.7.3.1 - Fotômetros de filtro
•• análise quantitativa de substâncias orgânicas na atmosfera
•• fonte ⇒⇒ hélice de Ni-Cr
•• detector ⇒⇒ piroelétrico
•• seleção do λ de trabalho ⇒⇒ filtros de interferência intercambiáveis (3000–750 cm-1)
Figura 5.19 – Fotômetro infravermelho portátil para análise de gases
•• amostra introduzida por bombeamento
•• percurso da radiação 0,50 m 20 m (espelhos no compartimento da amostra)
•• aplicação:
- análise de acrilonitrila, hidrocarbonetos clorados, CO, fosgênio e HCN
5.7.3.2 - Fotômetros sem filtro
•• monitoração de um único componente em correntes gasosas
•• Referência →→ gás que não absorve radiação
Figura 5.20 – Fotômetro no IV não dispersivo para monitorar CO
5.7.3.3 - Instrumento automático para análise quantitativa
•• Seleção de ν⇒⇒ filtros
- três placas espelhadas parcialmente transparentes separadas por camada de dielétrico em forma de cunha
- ν transmitido →→ varia com a espessura da cunha
•• Controlado por computador
- programado para medir absorbância de mistura de vários componentes em várias ν e depois computar a concentração de cada componente
5.8 - APLICAÇÕES DA ESPECTROMETRIA NO INFRAVERMELHO
Tabela 5.1 - Principais aplicações da espectrometria do infravermelho
Região espectral Tipo de medida Tipo de análise Tipo de amostras
reflectância difusa quantitativa produtos comerciais sólidos ou líquidos
IV próximo
absorção quantitativa misturas gasosas
qualitativa compostos sólidos, líquidos ou gasosos puros
quantitativa misturas complexas gasosas, sólidas ou líquidas absorção
cromatográfica misturas complexas gasosas, sólidas ou líquidas
reflectância qualitativa compostos puros sólidos ou líquidos
IV médio
emissão quantitativa amostras de ar atmosférico
IV afastado absorção qualitativa
Compostos inorgânicos puros ou espécies
5.8.1 - ESPECTROMETRIA DE ABSORÇÃO NO IV MÉDIO
Ä
Ä Determinação da estrutura de compostos orgânicos e bioquímicos
5.8.1.1 - Manuseio das amostras
Ø
Ø UV/VIS →→ ajuste do sinal de absorbância/transmitância
•• diluição das soluções
•• alteração da espessura das células
Ø
Ø IV →→ isto não pode ser feito →→ inexistência de solventes transparentes à RIV
Ø
Ø manuseio da amostra →→ parte mais difícil e demorada da análise
5.8.1.1.1 - Amostras gasosas
Ø
Ø gás ou líquido com baixo ponto de ebulição →→ expandir no interior da célula onde
previamente foi feito vácuo
•• células →→ janelas com transparência adequada
•• existem células com percurso ótico variando de poucos cm até metros (radiação pode ser refletida no interior da célula passando várias vezes através da amostra)
5.8.1.1.2 - Amostras líquidas
Ø
Ø Solventes →→ absorvem no IV
•• não existem solventes completamente transparentes ao IV
•• H2O e etanol evitados →→ atacam cloretos e brometos de metais alcalinos usados
nas janelas das células
Ø
Ø Células
•• mais estreitas que as de UV/VIS devido à absorção de energia pelo solvente
•• concentração das amostras: 0,1 a 10%
•• desmontáveis
Figura 5.23 – Vista expandida de célula desmontável para líquidos (espaçadores com espessura de 0,015 a 1 mm)
•• encher/esvaziar → seringa
•• material das janelas → NaCl ou KBr
Ø
Ø Determinação da espessura da célula
•• feixe de referência →→ direto para monocromador
•• feixe de amostra →→ célula vazia
Figura 5.24 – (a) Espectro da célula vazia atravessada pelo “feixe da amostra”; (b) cálculo do percurso “b’
∗∗ Interferência construtiva ⇒⇒
N
b
2
==
λλ
- selecionar dois λ →→ ∆N franjas entre eles
-
N
2
b
2
b
2
b
(
1 2)
2 1
νν
νν
λλ
λλ
∆
∆
==
−−
==
−−
∴
∴
)
(
2
N
b
2 1
νν
νν
∆
∆
−−
==
(29)OBS:
1- Franjas não são observadas em amostras líquidas ⇒⇒ ηlíquido≈ηjanela
2- Líquidos:
- pouca amostra
- não há solvente disponível
∗∗ problemático para análises quantitativas ( b varia)
∗∗ adequado para análises qualitativas
5.8.1.1.3 - Amostras sólidas
Ø
Ø Dispersas em matriz sólida ou líquida
•• 2 a 5 mg de amostra moída (dp < 2 µm) intimamente misturados com Nujol ou
Fluorolube (polímero halogenado)
- filme colocado entre placas de NaCl fixadas na célula
•• 1 a 3 mg de amostra moída intimamente misturada em gral com 100 mg de KBr previamente seco →→ mistura →→ molde →→ submetida a pressão →→ pastilha
→→ porta amostra
- se possível empastilhar sob vácuo para eliminar o ar ocluído
- espectros bandas a 3450cm-1 e a 1640 cm-1 umidade - para ν < 400 cm-1 →→ CsI
5.8.1.2 - Análises qualitativas
Ä
Ä Identificação dos compostos orgânicos ⇒⇒ processo em duas etapas:
Œ análise na região dos grupos funcionais ⇒⇒ 3600 – 1200 cm-1
- identificar os grupos funcionais presentes
• análise na região das impressões digitais 1200 – 600 cm-1
- comparação detalhada do espectro da amostra desconhecida com espectros de compostos puros conhecidos que contenha(m) aquele(s) grupo(s) funcional(is)
- nesta região é que pequenas diferenças na estrutura originam diferenças importantes no espectro
Ø
Ø Região dos grupos funcionais:
•• freqüência de vibração dos grupos funcionais (por exemplo: C = O, C = C, C – H, O – H, etc ⇒⇒ estimadas de modo aproximado pelo modelo do oscilador harmônico associado ao tratamento quântico das vibrações
∗∗ interações com outras vibrações envolvendo os átomos do grupo funcional ⇒⇒ variações dentro de faixa relativamente estreita
Tabela 5.2 – Freqüências (números de onda, νν) de alguns grupos funcionais orgânicos
Ligação Tipo de composto Faixa de νν (cm-1) Intensidade
2850 – 2970 forte
C – H Alcanos
1340 – 1470 forte
3010 – 3095 Média
C – H Alcenos
675 – 995 forte
C – H Alcinos 3300 Forte
3010 – 3100 média
C – H Anel aromático
690 – 900 forte
Álcoois, fenóis 3590 – 3650 variável
Álcoois, fenóis com ponte de H 3200 – 3600 variável
Ácidos carboxílicos 3500 – 3650 média
O - H
Ácidos carboxílicos com ponte de
H 2500 – 2700 larga
N – H Aminas, amidas 3300 – 3500 média
C = C Alcenos 1610 – 1680 variável
C = C Anéis aromáticos 1500 – 1600 variável
C ≡ C Alcinos 2100 – 2260 variável
C – N Aminas, amidas 1180 – 1360 forte
C ≡ N Nitrilas 2210 – 2280 forte
C - O Álcoois, éteres, ácidos
carboxílicos, ésteres 1050 – 1300 forte
C = O Aldeídos, cetonas, ácidos
carboxílicos, ésteres 1690 – 1760 forte
1500 – 1570 forte
NO2 Nitrocompostos
Ø
Ø Região das impressões digitais
• pequenas diferenças na molécula grandes diferenças no espectro
• semelhança grande entre dois espectros nessa região (composto conhecido e composto desconhecido, por exemplo ⇒⇒ evidência para identificação da substância
• complexidade dos espectros nessa região ⇒⇒ maior parte das ligações simples ⇒⇒ bandas de absorção nesta região ⇒⇒ freqüências semelhantes ⇒⇒ fortes interações entre ligações vizinhas
• Interpretação exata do espectro raramente é possível nesta região
Ex:
Figura 5.25 (a) e (b) ⇒⇒ efeito da presença de grupo metil ⇒⇒ diferenças nos espectros na região das impressões digitais
(a)
(c)
(d)
Figura 5.25 – Regiões das freqüências de grupo e das impressões digitais para alguns compostos orgânicos
Ø
Ø Uso das cartas de correlação
•• seu uso isolado raramente permite a identificação inequívoca dos espectro
- sobreposição das freqüências de grupo
- variações no espectro devido ao estado físico das amostras
- limitações instrumentais
•• usadas como base para uma análise mais detalhada
Ø
Ø Comparação de espectros
•• catálogos ⇒⇒ espectros dos mais diversos compostos puros ⇒⇒ busca e comparação manual ⇒⇒ demorada e cansativa
•• sistema de busca por computador
-posição e intensidade dos picos no espectro da espécie desconhecida ⇒⇒ determinados e armazenados ⇒⇒ comparados com dados armazenados de inúmeros compostosconhecidos
5.8.1.3 – Análises quantitativas
Ä
Ä Desvantagens e limitações ⇒⇒ Instrumentos dispersivos
•• maior complexidade dos espectros sobreposição de picos
•• bandas mais estreitas
•• limitações instrumentais ⇒ não obediência à Lei de Beer
- percurso ótico mais estreito →→ incertezas analíticas
- largura da banda ≈ largura da fenda
•• erros associados às medidas na região IV →→ maiores do que na região UV/VIS
Ä
Ä Desvios da lei de Beer
espectro no IV →→ bandas estreitas →→ favorecimento a desvios na Lei de Beer ↓
↓
instrumentos dispersivos →→ baixa intensidade da fonte + baixa sensibilidade dos detectores
↓ ↓
fendas largas ↓
↓
largura da fenda ≈ largura da banda ↓
↓
chegam ao detector vários λ nos quais as absortividades são muito diferentes ↓
↓
relação não linear entre A e C ↓
↓
Curva de Calibração ⇒⇒ determinação experimental com vários pontos
Ä
Ä Medidas de absorbância
Ø
Ø UV/VIS →→ cubeta →→ solvente (branco) →→ P0 cubeta →→ amostra →→ P
0
P
P
T
==
∴∴P
P
log
T
log
Ø
Ø IV →→ difícil utilizar esta metodologia
•• impossibilidade de se ter disponível células com características de absorção idênticas
•• percurso ótico pequeno →→ difícil de ser reproduzido
•• janelas facilmente atacadas →→ características se alteram com o uso
•• referência →→ feixe passando pelo ar ↓
↓
T < 100% mesmo quando a amostra não absorve
Ä
Ä Métodos para correção do espalhamento / absorção nas paredes da célula
Œ Obtenção do espectro do solvente (branco) e da amostra na mesma célula
Branco ⇒
r 0 0
P
P
T
==
Amostra ⇒r a a
P
P
T
==
Logo: 0 a 0 r r a 0 aP
P
P
P
x
P
P
T
T
T
==
==
==
•Método da linha de base
5.8.1.3.1 – Aplicações típicas
Ø
Ø Elevada especificidade
Œ Análise da mistura de HC aromáticos C8
C8H10⇒ etilbenzeno + p-xileno + m-xileno + o-xileno
(i) Espectros das espécies individualmente
Ø
Ø Seleção dos λ característicos
Ø
Ø Determinar ε nos quatro λ característicos
Figura 5.28 – Espectros no IV dos isômeros C8H10 em n-hexano
(ii) Mistura → sobreposição de bandas associadas aos grupos funcionais comuns
Ø
Ø Medida da absorvância nos quatro λ selecionados
Ø
Ø Solução de sistema de quatro equações e quatro incógnitas
• Determinação de contaminantes no ar atmosférico
Ø
Ø Fotômetros de filtro controlados ou não por computador
Tabela 5.3 – Exemplos de alguns contaminantes no ar atmosférico que podem ser analisados por IV atendendo a regulamentação da OSHA
Substância permitida (ppm) Exposição λλ (µµm) Concentração mínima detectável (ppm)
CS2 4 4,54 0,5
cloropreno 10 11,4 4
diborano 0,1 3,9 0,05
etilenodiamina 10 13,0 0,4
HCN 4,7 3,04 0,4
metil-mercaptano 0,5 3,38 0,4
nitrobenzeno 1 11,8 0,2
piridina 5 14,2 0,2
SO2 2 8,6 0,5
CH2=CHCl 1 10,9 0,3
Tabela 5.4 – Exemplo de análise por IV de contaminantes atmosféricos
Contaminantes C real (ppm) C medida (ppm) Er (%)
CO 50 49,1 1,8
metiletilcetona 100 98,3 1,7
CH3OH 100 99,0 1,0
óxido de etileno 50 49,9 0,2
5.8.2 – ESPECTROMETRIA DE ABSORÇÃO NO IV PRÓXIMO (IVP ou NIR)
Ä
Ä 770 – 2800 nm (13000 – 3500cm-1)
Ä
Ä Picos observados:
Ø
Ø Sobretons ou combinações envolvendo as bandas de estiramento fundamentais
(3500 – 1700 cm-1) ⇒⇒ C – H, N – H, O – H
Ø
Ø Picos pouco intensos →→ limite de detecção ≈ 0,1%
Ä
Ä Análises quantitativas de rotina:
Ø
Ø água, proteínas, HC com baixo peso molecular, gorduras em produtos agrícolas,
alimentícios e originários de indústrias químicas
Ä
Ä Tipo de medida mais usada ⇒⇒ reflectância difusa
Ä
ÄSolventes
5.8.2.2 - Instrumentação
Ø
Ø Espectrofotômetros similares aos usados na região UV/VIS
••Fonte ⇒⇒ lâmpada de W/I2 com janela de quartzo ••Cubetas de vidro ou quartzo
••Monocromador reticular
••Detector ⇒⇒ fotocondutor de PbS
Ø
ØEquipamentos comerciais ⇒⇒UV/VIS/NIR 180 – 2500nm (acessórios para medida de
reflectância)
5.8.2.3 - Aplicações
Ø
Ø Espectros ⇒⇒ maior utilidade para análises quantitativas ⇒⇒ C – H; N – H; O – H
•• Determinação de H2O em glicerina, hidrazina, filmes orgânicos e H2SO4 fumegante
•• Determinação de fenol, álcool e ácidos ⇒⇒sobreton do estiramento O – H (7100 cm-1)
••Determinação de ésteres, cetonas e ácidos carboxílicos ⇒⇒ absorção a 3300-3600cm-1
•• Determinação de aminas primárias e secundárias em presença de terciárias
5.8.3 – ESPECTROMETRIA DE REFLECTÂNCIA DIFUSA NO IV PRÓXIMO
Ä
Ä Aplicações:
Ø
ØDeterminação quantitativa rotineira de constituintes sólidos finamente divididos
•• Dosagem de proteínas, umidade, óleos, e celulose em grãos e sementes
Ä
ÄTécnica
Ø
ØAmostra de sólido finamente dividido →→ irradiada com uma ou mais bandas
Ä
Ä Reflectância difusa
Ø
Ø Radiação IV penetra na camada superficial das partículas provocando transições
vibracionais e sofre espalhamento em todas as direções
Ø
Ø Espectro de reflexão ⇒⇒ dependente da composição da amostra
Figura 5.30 – Espectro de reflectância difusa da amostra de trigo
Ä
Ä Reflectância ⇒⇒ razão entre a intensidade de radiação refletida pela amostra (I) e a
refletida por uma substância padrão (I0) (p.ex. BaSO4 finamente dividido)
0
I
I
R
==
(30)Ä
Ä Equipamentos específicos
Figura 5.31– Representação esquemática de um fotômetro de reflectância difusa
Ø
ØDesvantagem ⇒⇒ procedimento de calibração lento e demorado
Ø
5.8.4 – ESPECTROMETRIA NO IV AFASTADO (FAR IR)
Ä
Ä Aplicações
Ø
Ø Estudos de compostos inorgânicos:
•• absorção devida aos estiramentos e deformações das ligações entre os átomos metálicos e ligantes orgânicos ou inorgânicos (ν < 650 cm-1)
••informações sobre energias de retículo cristalino e sobre energias de transição em materiais semicondutores
Ø
ØEspectros rotacionais puros de espécies gasosas
Ä
Ä Equipamentos ⇒⇒ espectrômetros por transformada de Fourrier
5.8.5 - ESPECTROMETRIA DE REFLEXÃO NO IV MÉDIO
Ä
Ä Reflexão da radiação
- especular ⇒ i = r ⇒ superfície plana polida - difusa
- interna
- total atenuada
Seja a incidência de radiação no IV médio sobre uma amostra sólida que absorve radiaçãoIV
absorve IV
Variando-se os ν da radiação incidente observa-se que Ir é menor para os ν que
são absorvidos pelo meio
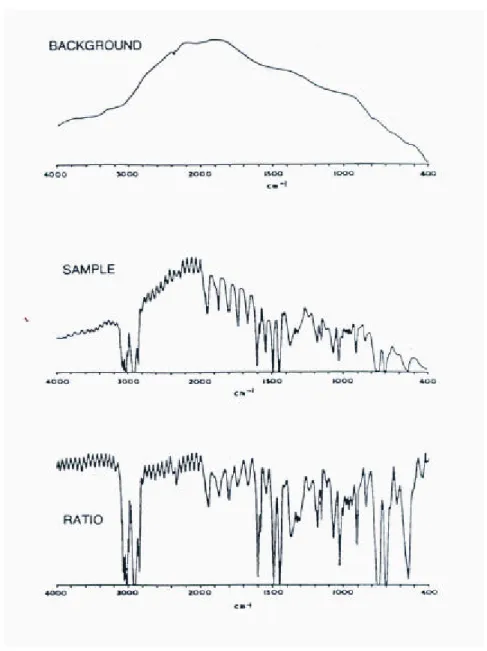
5.8.5.1 - Espectrometria de Reflectância Difusa (DRIFTS)
Ä
Ä Técnica que tem se desenvolvido em função da popularização do FTIR →
intensidade da radiação refletida pelo pó é baixa para ser detectada pelos instrumentos dispersivos
Ä
Ä Reflexão por amostras em forma de pó → processo complexo → cada superfície
plana → reflexão especular → como são várias superfícies → reflexão é orientada randomicamente em todas as direções → intensidade da radiação refletida independe do ângulo em que é observada
Ä
Ä Aplicações
Ø
Ø Análise de amostras em forma de pó com um mínimo de preparo
Ä
Ä Tratamento quantitativo
wKubelka e Munk
s
k
2R'
)
'
R
1
(
)
R
(
f
2==
−−
==
∞ ∞ ∞ ∞ ∞ ∞ (31)sendo: f(R'∞) = reflectância relativa
R'∞) = razão entre a intensidade da radiação refletida pela amostra e a refletida por padrão não absorvente (KCl)
k = coeficiente de absorção molar = 2,303εc s = coeficiente de espalhamento
Ä
Ä Instrumentação ⇒⇒ acessório para espectrômetro FTIR
Ä
Ä Espectro de reflectância ⇒ f(R’∞) x ν
(a) pastilha com KBr (b) 5% de amostra em KCl
- mesma posição dos picos
-intensidades relativas diferentes
Figura 5.33 – Comparação entre os espectros de absorção (a) e de reflectância difusa para o carbazol
5.8.5.2 - Espectrometria de Reflectância Total Atenuada (ATR)
Ä
Ä Princípio do método
η2
Ir/Ii ↑↑ i ↑↑
η1
η2 > η1
No processo de reflexão → feixe atua como se penetrasse uma pequena distância no meio menos denso antes da reflexão ocorrer
Ø
Ø profundidade de penetração
- varia de (λ/x) a xλ - depende de λ, η2, η1 e i
Ø
Ø radiação que penetra no meio ⇒ onda evanescente → sofre atenuação nos
Ä
Ä Aplicações
Ø
Ø amostras com manipulação difícil ⇒⇒ sólidos pouco solúveis, filmes, adesivos, pó,
pastas, fios
Ø
Ø Espectros ⇒⇒ semelhantes aos espectros de absorção convencionais
•• picos → mesmo ν, mas as intensidades relativas mudam •• sinal → depende de i
independe da espessura da amostra
Ø
Ø Vantagem
•• obtenção rápida de espectro de uma variedade de amostras com um mínimo de preparação
Ä
Ä Instrumentação
Figura 5.34 – Acessório para medida de reflectância total atenuada. (a) amostra posicionada na placa de reflexão
(b) sistema posicionado no espectrômetro
Ø
Ø Sólido com η elevado ⇒ TlBr/TlI ou GeSe/ZnSe
Ø
Ø Ângulo de incidência ajustado (i = 30o, 45o ou 60o) → múltiplas reflexões no interior do sólido
Ø