SEGURANÇA E EFICÁCIA CLÍNICA
NA SUBSTITUIÇÃO DE MEDICAMENTOS
BIOSSIMILARES: AVALIAÇÃO CRÍTICA
DA BIBLIOGRAFIA
SAFETY AND CLINICAL EFFICACY IN BIOSIMILAR SUBSTITUTION
Resumo
Os produtos biossimilares tornaram-se uma realidade nos últimos anos. Apesar de estar estabelecido na União Europeia desde 2005 um quadro legal para a aprovação dos biossimilares, bem como da crescente orientação regulamentar para o seu desenvolvimento e autorização, muitos profissionais de saúde continu-am relutantes em considerar estes produtos uma alternativa terapêutica para os seus doentes. Tcontinu-ambém a entrada dos biossimilares no mercado suscitou o debate sobre se a interpermutabilidade destes produtos com os seus respetivos medicamentos de referência se pode realizar de forma segura, sem um impacto nega-tivo na eficácia do tratamento ou um incremento no risco de imunogenicidade. O presente artigo apresenta uma revisão da informação disponível no que concerne à questão da substituição. Foi realizada uma pes-quisa por estudos clínicos pré e pós-aprovação nos quais se tenha procedido à substituição, em indivíduos doentes, entre um biofármaco inovador e um produto biossimilar aprovado e atualmente autorizado pela EMA. Presentemente, a informação disponível relativamente aos potenciais efeitos da substituição é escas-sa. Contudo, os dados obtidos a partir de estudos clínicos em que ocorreu substituição entre biofármacos inovadores e os seus respetivos biossimilares indicam que esta prática não tem impacto na eficácia, segu-rança e imunogenicidade do tratamento dos doentes.
Palavras-chave: Biossimilar, substituição, interpermutabilidade, imunogenicidade, segurança.
Abstract
Biosimilar medicinal products have become a reality in recent years. Despite an established legal pathway for biosimilars in the European Union since 2005 and increasing regulatory guidance on data requirements for their development and approval, many clinicians are still reluctant to consider biosimilars as a treatment option for their patients. Moreover, the approval of biosimilars has prompted the discussion of whether these products can be safely switched with the innovator biopharmaceuticals without a negative impact on the efficacy of the treatment or an increased risk of immunogenicity. This review gives an overview of the available data related to the switch between an originator product and a biosimilar. A search was conducted for pre- and post-approval clinical studies involving patients that were switched between innovator drugs and EMA approved biosimilars. Currently, there is limited clinical data that specifically investigates the effects of switching. Nevertheless, the data analyzed from clinical studies reporting on switching between originator products and their respective biosimilars indicates that switching has no impact on the efficacy, safety and immunogenicity of treatment in patients.
Keywords: Biosimilar, switch, interchangeability, immunogenicity, safety.
7 7
João Pedro Fernandes
Faculdade de Farmácia da Universidade de Lisboa
João Gonçalves
8
1. Introdução
O crescimento exponencial que a área da biotecnolo-gia conheceu nas últimas décadas tem-se refletido nos mais diversos campos da ciência, incluindo na medi-cina. O advento de processos biotecnológicos como a técnica do DNA recombinante ou a tecnologia do hibridoma possibilitou o desenvolvimento e produ-ção de biofármacos, que, por seu turno, vieram revo-lucionar a terapêutica de um conjunto variado de do-enças. Os biofármacos são medicamentos produzidos em organismos vivos, com recurso a métodos biotec-nológicos modernos, tendo a insulina humana sido o primeiro medicamento deste género a ser introduzido no mercado, em 1982. Desde então, mais de duas cen-tenas de outros medicamentos biotecnológicos foram desenvolvidos e comercializados1.
As patentes e os períodos de exclusividade dos pri-meiros medicamentos biotecnológicos já expiraram, abrindo caminho para a entrada no mercado de pro-dutos biossimilares. A EMA (European Medicines Agency) define biossimilares como biofármacos «que contêm uma versão da substância ativa de um medi-camento biológico inovador já autorizado no Espaço Económico Europeu (medicamento biológico de refe-rência), e com o qual demonstraram ter similaridade em termos de qualidade, atividade biológica, seguran-ça e eficácia, com base em estudos comparativos»2. Portanto, os produtos biossimilares são semelhantes, mas não exatamente idênticos, aos medicamentos bio-tecnológicos de referência1.
A penetração dos biossimilares no mercado tem sido mais lenta do que o esperado, o que, em parte, pode ser atribuído à desconfiança dos profissionais de saúde relativamente à qualidade, segurança e eficácia destes produtos. Nomeadamente, a aprovação dos biossimi-lares motivou o debate sobre se estes produtos podem ser utilizados interpermutavelmente com os seus res-petivos medicamentos de referência de forma segura, sem influenciar a eficácia do tratamento ou induzir uma eventual resposta imunitária. Consequentemen-te, levantou-se também a questão sobre se a substitui-ção automática será adequada no caso dos produtos biossimilares3-6. Interpermutabilidade e substituição automática são conceitos diferentes, embora se encon-trem intimamente relacionados6. Um medicamento é considerado interpermutável se for terapeuticamente equivalente a um outro medicamento e se a substitui-ção de um pelo outro, por iniciativa ou com o acordo do médico prescritor, permitir alcançar o mesmo efeito terapêutico, num dado contexto clínico e em qualquer doente. Por sua vez, substituição automática refere-se à prática de substituir, a nível da farmácia, um
medi-camento por outro considerado equivalente e inter-permutável, sem consentimento ou conhecimento do médico prescritor7,8. Atualmente, a larga maioria dos profissionais de saúde opõe-se à substituição entre um medicamento biotecnológico de referência e o seu respetivo biossimilar, por receio dos potenciais efeitos adversos que poderão advir desta prática4,5,9.
O presente artigo tem como propósito analisar estu-dos clínicos disponíveis até à presente data, nos quais se tenha procedido, no decurso de um tratamento, à substituição entre um biofármaco inovador e o seu res-petivo biossimilar. Pretende-se deste modo averiguar se a substituição tem um impacto negativo na eficácia de um tratamento e se está associada a um aumento da incidência de efeitos adversos, e em particular a um incremento do risco de imunogenicidade.
2. Métodos
Foi conduzida uma pesquisa pela literatura biomé-dica, por estudos clínicos em que, no decurso de um tratamento, se tenha procedido à substituição entre um biofármaco inovador e um produto biossimilar aprovado e atualmente autorizado pela EMA. Foram incluídos estudos pré e pós-aprovação nos quais ape-nas tenham participado populações de doentes. A pes-quisa foi realizada eletronicamente no motor de busca Google Scholar e na base de dados PubMed (2004 até ao presente). Esta estratégia de pesquisa foi igualmen-te utilizada para a obigualmen-tenção de documentos relevan-tes acerca de produtos biossimilares. Adicionalmente, acedeu-se aos sítios eletrónicos da EMA e FDA, de modo a consultar guidelines e decisões regulatórias.
Foram também consultados os EPAR dos biossimi-lares atualmente autorizados na União Europeia. Os parâmetros de pesquisa utilizados incluíram «biosimi-lar», «biotechnology», «immunogenicity», «switch» e «interchangeability», assim como a respetiva DCI (de-nominação comum internacional) dos diferentes bios-similares e medicamentos biotecnológicos.
3. Biossimilares Não São Genéricos
As autoridades competentes cedo reconheceram que o conceito de medicamento genérico não poderia ser aplicado de forma análoga aos biofármacos1. Enquanto os fármacos de síntese química são substâncias de baixo peso molecular, estáveis e com uma estrutura bem defi-nida, os medicamentos biotecnológicos apresentam um elevado peso molecular (normalmente compreendido entre os 10 mil e os 150 mil Da), possuem uma estru-tura complexa e são moléculas heterogéneas e relativa-mente instáveis. Como resultado desta complexidade que caracteriza os biofármacos, os métodos analíticos
9 atuais não são suficientemente sensíveis para permitir
a completa caracterização físico-química destes produ-tos. Por este motivo, e ao invés do que se verifica com os fármacos de síntese química, não é possível demonstrar que a substância ativa de um biossimilar seja idêntica à do medicamento biotecnológico de referência. Acresce ainda o facto da substância ativa de um biofármaco não ser constituída por uma única entidade molecular, mas sim por um conjunto de moléculas muito semelhantes entre si (isoformas)4,7,10-12.
A heterogeneidade que caracteriza os medicamentos biotecnológicos é consequência de um intricado pro-cesso de fabrico. Os medicamentos biotecnológicos, devido à sua inerente complexidade e ao facto de serem produzidos em sistemas vivos, são moléculas extrema-mente sensíveis ao seu processo de produção. Pequenas variações em alguma das fases deste processo podem influenciar as propriedades do produto final, e por-tanto afetar a sua atividade, estabilidade e qualidade. A suscetibilidade dos biofármacos às condições de pro-dução torna inevitável que lotes de um mesmo produto apresentem diferenças entre si. A variabilidade é ain-da mais acentuaain-da em medicamentos biotecnológicos produzidos por dois fabricantes distintos, devido às diferenças entre os seus processos de produção. Este é exatamente um dos grandes desafios que se colocam na produção de biossimilares. Um fabricante que preten-da comercializar um biossimilar não poderá reproduzir integralmente o processo de fabrico do biofármaco ino-vador, uma vez que não tem acesso nem aos materiais do fabricante original (como bancos celulares) nem à informação respeitante a determinadas componentes do processo em si, que é conhecimento do proprietário. Deste modo, um fabricante de biossimilares não poderá replicar com exatidão o medicamento biotecnológico de referência. Consequentemente, os biossimilares po-derão exibir perfis de segurança e de eficácia distintos relativamente aos medicamentos inovadores5,7,10,13,14.
4. Preocupações dos Profissionais
de Saúde com os Biossimilarese
a Questão da Interpermutabilidade
A comunidade médica tem-se mostrado relutante em considerar os biossimilares como opção para o trata-mento dos seus doentes, não obstante o menor custo no acesso a estes produtos comparativamente com os medicamentos biotecnológicos inovadores. Uma vez que os biossimilares não são exatamente idênticos aos biofármacos de referência, dúvidas persistem quanto à sua qualidade, segurança e eficácia, apesar da exigen-te regulamentação a que esexigen-tes medicamentos estão sujeitos3,15.
Um dos principais motivos de preocupação dos profis-sionais de saúde com os biossimilares prende-se com a sua imunogenicidade3,10. As questões suscitadas pela comunidade médica baseiam-se na possibilidade des-tas moléculas poderem vir a exibir um padrão de imu-nogenicidade distinto do biofármaco inovador, como resultado das pequenas diferenças subjacentes a estes produtos4. De um modo geral, o desencadeamento de uma resposta imunitária contra os produtos biofarma-cêuticos raramente tem um impacto clínico significa-tivo16. Normalmente, os anticorpos produzidos têm carácter não neutralizante, pelo que não afetam a ativi-dade biológica da proteína terapêutica. No entanto, por vezes, estes anticorpos podem alterar as propriedades farmacocinéticas dos produtos biofarmacêuticos16-18. As implicações clínicas podem ser mais severas quan-do ocorre a produção de anticorpos neutralizantes, os quais podem levar à perda de eficácia dos biofármacos. A consequência mais grave dos anticorpos neutrali-zantes resulta da sua capacidade de também se ligarem às proteínas endógenas, com efeitos extremamente ne-fastos para os doentes. A situação mais comummente descrita é a dos casos de PRCA (pure red cell aplasia) que
se observaram entre 1998 e 2003 em indivíduos tra-tados com uma nova formulação do Eprex®/Erypo® (eritropoietina recombinante), a qual desencadeou a produção de anticorpos neutralizantes também dirigi-dos contra a eritropoietina endógena, provocando uma anemia grave nestes doentes16,18.
Também o facto de os biossimilares serem autoriza-dos segundo uma via de aprovação abreviada, aliado à reduzida experiência clínica com estes produtos, con-tribui para a desconfiança em torno dos mesmos4. De facto, a informação disponível no momento da aprova-ção dos biossimilares é escassa, uma vez que os dados existentes estarão limitados aos estudos de compara-bilidade até então realizados3,4,15.
A comunidade médica tem igualmente levantado ques-tões acerca da possibilidade dos biossimilares poderem ser utilizados de forma interpermutável com os respe-tivos medicamentos biotecnológicos de referência3-6. Atualmente, a larga maioria dos profissionais de saúde opõe-se à substituição entre biofármacos inovadores e biossimilares na prática clínica4,5,9. Esta posição é fundamentada pelas incertezas quanto à equivalência terapêutica dos biossimilares em relação aos medi-camentos de referência, como também pelo receio de que a administração de diferentes produtos biofarma-cêuticos durante o tratamento possa aumentar o risco de ocorrência de efeitos adversos, nomeadamente o desenvolvimento de uma resposta imunitária5,6,15. De facto, pensa-se que um doente previamente exposto
10
a uma determinada proteína terapêutica possa de-senvolver uma pré-sensibilização que influenciará o desencadeamento de uma resposta imunitária se, em terapêuticas subsequentes, lhe for administrado um produto biofarmacêutico similar, mas não exatamen-te idêntico, àquele que anexatamen-teriormenexatamen-te foi utilizado no tratamento19. Uma vez que são escassos os estudos científicos em que esta questão tenha sido investiga-da, certas sociedades médicas recomendam a adminis-tração do mesmo biofármaco durante a totalidade do tratamento de um determinado doente5.
5. Regulamentação
dos Biossimilares
5.1. European Medicines Agency
A União Europeia foi pioneira no estabelecimento de um quadro legal para uma via de aprovação abre-viada para os produtos biossimilares. As autoridades reguladoras rapidamente reconheceram que, devido à complexidade que caracteriza os biofármacos, a via de aprovação estabelecida para os medicamentos genéri-cos não poderia ser aplicada aos biossimilares, tendo, por esta razão, sido instituída uma via de aprovação diferenciada para estes produtos10.
A EMA desenvolveu e publicou guidelines onde constam
os requisitos que os biossimilares devem cumprir de modo a serem aprovados no espaço europeu. Um pro-duto biossimilar deverá demonstrar ser semelhante ao medicamento de referência em termos de qualidade, segurança e eficácia através de um extenso exercício de comparabilidade, o qual inclui uma vasta bateria de testes pré-clínicos e estudos clínicos. Será necessário comprovar, com recurso a métodos analíticos apro-priados, que as propriedades físico-químicas, o perfil de pureza e a atividade biológica in vitro do biossimilar
são semelhantes aos do biofármaco inovador7,12,20. Os resultados obtidos na avaliação da qualidade do bios-similar irão determinar a extensão e natureza dos estu-dos não clínicos in vivo e estudos clínicos7,20,21. Assim, a informação necessária a submeter para a aprovação de um biossimilar será menos extensa do que a necessária para a aprovação de um biofármaco inovador7.
Os estudos realizados até ao momento da aprovação são insuficientes para identificar todos os potenciais efeitos adversos de um produto biossimilar. Princi-palmente, estes estudos não permitem prever com certeza a imunogenicidade destes medicamentos. Por este motivo, o requerente de uma AIM (autorização de introdução no mercado) para um produto biossi-milar deve, juntamente com a sua aplicação, apresen-tar um risk management plan, no qual são estabelecidas as
medidas a adotar no sentido de prevenir e minimizar
potenciais riscos de segurança relacionados com o me-dicamento. O requerente compromete-se a estabelecer um sistema de farmacovigilância com o propósito de monitorizar a segurança e a eficácia do produto bios-similar após a sua aprovação8,9,22.
A EMA remete qualquer decisão sobre a interpermu-tabilidade ou a substituição automática entre os me-dicamentos biotecnológicos de referência e respetivos biossimilares para as autoridades competentes de cada Estado-membro. Para efeitos de aprovação do bios-similar, a EMA não exige a realização de estudos em que seja avaliada a interpermutabilidade entre estes produtos e o medicamento de referência. É recomen-dado, contudo, que a decisão de tratar um doente com o biofármaco inovador ou com um produto biossimilar seja tomada por um profissional de saúde qualificado8. É importante salientar que vários Estados-membros aprovaram legislação que proíbe expressamente a tro-ca de um meditro-camento biotecnológico inovador pelo biossimilar ao nível da farmácia20.
Até ao momento, a EMA aprovou as seguintes classes de biossimilares: somatropina, eritropoietina, filgrastim, in-fliximab, folitropina-alfa e insulina glargina (Tabela 1). 5.2. Food and Drug Administration
Nos Estados Unidos da América, somente em março de 2010 foi implementada legislação que estabelece uma via de aprovação abreviada para os produtos biossimilares. Esta peça legislativa é conhecida por «The Biologics Price Competition and Innovation Act» (BPCI)23.
A Food and Drug Administration (FDA) é a entidade responsável pela avaliação do pedido de AIM do can-didato a biossimilar11,23-25. Em 2012, foram publicados pela FDA os esboços de três guidelines com o
propósi-to de facultar à indústria orientações e recomendações para o desenvolvimento de produtos biossimilares. Es-tas guidelines baseiam-se nos mesmos princípios
cientí-ficos estabelecidos nas guidelines da EMA11,23.
Ao abrigo do BPCI, a FDA tem autoridade para definir se um produto biossimilar pode ser utilizado em vez do biofármaco inovador na prática clínica23. De acor-do com o BPCI, um produto biossimilar poderá ser utilizado de forma interpermutável com o seu medi-camento de referência caso se cumpram os seguintes critérios: o biossimilar produz o mesmo efeito clínico que o medicamento de referência em qualquer doen-te; um tratamento em que se alterne entre a adminis-tração do biofármaco inovador; e a adminisadminis-tração do biossimilar num mesmo doente tem de ter igual segu-rança e eficácia que um tratamento em que somente se tenha administrado o biofármaco inovador. O BPCI
11 Tabela 1 – Biossimilares atualmente autorizados na Europa
Somatropina
Classe de biofármaco Biossimilar
Nomes comerciais Data de aprovação Biofármaco de referência
Omnitrope Omnitrope® (Sandoz) 2006 Genotropin® (Pfizer)
Eritropoietina
HX575 Abseamed® (Medice Arzneimittel
Pütter), Binocrit® (Sandoz), Epoetin-alfa Hexal® (Hexal)
2007 Eprex/Erypo® (Janssen-Cilag)
SB309 (epoetina-zeta) Retacrit® (Hospira), Silapo®
(Stada Arzneimitte) 2007 Eprex/Erypo® Filgrastim XM02 Biograstim® (AbZ-Pharma), Ratiograstim® (Ratiopharm), Tevagrastim® (Teva) 2008 Neupogen® (Amgen)
EP2006 Zarzio® (Sandoz), Filgrastim
Hexal® (Hexal)
2009 Neupogen®
PLIVA/MayneFilgrastim Nivestim® (Hospira) 2010 Neupogen®
Apo-Filgrastim Gastrofil® (Apotex Europe) 2013 Neupogen®
Infliximab CT-P13 Inflectra® (Hospira) e Remsima® (Celltrion Healthcare)
2013 Remicade®
(Janssen Biologics)
Folitropina-alfa
XM17 Ovaleap® (Teva Pharma) 2013 Gonal-F® (Merck Serono
Europe)
Insulina glargina LY2963016 Abasria® (Eli Lilly) 2014 Lantus® (Sanofi-Aventis)
determina ainda que um produto biossimilar consi-derado interpermutável com o seu medicamento de referência poderá ser utilizado em substituição deste, sem ser necessária a intervenção ou notificação do mé-dico prescritor. Contudo, a FDA não publicou ainda
guidelines que abordem a questão da substituição, nem
estabeleceu métodos e critérios para a realização de estudos que possibilitem avaliar os eventuais efeitos adversos que possam advir desta prática23,26.
6. Estudos Clínicos
No total, foram identificados 16 estudos clínicos nos quais se procedeu à substituição entre um medica-mento biotecnológico de referência e o seu respetivo biossimilar. Destes, três estudos são referentes à subs-tituição entre a somatropina original e o Omnitrope® e oito estudos são relativos a produtos contendo como substância ativa a eritropoietina recombinante (três estudos em que se procedeu à substituição da epoetina inovadora pelo biossimilar HX575 e cinco estudos em que houve substituição entre a epoetina original e o biossimilar SB309). De igual modo, foram
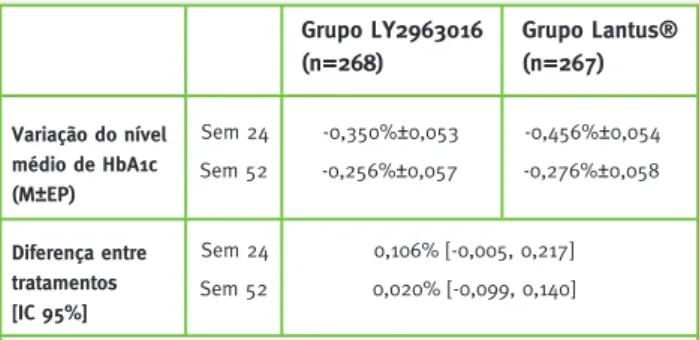
analisados dois estudos em que o filgrastim original foi substituído por um produto biossimilar (em am-bos os estudos o biossimilar avaliado foi o XM02) e dois estudos nos quais o infliximab foi substituído pelo biossimilar CT-P13. É também analisado um es-tudo em que a insulina glargina inovadora foi subs-tituída pelo biossimilar LY2963016. Relativamente à folitropina-alfa, não foram identificados estudos que se inserissem no âmbito deste artigo. Os estudos são descritos em termos de objetivos, métodos, popula-ção de doentes estudada e resultados.
6.1. Somatropina
6.1.1. Estudo AQ, estudo Lyo e estudo López- -Siguero et al (análise post-hoc)27-29
Objetivos: Pretendeu-se com esta análise post-hoc avaliar o impacto da substituição da somatropina original pelo Omnitrope® na eficácia e segurança do tratamento em doentes com deficiência na hormona de crescimento. Métodos: A análise englobou os dados de três ensaios clínicos de fase III: estudo AQ, estudo Lyo e estudo López-Siguero et al.
12
O estudo AQ foi um ensaio clínico de fase III aberto, multicêntrico, randomizado e controlado (15 meses), que integrou o dossiê submetido à EMA para a ob-tenção da AIM do Omnitrope®, e no qual participa-ram inicialmente 89 doentes. Este ensaio foi subdi-vidido em três fases diferentes: numa primeira fase, os participantes foram aleatorizados para receberem tratamento com Omnitrope® pó para solução injetá-vel (n=44) ou com Genotropin® (n=45), durante um período de nove meses, com o propósito de avaliar as potenciais diferenças entre os dois produtos em termos de eficácia e segurança. Numa fase subsequente, os par-ticipantes a quem foi administrado o Genotropin® pas-saram a fazer tratamento com Omnitrope® solução (n=44), enquanto os restantes continuaram a efetuar tratamento com Omnitrope® pó (n=42), durante um período de seis meses. O principal objetivo desta fase foi determinar se existiam diferenças a nível de seguran-ça e eficácia entre as duas formulações de Omnitrope®. Na fase final do estudo, que se estendeu por 69 meses e cujo objetivo era avaliar a segurança do Omnitrope® a longo prazo, todos os participantes passaram a efetuar tratamento com Omnitrope® solução.
O estudo Lyo foi um ensaio clínico de fase III, aberto, multicêntrico e não controlado, que também integrou o dossiê submetido à EMA para a obtenção da AIM do Omnitrope®, e cujo objetivo foi avaliar a segurança e eficácia a longo prazo deste biossimilar. Neste estu-do, 51 doentes receberam tratamento com Omnitrope® pó para solução injetável durante 54 meses.
Por último, o estudo López-Siguero et al, à semelhan-ça do estudo Lyo, foi um estudo clínico aberto, mul-ticêntrico e não controlado, que teve como propósi-to avaliar a eficácia e segurança do tratamenpropósi-to com Omnitrope® solução a longo prazo em 70 doentes, no decurso de 60 meses.
Na análise post-hoc, apenas foram considerados os
pri-meiros 18 meses de cada estudo anteriormente des-crito. A análise consistiu em estabelecer uma compa-ração entre a curva de crescimento médio observada para o grupo Genotropin®/Omnitrope® (estudo AQ) e a curva de crescimento médio dos doentes que fi-zeram tratamento apenas com o Omnitrope® (estu-dos Lyo e López-Siguero et al). Parâmetros como o HSDS (height standard deviation score), HV (height ve-locity) e HVSDS (height velocity standard deviation sco-re) foram igualmente comparados entre os
dife-rentes grupos de tratamento. As concentrações de IGF-1 (insulin-like growth factor 1) e de IGFBP-3 (IGF binding protein 3) foram definidas como endpoints
secun-dários. Para o grupo do estudo AQ, foi ainda elabo-rado um modelo ajustado ao crescimento
observa-do nos primeiros nove meses, no decurso observa-dos quais o Genotropin® foi administrado aos doentes. Este modelo foi utilizado para prever a trajetória de cresci-mento dos participantes para os nove meses seguintes, como se estes tivessem continuado a fazer tratamento com Genotropin®. Os valores de altura previstos pelo modelo foram depois comparados com os resultados observados durante o tratamento com Omnitrope®. A segurança do tratamento foi avaliada em termos de in-cidência de efeitos adversos e imunogenicidade. Nos três estudos, o tratamento com hormona de cresci-mento recombinante (rhGH) consistiu em 0,03 mg/kg por dia, administrado por via subcutânea (SC). Participantes: Foram avaliados nesta análise os da-dos do grupo de doentes do estudo AQ que, após lhes ter sido administrado o Genotropin® durante nove meses, passaram a ser tratados com o Omnitrope® solução (n=45), como também os dados dos partici-pantes dos restantes dois estudos (n=51 e n=70, para o estudo Lyo e estudo López-Siguero et al, respetiva-mente). Eram elegíveis para os estudos anteriormen-te descritos apenas crianças pré-adolescenanteriormen-tes com deficiência na hormona de crescimento, cuja altura e taxa de crescimento (avaliada num intervalo de pelo menos seis meses antes da participação no estudo) fossem, no mínimo, de dois desvios padrão (DP) e de um DP, respetivamente, abaixo da média para o seu sexo e idade. As características demográficas e antro-pométricas dos doentes que participaram nestes três estudos eram, no geral, semelhantes entre si, apesar de ligeiras diferenças na idade e género entre parti-cipantes dos diferentes estudos. Nenhum doente in-cluído nestes ensaios tinha efetuado tratamento com rhGH anteriormente ao início dos estudos.
Resultados
Eficácia: As curvas de crescimento médio para os di-ferentes grupos de tratamento foram semelhantes en-tre si. De igual modo, a evolução dos valores médios de HV e HVSDS registados ao longo do estudo foi comparável entre o grupo Genotropin®/Omnitrope® e os doentes que fizeram tratamento continuado com Omnitrope®, apesar de se terem observado diferen-ças não significativas no pico de HV e HVSDS. Estas diferenças podem ser atribuídas aos distintos perfis de idade e género entre os participantes dos vários estudos. Também os valores médios de HSDS e as concentrações de IGF-1 e IGFBP-3 foram semelhan-tes entre os diferensemelhan-tes grupos de tratamento.
No grupo do estudo AQ, o crescimento observado durante o período de tratamento com Omnitrope® foi consistente com o crescimento previsto pelo modelo. Segurança: Não foram reportadas quaisquer reações
13 adversas relacionadas com a substituição. Também
não se detetou um incremento no risco de se desen-volverem anticorpos anti-GH como consequência da substituição do Genotropin® pelo Omnitrope®. De facto, a presença de anticorpos anti-GH em doentes do grupo Genotropin®/Omnitrope® só foi deteta-da em dois ou mais testes consecutivos num único participante durante o período de tratamento com Omnitrope®. Deve-se igualmente salientar que o tra-tamento com Omnitrope® no estudo Lyo e no estudo López-Siguero et al foi bem tolerado pelos doentes. Relativamente a esta análise post-hoc, contudo, é
im-portante referir que, para o fim a que se propunha, teria sido mais apropriada a comparação entre um tratamento em que houve substituição com um trata-mento contínuo com a somatropina original.
6.1.2. Estudo Flodmark et al30
Objetivos: Este estudo investigou o impacto de subs-tituir a somatropina inovadora pelo Omnitrope® na segurança e eficácia do tratamento em doentes com diversos distúrbios de crescimento.
Métodos: A altura e o HSDS foram monitorizados in-dividualmente em cada doente durante o decurso do tratamento. Adicionalmente, foi utilizado um modelo para prever a trajetória de crescimento de cada par-ticipante, como se estes tivessem continuado o tra-tamento com a somatropina inovadora. As previsões efetuadas pelo modelo foram posteriormente com-paradas com o crescimento efetivamente observado após a substituição para o Omnitrope®.
Participantes: Participaram no estudo 98 doentes com uma idade compreendida entre um e 15 anos e os seguintes diagnósticos: deficiência na hGH (n=40); sín-drome de Turner (n=9); sínsín-drome de Prader-Willi (n=6); pequena estatura para a idade gestacional (n=11); ou- tros (n=36). Portanto, a população analisada neste es-tudo era relativamente heterogénea quanto ao distúr-bio de crescimento e estádio pubertário.
Resultados
Eficácia: A análise da altura e do HSDS obtidos du-rante o período do tratamento com Omnitrope® permitiu concluir que a substituição não teve um impacto negativo na velocidade de crescimento dos participantes. Também o crescimento observado após a substituição pelo Omnitrope® foi consistente com o crescimento previsto pelo modelo.
Segurança: Não foram reportados eventos adversos severos ou inesperados após a substituição. Porém, 18 doentes queixaram-se de dores no local da injeção, tendo inclusive um deles desenvolvido um edema lo-cal. Destes, seis regressaram ao tratamento inicial.
Não existe referência sobre se a imunogenicidade foi avaliada no decurso deste estudo.
6.1.3. Estudo Ullah et al31
Objetivos: Neste estudo aberto e de braço único pretendeu-se avaliar o custo, segurança e eficácia do tratamento com Omnitrope® comparativamente com o tratamento com Genotropin®.
Métodos: O Genotropin® foi substituído pelo Omni-trope® em indivíduos adultos com deficiência na hor-mona de crescimento. O tratamento com Omnitrope® foi avaliado durante um período de nove meses. Fo-ram comparados os valores do score do AGHDA (adult growth hormone deficiency quality of life questionnaire) e os
níveis de IGF-1 obtidos antes e após a substituição do Genotropin® pelo Omnitrope®. Outros parâme-tros avaliados incluíram a massa gorda dos doentes, a massa magra, o peso, a circunferência da cintura e a pressão arterial.
Participantes: Participaram no estudo 14 indivíduos adultos com deficiência na hormona de crescimento. Resultados
Eficácia: Os doentes mantiveram a mesma dose que se encontravam a fazer com Genotropin®, após a substituição para o Omnitrope®. O score do AGHDA
e os níveis de IGF-1 obtidos após o tratamento com Omnitrope® foram semelhantes aos valores verifica-dos durante o tratamento com o Genotropin® (score
do AGHDA foi de 13,8±6,5 e 11,9±6,3 e as concentra-ções de IGF-1 foram 37,6±16,7 e 38,3±15,9 nmol/L, para o tratamento com Genotropin® e Omnitrope®, respetivamente). Também nos restantes parâmetros avaliados neste estudo, os resultados obtidos após a substituição foram semelhantes àqueles que se obti-veram com a somatropina original.
Segurança: Não foram reportados efeitos adversos relacionados com a substituição. Contudo, não existe qualquer indicação da imunogenicidade ter sido ava-liada no decorrer deste estudo.
6.2. Eritropoietina 6.2.1. HX575
6.2.1.1. Estudo INJ-932,33
Objetivos: O seguinte ensaio clínico de fase III, mul-ticêntrico, randomizado, controlado, de grupos para-lelos e em dupla ocultação (28 semanas), teve como propósito averiguar se o HX575 apresentava uma eficácia e uma segurança comparáveis às do medica-mento de referência Eprex®/Erypo®, em doentes com anemia devido a IRC (insuficiência renal crónica). Este estudo foi parte integrante do dossiê submetido à EMA para obtenção da AIM do HX575.
14
Métodos: Neste estudo, 478 doentes que se encon-travam a fazer tratamento com Eprex®/Erypo® foram aleatorizados para continuarem a receber tratamento com este produto (n=164) ou para passarem a fazer o HX575 (n=314). O estudo teve uma duração total de 56 semanas e foi subdividido em duas partes distintas: a primeira parte do estudo consistiu numa fase inicial de ajustamento da dose e manutenção dos valores de concentração de hemoglobina (Hb) nos participantes (semanas um a 24), à qual se seguiu um período de avaliação (semanas 25 a 28), em que foi determinado o endpoint de eficácia primário. Na segunda parte do
estudo (semanas 29 a 56), todos os participantes pas-saram a fazer tratamento com HX575, com o objetivo de avaliar a segurança a longo prazo deste produto. O tratamento foi administrado a cada doente por via intravenosa (IV), três vezes por semana.
O endpoint de eficácia primário foi definido como a
di-ferença entre a média dos valores de Hb registados no início do estudo e a média dos valores de Hb regista-dos durante o período de avaliação, para cada braço de tratamento. A segurança dos diferentes tratamen-tos foi avaliada em termos da incidência de eventratamen-tos adversos e imunogenicidade.
Participantes: Participaram inicialmente no estudo 478 indivíduos com anemia devido a IRC, a fazer he-modiálise e tratamento com Eprex®/Erypo®. De modo a serem elegíveis para este ensaio, os participantes ti-nham de apresentar um valor de Hb compreendido entre 10 e 13 g/dL nas 12 semanas que antecederam o início do estudo. Outros critérios de inclusão relevan-tes incluíam uma dose IV de Eprex®/Erypo® estável, no mínimo, durante 14 semanas antes do início do es-tudo, a qual não poderia ultrapassar os 300 UI/kg por semana e os doentes tinham de se encontrar a fazer hemodiálise, três vezes por semana, há pelo menos seis meses. As características dos doentes de ambos os braços de tratamento eram semelhantes entre si. Resultados
Eficácia: A avaliação da eficácia do HX575 relativa-mente ao Eprex®/Erypo® foi conduzida na população PP (per protocol), a qual era constituída por 207
par-ticipantes no grupo HX575 e por 118 parpar-ticipantes no grupo Eprex®/Erypo®. A variação observada (em valor absoluto) no nível médio de Hb desde do iní-cio do estudo até ao período de avaliação foi seme-lhante para ambos os braços de tratamento. Também o IC a 95 por cento obtido para a diferença entre os dois tratamentos quanto a este parâmetro se inseriu dentro da margem de equivalência, estipulada em ±0,5 g/dL (Tabela 2). Foram igualmente avaliados outros endpoints secundários nesta primeira parte do
estudo, para os quais o tratamento com HX575 ob-teve resultados semelhantes àqueles que foram re-gistados com o tratamento com Eprex®/Erypo®. Na segunda parte deste estudo, na qual todos os parti-cipantes fizeram tratamento com HX575, os valores médios de Hb para o grupo HX575 (11,6-11,9 g/dL) fo-ram semelhantes aos valores registados para o grupo Eprex®/Erypo®-HX575 (11,5-12,1 g/dL). Do mesmo modo, a percentagem de doentes cujos valores de Hb se mantiveram dentro do intervalo aceitável de 10-13 g/dL foi semelhante para ambos os grupos. Não se registou, igualmente, uma diferença estatistica-mente significativa na dose média semanal de epoe-tina entre os dois braços de tratamento, ao longo do estudo.
Tabela 2 – Resultados para o endpoint primário
do estudo INJ-9 Variação do nível médio de Hb (g/dL) (M±EP) Grupo HX575 (n=207) 0,147±0,092 Diferença entre tratamentos [IC 95%] Grupo Eprex®/Erypo® (n=118) 0,063±0,117 0,084 [-0,170; 0,338]
Abreviaturas: M – média; EP – erro padrão; IC – intervalo de confiança.
Segurança: O perfil de eventos adversos regis-tado foi, na globalidade, semelhante para os dois grupos de tratamento e consistente com o expec-tável para a população de doentes estudada. Dois doentes do grupo HX575 e quatro doentes do grupo Eprex®/Erypo®-HX575 apresentaram níveis tran-sientes de anticorpos não neutralizantes acima do valor limite no ensaio RIP (ribonucleoprotein immuno-precipitation), nas semanas 28 e 42. Os dois doentes do
grupo HX575 já apresentavam níveis de anticorpos elevados desde do início do estudo, indiciando que tinha sido o tratamento prévio com Eprex®/Erypo® a induzir a produção desses anticorpos. Um destes participantes obteve ainda um resultado no limiar do positivo para anticorpos neutralizantes no ensaio NAB (neutralizing antibodies). Contudo, este doente
não apresentou outros sinais sugestivos de PRCA, tendo apenas necessitado de doses mais elevadas de HX575 durante o seu tratamento. Do mesmo modo,
15 os restantes participantes com títulos de
anticor-pos elevados não apresentaram também sinais indi-cativos de PRCA. Um participante, que na segun-da parte do estudo interrompeu o tratamento com Eprex®/Erypo® para passar a ser-lhe administrado o HX575, teve o primeiro resultado positivo no ensaio RIP à semana 56. No entanto, não foi possível apro-fundar qual a causa que conduziu ao aparecimento dos níveis elevados de anticorpos, uma vez que este doente faleceu poucos meses depois. A taxa de mor-talidade foi semelhante para os dois grupos de trata-mento e condizente com a condição médica grave dos participantes. Nenhum óbito foi atribuído às duas medicações em estudo.
6.2.1.2. Estudo Turner et al34
Objetivos: Este estudo prospetivo, observacional e multicêntrico teve como propósito alargar a experi-ência clínica com o HX575 em indivíduos com ane-mia devido a IRC.
Métodos: Os participantes que se encontravam a re-ceber tratamento com outras eritropoietinas recom-binantes passaram a fazer tratamento com HX575, por via IV. O estudo teve uma duração total de seis meses. A eficácia do tratamento com HX575 foi ava-liada segundo a mudança observada no valor médio de Hb e no valor da dose média de HX575 registados no início do estudo, comparativamente com os respe-tivos valores registados no final do mesmo.
Participantes: Foram incluídos no estudo 326 doen-tes com anemia devido a IRC a fazerem hemodiálise e tratamento prévio com epoetinas. Destes 326 parti-cipantes, 63 já se encontravam a receber tratamento com o HX575.
Resultados
Eficácia: O valor médio de Hb aumentou de 11,2 g/dL no início do estudo para 11,52 g/dL ao fim dos seis meses. Registou-se um aumento da dose média de HX575 administrada aos participantes, de 7795 UI no início do estudo para 8246 UI no final do mesmo. A eficácia global do HX575 foi classificada como «mui-to boa»/«boa» por 88,1 por cen«mui-to dos investigadores. Segurança: O tratamento com HX575 foi bem tole-rado e não foi reportada a ocorrência de anticorpos neutralizantes.
6.2.1.3. Estudo EPO-PASS32,35,36
Objetivos: O estudo EPO-PASS foi um ensaio clínico de segurança pós-aprovação, prospetivo, multicên-trico e de braço único, realizado no âmbito do RIP submetido à EMA para o HX575. O principal objetivo deste estudo era monitorizar a incidência de eventos
adversos relacionados com o tratamento com HX575 em doentes com anemia devido a IRC, com especial enfoque na imunogenicidade, no desenvolvimento de tumores e na ocorrência de episódios de trombose vascular. Pretendeu-se igualmente avaliar a eficácia do tratamento com HX575 nos doentes que, previa-mente ao início do estudo, se encontravam a fazer tratamento com outras epoetinas.
Métodos: Todos os participantes que se encontra-vam a receber tratamento com outras eritropoieti-nas recombinantes passaram a fazer tratamento com HX575, por via IV, três vezes por semana. O estudo teve uma duração total de seis meses. Os endpoints de
eficácia estipulados foram a proporção de doentes com um valor de Hb entre 10 e 12 g/dL ao longo do estudo, a dose média semanal de HX575 e a evolução do valor médio de Hb durante o estudo. No decurso do ensaio foram efetuados ajustamentos na dose de HX575, de modo a manter os valores de Hb dos parti-cipantes dentro do intervalo 10-12 g/dL.
Participantes: Participaram inicialmente neste estu-do 1695 estu-doentes, sujeitos ou não a diálise. Do número total de participantes, 98,1 por cento encontravam-se a fazer hemodiálise e 92,3 por cento estavam a fazer tratamento com uma determinada eritropoietina re-combinante (incluindo epoetina-alfa, epoetina-beta e darbepoetina) previamente ao início do estudo, por via IV ou por via SC. Destes, 14,9 por cento estavam já a ser tratados com o HX575. Os doentes eram elegí-veis para participarem no estudo mesmo se os valores de Hb e as doses de eritropoietina recombinante uti-lizadas no seu tratamento não se encontrassem está-veis antes do seu início.
Do número inicial de participantes, 1392 completaram o estudo. Destes, 1143 constituíram a população PP. Resultados
Eficácia: A proporção de doentes com um valor de Hb compreendido entre 10 e 12 g/dL aumentou de 57,5 por cento no início do estudo para 66,8 por cento ao fim dos seis meses. Contudo, os autores do estudo não concluíram se estes resultados eram estatistica-mente significativos. O valor médio de Hb manteve-se estável durante todo o período do estudo (11,2 g/dL no início do estudo e 11,3 g/dL aos dois, quatro e seis me-ses). Do mesmo modo, também a dose semanal média de HX575 se manteve estável ao longo do ensaio. Para os 764 participantes que anteriormente ao início do estudo fizeram tratamento com epoetinas por via IV (incluindo o HX575), verificou-se, durante o es-tudo, um aumento pouco significativo (5 por cento) na dose média de HX575, comparativamente com a última dose administrada das outras epoetinas.
16
tudo, nos participantes que fizeram tratamento com outras epoetinas por via SC, o aumento de 13 por cen-to desde da última dose administrada dessas epoeti-nas para a dose de HX575 registada no final do estudo foi já considerado significativo. No entanto, o número de participantes neste subgrupo (n=266) era demasia-do reduzidemasia-do para se poderem retirar quaisquer conclu-sões fundamentadas relativamente à eficácia do HX575. Todos os participantes que fizeram tratamento com epoetinas antes do início do estudo mantiveram o nível médio de Hb dentro do intervalo 10-12 g/dL durante os seis meses. É importante ainda referir que, nos seis meses que antecederam o estudo, vários participan-tes alternaram pelo menos uma vez entre diferenparticipan-tes epoetinas durante o seu tratamento. O tratamento com HX575 obteve resultados semelhantes nos sub-grupos de doentes que experimentaram uma, duas ou mais que duas substituições (incluindo a substitui-ção para o HX575 no início do estudo).
Segurança: Os eventos adversos observados foram condizentes com o expectável para a população em estudo e com o perfil de segurança conhecido para as epoetinas. Foram reportados episódios de trombose venosa, relacionados e não relacionados com o trata-mento, em 11,9 por cento dos participantes. Nenhuma das neoplasias diagnosticadas durante o estudo (1,4 por cento) foi atribuída ao HX575. Do mesmo modo, nenhum dos óbitos registados durante o estudo se deveu ao HX575, no entendimento dos investigado-res. Não foram reportados casos de participantes que tenham desenvolvido anticorpos anti-epoetina. 6.2.2. SB309
6.2.2.1. Estudos 04-05, 04-04 e 04-14 (análise post-hoc)37-40
Objetivos: Pretendeu-se com esta análise post-hoc avaliar o impacto da substituição da epoetina-alfa inovadora pela epoetina-zeta (ou vice-versa) na efi-cácia e segurança do tratamento em doentes com ane-mia causada por IRC.
Métodos: A análise englobou os dados obtidos em três ensaios clínicos de fase III (dois ensaios de efi-cácia e um ensaio de segurança), os quais integraram o dossiê submetido à EMA para a obtenção da AIM do SB309.
O primeiro ensaio clínico de eficácia (04-05) foi um estudo multicêntrico, randomizado, controlado, de grupos paralelos e com dupla ocultação, que teve como objetivo estabelecer a equivalência terapêu-tica da epoetina-zeta relativamente ao medicamen-to biotecnológico de referência, Eprex®/Erypo®, na correção dos níveis de Hb em 609 doentes anémicos.
Os participantes foram aleatorizados para serem tra-tados com SB309 (n=305) ou com Eprex®/Erypo® (n=304), uma a três vezes por semana por via IV, du-rante 24 semanas. As doses foram ajustadas ao longo do estudo de modo a obter nos participantes um va-lor de Hb estável, compreendido entre 11 e 12 g/dL. O segundo ensaio clínico de eficácia (04-04), por sua vez, foi um estudo multinacional, randomizado, controlado, de crossover e com dupla ocultação,
reali-zado com o propósito de demonstrar a equivalência terapêutica entre a epoetina-zeta e o Eprex®/Erypo® na manutenção dos níveis de Hb em 313 doentes. O estudo compreendeu inicialmente um período de
run-in, com uma duração de 12 a 16 semanas, durante
o qual foi administrado o Eprex®/Erypo® a todos os participantes. Em seguida, os doentes foram aleatori-zados para receberem tratamento com SB309 (n=155) ou com Eprex®/Erypo® (n=158), administrado por via IV, uma a três vezes por semana, durante 12 semanas. Após estas 12 semanas, os participantes trocaram de tratamento e fizeram a outra epoetina durante mais 12 semanas. Não houve período de washout entre os
dois tratamentos.
Todos os doentes que completaram o estudo de corre-ção e o estudo de manutencorre-ção tinham a possibilidade de entrar no estudo de segurança (04-14). Este estu-do aberto e não controlaestu-do teve como principal obje-tivo avaliar a segurança a longo prazo do tratamen-to com epoetina-zeta. Foram incluídos neste ensaio de segurança 745 dos 922 doentes que inicialmente tinham participado nos dois estudos anteriores. A epoetina-zeta foi administrada uma a três vezes por semana, durante 56 semanas (grupo geral) ou duran-te 108 semanas (subgrupo de participanduran-tes). Apenas as primeiras 12 semanas de tratamento deste estudo foram abrangidas na análise.
Apenas foram considerados para a análise post-hoc os
participantes dos três ensaios clínicos anteriormente referidos que completaram os respetivos tratamentos sem desvios significativos do protocolo. Deste modo, foram analisados os dados de 481 doentes. Para o pro-pósito desta análise, os participantes foram divididos em quatro grupos diferentes: o grupo 1 (n=118) foi for-mado pelos participantes do estudo de manutenção que nas primeiras 12 semanas fizeram tratamento com Eprex®/Erypo® e que nas restantes 12 semanas do es-tudo passaram a fazer tratamento com epoetina-zeta; o grupo 2 (n=121) foi constituído, por sua vez, pelos participantes que iniciaram o estudo de manutenção com epoetina-zeta e que depois alternaram para o tra-tamento com Eprex®/Erypo®; o grupo 3 (n=101) con-sistiu nos participantes que nas 12 últimas semanas
17 do estudo de manutenção fizeram tratamento com
Eprex®/Erypo® e que em seguida, no ensaio clínico de segurança, cumpriram 12 semanas de tratamento com epoetina-zeta; o grupo 4 (n=242) foi formado por participantes do estudo de correção que fizeram trata-mento com Eprex®/Erypo® (24 semanas) e que depois cumpriram 12 semanas de tratamento com epoetina-zeta no ensaio clínico de segurança.
É importante notar que os participantes que ini-ciaram o estudo de manutenção com epoetina-zeta e que depois passaram a fazer tratamento com o Eprex®/Erypo® (grupo 2), voltaram, no estudo de se-gurança, a mudar de tratamento para a epoetina-zeta (grupo 3), razão pela qual foram avaliados duas vezes para cada endpoint.
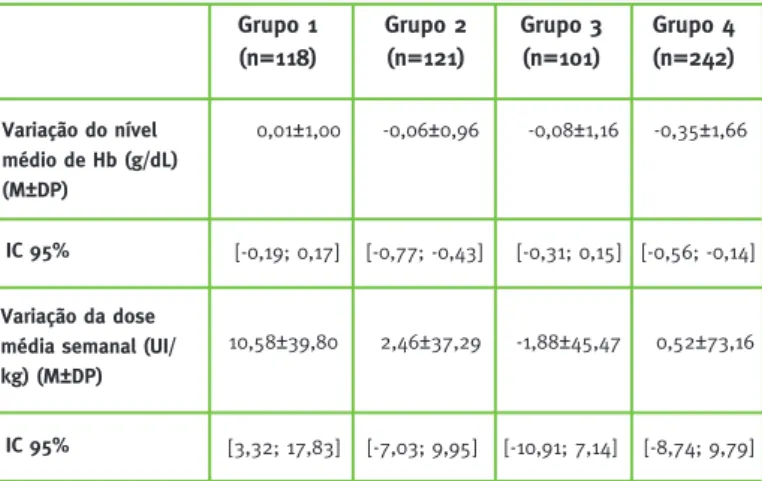
Foram estipulados como endpoints a variação no valor
médio de Hb e a variação na dose média semanal de epoetina, após cada substituição. Também os valores médios de Hb deveriam manter-se compreendidos entre 10,5 e 12,5 g/dL após cada troca de tratamento. Estes parâmetros foram avaliados, em cada grupo de participantes, nas quatro semanas que antecederam cada substituição e nas últimas quatro semanas do período de 12 semanas que se seguiu a cada troca de epoetina. A incidência de eventos adversos nos parti-cipantes foi igualmente avaliada nestes períodos. Os dados obtidos nas primeiras oito semanas após cada substituição não foram, portanto, considerados na aná-lise, uma vez que estes poderiam ter sido influenciados pelo efeito carry-over dos tratamentos anteriores.
Participantes: Os 481 doentes incluídos nesta análise apresentavam anemia resultante de uma IRC e encon-travam-se a fazer hemodiálise. Eram elegíveis para o estudo de correção (04-05) apenas doentes com va-lores de Hb <9 g/dL, quer tivessem ou não efetuado anteriormente tratamento com epoetinas. De igual modo, poderiam participar no estudo de manutenção (04-04) somente os doentes cujos valores de Hb se en-contrassem compreendidos entre 10,5 e 12,5 g/dL (com uma dose de epoetina estável) no final do período de
run-in e sem uma variação intra-individual do nível de
Hb >0,6 g/dL nas quatro semanas que antecederam a randomização. Os critérios de exclusão relevantes para o estudo de segurança (04-14) incluíam níveis detetáveis de anticorpos neutralizantes ou presença de tumores malignos. Não existiam diferenças signi-ficativas nas características demográficas dos doen-tes de cada grupo.
Resultados
Eficácia: Os resultados de cada grupo de participan-tes são apresentados na Tabela 3. Os valores de Hb mantiveram-se dentro do intervalo estabelecido de
10,5-12,5 g/dL após cada substituição efetuada. O IC a 95 por cento para variação no valor médio de Hb não ultrapassou, em cada grupo de participantes, os limi-tes da margem de equivalência, definida em ±1,0 g/dL. Do mesmo modo, o IC a 95 por cento obtido para a diferença entre as doses médias semanais de epoetina antes e após cada substituição, inseriu-se dentro da margem de equivalência, estipulada em ±45 UI/kg. É também importante referir os resultados obtidos após a conclusão do ensaio de segurança. Os parti-cipantes que completaram as 56 (grupo geral) ou 108 (subgrupo de participantes) semanas deste es-tudo mantiveram os níveis médios de Hb dentro do intervalo aceitável, não se tendo observado perda ou falta de eficácia do tratamento.
Segurança: O perfil de eventos adversos registado foi semelhante para os quatro grupos de participan-tes. Não existiu qualquer evidência que sugerisse que algum dos eventos adversos observados tivesse ocor-rido como consequência da alternância entre as di-ferentes epoetinas durante o tratamento. Não foram reportados casos de participantes que tenham desen-volvido anticorpos anti-epoetina ou PRCA.
De igual modo, o tipo e frequência de eventos adver-sos observados até ao final do ensaio de segurança foi consistente com o perfil de segurança já conhecido das epoetinas, não tendo também sido reportados quaisquer casos de imunogenicidade.
6.2.2.2. Estudo 07-0837,41,42
Objetivos: O estudo 07-08 foi um ensaio clínico de fase III, multicêntrico, randomizado, controlado, de grupos paralelos e com ocultação do observador, que teve como propósito avaliar se o tratamento com epoetina-zeta, quando administrado por via SC, tem semelhante eficácia e segurança comparativamen-te ao tratamento com o medicamento de referência, Eprex®/Erypo®, na manutenção dos níveis de Hb em doentes com anemia devido a IRC. Os resultados des-te estudo foram submetidos à EMA com o objetivo de se obter a aprovação da via SC para o tratamento da anemia na população insuficiente renal crónica. Métodos: O ensaio compreendeu inicialmente um período de run-in com uma duração de 12 a 16 semanas,
durante o qual a epoetina-zeta foi administrada a todos os participantes por via SC. Em seguida, um total de 462 participantes foi aleatorizado para receber tratamento com SB309 (n=232) ou com Eprex®/Erypo® (n=230), ad-ministrado por via SC, uma a três vezes por semana, durante 28 semanas. Após o fim deste período, todos os participantes poderiam continuar a receber trata-mento com epoetina-zeta durante mais 54 semanas
18
Tabela 3 – Variação dos níveis de Hb e da dose média semanal após a substituição e respetivos IC
Variação do nível médio de Hb (g/dL) (M±DP) Grupo 1 (n=118) 0,01±1,00 -0,06±0,96 -0,08±1,16 -0,35±1,66 IC 95% Grupo 2 (n=121)
Abreviaturas: M – média; DP – desvio padrão; IC – intervalo de confiança.
Grupo 3 (n=101) Grupo 4 (n=242) [-0,19; 0,17] [-0,77; -0,43] [-0,31; 0,15] [-0,56; -0,14] Variação da dose média semanal (UI/ kg) (M±DP)
10,58±39,80 2,46±37,29 -1,88±45,47 0,52±73,16
IC 95% [3,32; 17,83] [-7,03; 9,95] [-10,91; 7,14] [-8,74; 9,79]
como parte de uma fase de extensão aberta e não con-trolada, cujo principal objetivo foi avaliar a segurança a longo prazo deste produto.
A avaliação da eficácia da epoetina-zeta relativa-mente ao Eprex®/Erypo® foi conduzida na população PP, a qual era constituída por 154 participantes no grupo epoetina-zeta e por 165 participantes no grupo Eprex®/Erypo®. Estipularam-se como endpoints de
efi-cácia primários o valor médio de Hb dos participantes e a dose média semanal de epoetina durante as últimas quatro semanas de tratamento da fase principal do es-tudo. Os endpoints de segurança incluíram a incidência
de eventos adversos, a ocorrência de anticorpos anti- -epoetina e a tolerabilidade dos doentes ao tratamento. Participantes: Foram incluídos inicialmente no es-tudo 679 doentes. Apenas eram elegíveis para partici-par no ensaio doentes com anemia devido a IRC cujos regimes de diálise se mantivessem estáveis nos três meses que precederam o período de run-in e que se
en-contrassem a receber tratamento com epoetinas tam-bém há pelo menos três meses antes do início desta primeira fase do estudo. De modo a entrarem na fase principal do ensaio, os valores de Hb dos doentes, no final do período de run-in, deveriam estar
compreen-didos entre 10 e 12 g/dL (com uma dose de epoetina estável) e a variação intra-individual do nível de Hb não poderia ser superior a 0,5 g/dL nas quatro sema-nas que antecederam a randomização. Participaram na fase principal do estudo, no total, 486 doentes. Por sua vez, a fase de extensão incluiu 346 participantes. Resultados
Eficácia: O valor médio de Hb durante as últimas quatro semanas de tratamento foi de 10,94±0,84 g/dL e de 11,02±0,94 g/dL para o grupo epoetina-zeta e
Eprex®/Erypo®, respetivamente. O IC a 95 por cento para a diferença entre os níveis mé-dios de Hb registados para cada tratamento foi [-0,28 g/dL; 0,12 g/dL], o qual se encontra-va dentro da margem de equiencontra-valência, definida em ±0,5 g/dL. Por sua vez, a dose média sema-nal de epoetina durante o mesmo período foi de 97,0±94,3 UI/kg por semana e de 86,0±78,0 UI/kg por semana para o grupo epoetina-zeta e Eprex®/Erypo®, respetivamente. O IC a 95 por cento para a diferença entre as doses mé-dias semanais de epoetina registados para cada tratamento foi [-8,06 UI/kg por semana; 29,96 UI/kg por semana], o qual se encontrava dentro da margem de equivalência, definida em ±45 UI/kg por semana. Foram avaliados outros
endpoints secundários, para os quais não foram
observadas diferenças estatisticamente signifi-cativas entre os dois braços de tratamento.
Durante a fase de extensão, a eficácia do tratamento com epoetina-zeta foi igualmente avaliada. No decurso das 54 semanas, o valor médio de Hb dos participantes, avaliado mensalmente, oscilou entre 10,7 e 11,2 g/dL, mantendo-se, portanto, dentro do intervalo aceitável de 10-12 g/dL. Também a proporção de doentes cujo valor médio de Hb ultrapassou os limites deste intervalo foi semelhante à registada durante a fase principal do estudo.
Segurança: O perfil de eventos adversos observado foi semelhante para os dois grupos de tratamento e consis-tente com o perfil de segurança já conhecido das epo-etinas. Não foram detetados anticorpos anti-epoetina nem observados quaisquer sinais clínicos de PRCA nos participantes durante o decurso de todo o ensaio, incluindo a fase de extensão. A tolerabilidade dos do-entes para ambas as medicações em estudo foi, na larga maioria dos casos, reportada como boa ou excelente. Deve-se salientar que se verificou, na fase principal do estudo, uma disparidade no número de óbitos entre os diferentes grupos de tratamento. Registaram-se nes-ta fase do ensaio 23 óbitos, dos quais 16 ocorreram no grupo epoetina-zeta e sete no grupo Eprex®/Erypo®. Contudo, apenas dois óbitos (um em cada grupo de tratamento) foram considerados como estando rela-cionados com a medicação em estudo.
6.2.2.3. Estudo Bajraktar et al43
Objetivos: Este estudo, conduzido em dois centros de diálise, avaliou a segurança e eficácia do tratamen-to com epoetina-zeta, na manutenção dos níveis de Hb em doentes com anemia devido a IRC.
Métodos: O estudo consistiu num período de run-in com uma duração de dois meses, após o qual se
dos participantes compreendidos entre 10,5 e 12 g/dL. A ocorrência de eventos adversos e os níveis de Hb foram monitorizados mensalmente.
Participantes: Participaram no estudo 33 doentes com anemia devido a IRC e a fazerem hemodiálise. An-teriormente ao início do estudo, 30 dos 33 participan-tes estavam a fazer tratamento com outras epoetinas. Resultados
Eficácia: O tratamento com epoetina-zeta revelou-se eficaz na manutenção dos níveis de Hb dos par-ticipantes dentro do intervalo alvo. A dose média de epoetina-zeta permaneceu estável ao longo dos seis meses de tratamento.
Segurança: Os eventos adversos reportados foram consistentes com o expectável para a população em estudo e com o perfil de segurança conhecido das epo-etinas. Não foram detetados anticorpos anti-epoetina nem observados quaisquer sinais clínicos de PRCA nos participantes deste estudo.
6.2.2.4. Estudo Lonnemann et al44
Objetivos: Este estudo observacional, conduzido num centro clínico alemão, teve como objetivo deter-minar a eficácia da epoetina-zeta na manutenção dos níveis de Hb em doentes com anemia devido a IRC. Métodos: Após um período de run-in com a duração de dois meses, a epoetina-zeta foi administrada a to-dos os participantes, via IV, durante seis meses. Os níveis de Hb e a ocorrência de eventos adversos foram monitorizados mensalmente.
Participantes: Participaram no estudo 18 doentes com anemia devido a IRC e a fazer hemodiálise. Destes, 17 encontravam-se a receber tratamento com outras epoetinas anteriormente ao início do estudo.
Resultados
Eficácia: Não se observaram diferenças significativas entre os valores médios de Hb registados antes do início do tratamento com epoetina-zeta (11,72±0.64 g/dL) e os valores registados no final dos seis meses do estudo (11,62±0,70 g/dL). Do mesmo modo, não existiu uma diferença estatisticamente significativa entre a dose média semanal das epoetinas administradas aos parti-cipantes antes do início do estudo (79,4±57,7 UI/kg por semana) e a dose média semanal de epoetina-zeta ad-ministrada no final do mesmo (91,8±65,4 UI/kg por semana).
Objetivos: No seguinte estudo observacional, con-duzido num centro clínico em Itália, pretendeu-se avaliar o impacto da substituição de CERA® (meto-xi-polietilenoglicolepoetina-beta) pela epoetina-zeta no custo, eficácia e segurança do tratamento em do-entes com anemia devido a IRC.
Métodos: Os participantes foram submetidos a tra-tamento com epoetina-zeta, por via IV, após terem sido tratados com CERA® durante um período de pelo menos dez meses. Os níveis de Hb foram moni-torizados mensalmente.
Participantes: Participaram no estudo 12 doentes com anemia devido a IRC.
Resultados
Eficácia: Não foram observadas diferenças estatistica-mente significativas entre o tratamento com CERA® e o tratamento com epoetina-zeta no que respeita aos valores de Hb registados. Contudo, foi necessário um aumento da dose de epoetina-zeta durante o trata-mento, de modo a manter estáveis os níveis de Hb. A dose média de epoetina-zeta aumentou dos 6500 UI por semana iniciais para os 7000 UI por semana. Segurança: Não foram reportados quaisquer eventos adversos relacionados com a substituição nem casos de imunogenicidade.
6.3. Filgrastim
6.3.1. Estudo XM02-03-INT46-48
Objetivos: O estudo XM02-03-INT foi um ensaio clínico de fase III, multicêntrico, multinacional, ran-domizado e controlado, que teve como objetivo de-monstrar que o tratamento com o XM02 tem uma eficácia e segurança semelhante ao tratamento com o medicamento de referência, Neupogen®. Este estudo foi parte integrante do dossiê submetido à EMA para obtenção da AIM do XM02.
Métodos: Neste estudo, 240 doentes com cancro do pulmão foram aleatorizados para fazerem tratamen-to com XM02 (n=160) ou com Neupogen® (n=80) no primeiro ciclo de quimioterapia. Nos ciclos subse-quentes, todos os participantes fizeram tratamento com XM02, até um total de seis ciclos. O XM02 ou o Neupogen® (no primeiro ciclo) foram administra-dos por via SC (5 µg/kg por dia), durante pelo menos cinco dias, até um máximo de 14 dias. O tratamento com G-CSF (fator estimulante do crescimento das colónias de granulócitos) era interrompido quando
20
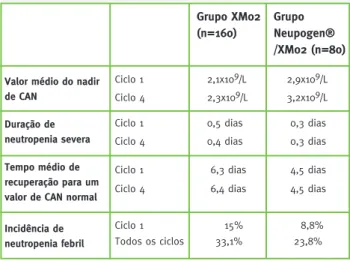
se atingisse um valor de CAN >10x109/L (contagem absoluta de neutrófilos), após o nadir. A eficácia dos tratamentos foi avaliada segundo o valor mé-dio do nadir de CAN, a duração de neutropenia se-vera nos doentes (definida como o número de dias em que os participantes apresentavam um valor de CAN <0,5x109/L) e o tempo médio de recuperação para um valor de CAN normal. Estes parâmetros foram avaliados no ciclo 1 e no ciclo 4. Foi ainda estipulado como endpoint de eficácia a incidência de neutropenia
febril nos participantes durante o estudo. Contudo, o objetivo primário deste estudo era avaliar a seguran-ça do tratamento com XM02, quanto à incidência de eventos adversos e imunogenicidade.
Participantes: Foram incluídos no estudo 240 doen-tes com cancro do pulmão. A população de doendoen-tes que participou neste estudo era relativamente hetero-génea quanto às suas características. Nomeadamente, foram incluídos no estudo participantes que nunca fizeram quimioterapia, como também participantes que já tinham anteriormente efetuado um regime de quimioterapia. Deve-se ainda referir que os partici-pantes neste estudo nunca tinham efetuado anterior-mente tratamento com G-CSF recombinantes. Resultados
Eficácia: Os resultados são apresentados na Tabela 4. Registaram-se diferenças estatisticamente signifi-cativas entre os diferentes braços de tratamento no valor médio do nadir de CAN no ciclo 1 de quimiote-rapia e no tempo médio de recuperação para um valor de CAN normal, também no ciclo 1.
Segurança: O perfil de eventos adversos registados foi semelhante para os dois grupos de tratamento e consistente com o perfil de segurança já conhecido dos G-CSF. Não foram reportados quaisquer casos de imunogenicidade. No entanto, é necessário ter em consideração que este estudo foi conduzido numa população de doentes imunodeprimidos, pelo que a ausência de casos de imunogenicidade deve ser inter-pretada com cuidado.
6.3.2. Estudo XM02-04-INT46,48
Objetivos: O estudo XM02-04-INT foi um ensaio clínico de fase III, multicêntrico, multinacional, ran-domizado e controlado, cujo objetivo foi demonstrar que o tratamento com XM02 tem uma eficácia e se-gurança semelhante ao tratamento com o biofármaco inovador, Neupogen®. Este estudo foi também parte integrante do dossiê submetido à EMA para obten-ção da AIM do XM02.
Métodos: Neste estudo, 92 doentes com linfoma não Hodgkin agressivo foram aleatorizados para fazerem
Tabela 4 – Resultados para os endpoints de eficácia do
estudo XM02-03-INT
Valor médio do nadir de CAN Duração de neutropenia severa Grupo XM02 (n=160)
Grupo Neupogen® /XM02 (n=80) Ciclo 1 2,1x109/L 2,9x109/L Tempo médio de recuperação para um valor de CAN normal
Ciclo 1 6,3 dias 4,5 dias
Incidência de neutropenia febril
Ciclo 1 15% 8,8%
Ciclo 4 2,3x109/L 3,2x109/L
Ciclo 1 0,5 dias 0,3 dias
Ciclo 4 0,4 dias 0,3 dias
Ciclo 4 6,4 dias 4,5 dias
Todos os ciclos 33,1% 23,8%
tratamento com XM02 (n=63) ou com Neupogen® (n=29) no primeiro ciclo de quimioterapia. Nos ci-clos subsequentes, todos os participantes fizeram tratamento com XM02, até um total de seis ciclos. O XM02 ou o Neupogen® (no primeiro ciclo) foram administrados por via SC (5 µg/kg por dia), durante pelo menos cinco dias, até um máximo de 14 dias. O tratamento com G-CSF era interrompido quando se atingisse um valor de CAN >10x109/L, após o nadir. À semelhança do estudo XM02-03-INT, o objetivo principal deste ensaio clínico era avaliar a segurança do tratamento com XM02. De igual modo, os endpoints
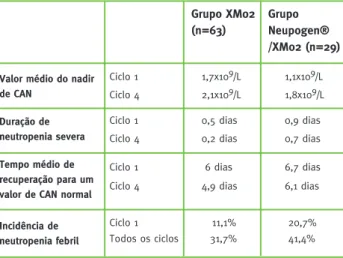
de eficácia estipulados para o estudo XM02-04-INT foram os mesmos que os estabelecidos no estudo XM02-03-INT.
Participantes: Foram incluídos no estudo 92 doen-tes com linfoma não Hodgkin agressivo. As caracte-rísticas dos participantes de cada grupo eram seme-lhantes entre si. Nenhum participante tinha efetuado quimioterapia anteriormente ao início do estudo nem tratamento com G-CSF recombinantes.
Resultados
Eficácia: Os resultados são apresentados na Tabela 5. Não foram registadas diferenças estatisticamente significativas entre os dois braços de tratamento para nenhum dos endpoints de eficácia.
Segurança: O perfil de eventos adversos registados foi semelhante para os dois grupos de tratamento e consistente com o perfil de segurança já conhecido dos G-CSF. Não foram reportados casos de imunoge-nicidade. Contudo, este estudo foi conduzido numa população de doentes imunodeprimidos, pelo que a ausência de casos de imunogenicidade deve ser inter-pretada com cuidado.
21 Tabela 5 – Resultados para os endpoints de eficácia
do estudo XM02-04-INT
Valor médio do nadir de CAN Duração de neutropenia severa Grupo XM02 (n=63)
Grupo Neupogen® /XM02 (n=29) Ciclo 1 1,7x109/L 1,1x109/L Tempo médio de recuperação para um valor de CAN normal
Ciclo 1 6 dias 6,7 dias
Incidência de neutropenia febril
Ciclo 1 11,1% 20,7%
Ciclo 4 2,1x109/L 1,8x109/L
Ciclo 1 0,5 dias 0,9 dias
Ciclo 4 0,2 dias 0,7 dias
Ciclo 4 4,9 dias 6,1 dias
Todos os ciclos 31,7% 41,4%
Resultados
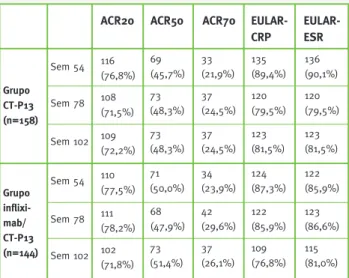
Eficácia: No final do estudo PLANETAS (semana 54), a proporção de doentes que registaram melhorias, se-gundo os critérios de resposta ASAS partial remission,
ASAS20 e ASAS40, foi semelhante para o tratamen-to com CT-P13 e para o tratamentratamen-to com Remicade®. Do mesmo modo, a taxa de participantes que atingiu cada um dos critérios de resposta, às semanas 78 e 102 da fase de extensão, foi semelhante para o grupo CT-P13 e para o grupo infliximab/CT-P13 (Tabela 6).
Tabela 6 – Resultados de cada grupo de tratamento para os critérios de resposta ASAS partial remission,
ASAS20 e ASAS40 às semanas 54, 78 e 102
Grupo CT-P13 (n=88) ASAS partial remission
ASAS20 Sem 54 18 (20,5%) 62 (70,5%) 51 (58,0%) Sem 78 19 (21,8%) 61 (70,1%) 50 (57,5%) Sem 102 23 (27,7%) 67 (80,7%) 53 (63,9%) ASAS40 Grupo infliximab/CT-P13 (n=86) Sem 54 17 (19,8%) 65 (75,6%) 46 (53,5%) Sem 78 18 (21,7%) 64 (77,1%) 43 (51,8%) Sem 102 22 (28,2%) 60 (76,9%) 48 (61,5%)
Segurança: A incidência de eventos adversos foi mais elevada no grupo infliximab/CT-P13 do que no grupo CT-P13. Contudo, a proporção de doentes em que foram reportados eventos adversos severos foi seme-lhante em ambos os grupos de tratamento. Também a incidência de reações adversas relacionadas com a infusão foi semelhante para ambos os grupos de do-entes. É de salientar ainda a ocorrência de um caso de tuberculose em cada braço de tratamento.
O número de doentes nos quais foram detetados an-ticorpos direcionados contra a medicação em estudo foi semelhante para ambos os braços de tratamento. No grupo CT-P13 foram detetados anticorpos em 20 doentes à semana 54 e em 21 doentes nas semanas 78 e 102. Por sua vez, no grupo infliximab/CT-P13, 22, 25 e 23 doentes apresentaram resultados positivos para anticorpos às semanas 54, 78 e 102, respetivamente. 6.4.2. Estudo PLANETRA (fase de extensão)51,52
Objetivos: Este estudo de extensão aberto teve como objetivo avaliar o impacto da substituição do inflixi-mab pelo CT-P13 na eficácia e segurança do tratamen-to em doentes com artrite reumatratamen-toide, que tenham completado o ensaio PLANETRA. Pretendeu-se igualmente avaliar a segurança e eficácia a longo pra-zo do tratamento com CT-P13.
6.4. Infliximab
6.4.1. Estudo PLANETAS (fase de extensão)49,50
Objetivos: Este estudo de extensão aberto teve como objetivo avaliar o impacto da substituição do infli-ximab pelo CT-P13 na eficácia e segurança do tra-tamento em doentes com espondilite anquilosante, que tenham completado o estudo PLANETAS. Pre-tendeu-se igualmente avaliar a segurança e eficácia a longo prazo do tratamento com CT-P13.
Métodos: O estudo PLANETAS foi um ensaio clíni-co de fase I, prospetivo, multicêntriclíni-co, multinacio-nal, randomizado, de grupos paralelos e com dupla ocultação, com uma duração de 54 semanas, reali-zado com o propósito de demonstrar que CT-P13 é semelhante ao infliximab em termos de farmacoci-nética, segurança e eficácia. Neste estudo, os parti-cipantes foram aleatorizados para receberem trata-mento com CT-P13 ou para fazerem tratatrata-mento com Remicade® (5 mg/kg, via IV).
Após completarem o estudo PLANETAS, 174 doen-tes entraram na fase de extensão: 88 doendoen-tes conti-nuaram a fazer tratamento com CT-P13, enquanto os restantes 86 doentes interromperam o tratamento com Remicade® para passarem a receber também o CT-P13. Esta fase de extensão teve uma duração to-tal de 48 semanas e a medicação em estudo foi ad-ministrada a cada oito semanas. Estipulou-se como
endpoint de eficácia a proporção de doentes que
res-ponderam ao tratamento segundo os critérios da Assessment of SpondyloArthritis International Society (ASAS partial remission, ASAS20 e ASAS40). A
segurança do tratamento foi avaliada quanto à inci-dência de eventos adversos e imunogenicidade. Participantes: Participaram nesta fase de extensão 174 doentes com espondilite anquilosante.
![Tabela 2 – Resultados para o endpoint primário do estudo INJ-9 Variação do nível médio de Hb (g/dL) (M ± EP) Grupo HX575(n=207)0,147±0,092 Diferença entre tratamentos [IC 95%] Grupo Eprex®/Erypo®(n=118) 0,063±0,1170,084 [-0,170; 0,338]](https://thumb-eu.123doks.com/thumbv2/123dok_br/17024593.766035/8.892.457.808.559.718/tabela-resultados-endpoint-primário-estudo-variação-diferença-tratamentos.webp)