Métodos Computacionais Aplicados no Estudo de
Fármacos
Métodos Computacionais Aplicados no Estudo de
Fármacos
Tese submetida à Coordenação do Curso de Pós-Graduação em Física, da Universidade Fe-deral do Ceará, como requisito parcial para a obtenção do grau de Doutor em Física
Orientador:
Alejandro Pedro Ayala
UNIVERSIDADE FEDERAL DO CEARÁ - DEPARTAMENTO DE FÍSICA
Fortaleza
Métodos Computacionais Aplicados no Estudo de
Fármacos
Tese submetida à Coordenação do Curso de Pós-Graduação em Física, da Universidade Fe-deral do Ceará, como requisito parcial para a obtenção do grau de Doutor em Física
Aprovada em 16/09/2011
BANCA EXAMINADORA
Profa. Dra. Silvete Coradi Guerini
Departamento de Física, Universidade Federal do Maranhão
Prof. Dr. Waldeci Paraguassu Feio
Departamento de Física, Universidade Federal do Pará
Prof. Dr. Paulo de Tarso Cavalcante Freire
Departamento de Física, Universidade Federal do Ceará
Prof. Dr. José Alves de Lima Júnior
A formação de um profissional da área de física demanda muito esforço. Não somente do estudante, mas também de todas as pessoas envolvidas, órgãos financiadores e universidade. Tendo em vista as condições sócio-econômicas da maior parte da população de nosso país, acredito que a formação de nível superior e principalmente de nível de pós-graduação é um privilégio de poucos.
Classifico o ato de agradecer todos que de alguma forma contribuíram para minha for-mação profissional como extremamente prazeroso, já que ao longo dos anos, muito do que sou e tenho é devido em grande parte ao esforço de outros.
Agradeço à CAPES pela provisão da bolsa de doutorado. Também agradeço ao Grupo de Espalhamento de Luz da UFC pelo suporte, pela disposição dos laboratórios, equipamentos e pessoal, que foram essenciais durante a realização desse trabalho.
Também agradeço ao professor Josué Mendes Filho, por ser um grande incentivador, por compartilhar a sua vasta experiência científica e por seu esforço incessante de proporcionar o ambiente adequado para a proliferação do conhecimento científico no Departamento de Física da Universidade Federal do Ceará.
Não me permito esquecer de agradecer queridos professores que tanto contribuíram durante esses mais de 10 anos que me dedico à Física: Francisco Erivan de Abreu Melo, Paulo de Tarso Cavalcante Freire, Maria Marlúcia Freitas Santiago, Gil de Aquino Farias, Eloneid Felipe Nobre e Antônio Gomes Souza Filho.
Agradeço em especial ao professor Alejandro Pedro Ayala por acreditar no meu potencial, pela orientação acadêmica, pela dedicação constante e concelhos valiosos.
Nesse trabalho realizamos cálculos ab initio em dois fármacos, benzonidazol e
Lista de Abreviaturas
Lista de Figuras
Lista de Tabelas
1 INTRODUÇÃO p. 16
2 CÁLCULOS DE PRIMEIROS PRINCÍPIOS p. 21
2.1 DFT . . . p. 21
2.1.1 Aproximação de Born-Oppenheimer . . . p. 21
2.1.2 Equação de Schrödinger . . . p. 22
2.1.3 Teorema de Kohn e Hohenberg . . . p. 23
2.1.4 Funcional de Correlação e Troca . . . p. 26
2.1.5 Cálculo de Frequências . . . p. 26
2.1.6 Intensidade dos Espectros Raman e Infravermelho . . . p. 29
2.2 Dinâmica Molecular . . . p. 30
2.2.1 Teoria de Car-Parrinello . . . p. 31
3 MODOS NORMAIS DE VIBRAÇÃO p. 34
3.3 Coordenadas Internas . . . p. 38
3.3.1 Estiramento . . . p. 39
3.3.2 Deformação no Plano . . . p. 40
3.3.3 Torção . . . p. 42
3.3.4 Deformação Fora do Plano . . . p. 43
3.4 Equação Secular em Coordenadas Internas . . . p. 43
3.5 Aplicação da Matriz L para Obter a Distribuição da Energia Potencial . . . . p. 46
3.5.1 Mudança de Sistema de Coordenadas . . . p. 48
3.6 PEDCALC . . . p. 48
3.6.1 Coordenadas Naturais . . . p. 49
3.6.2 Identificação de Coordenadas Redundantes . . . p. 52
3.6.3 Constantes de Força . . . p. 53
3.6.4 Esquema de Cores para oPED. . . p. 55
3.6.5 Esquema do cálculo doPED . . . p. 57
4 BENZONIDAZOL p. 59
4.1 Introdução . . . p. 59
4.2 Estrutura Cristalina . . . p. 62
4.3 Conformação Geométrica . . . p. 64
4.4 Espectroscopia Vibracional . . . p. 65
4.4.1 Procedimento de Classificação dos Modos Normais de Vibração . . . p. 66
4.5 Distribuição da Energia Potencial . . . p. 78
5 MEBENDAZOL p. 89
5.1 Introdução . . . p. 89
5.2 Otimização da Geometria . . . p. 92
5.2.1 Introdução . . . p. 92
5.2.2 Investigações Sobre a Geometria do Mebendazol-C . . . p. 93
5.2.3 Varreduras . . . p. 94
5.2.4 Nova Geometria . . . p. 98
5.2.5 Espectro Vibracional do Mebendazol . . . p. 101
5.3 PEDdo Mebendazol . . . p. 113
5.4 Dinâmica Molecular . . . p. 127
5.4.1 Dinâmica Molecular - Dímero Livre . . . p. 128
5.4.2 Dinâmica Molecular - Novas Condições de Contorno . . . p. 130
5.5 Conclusão . . . p. 132
6 CONCLUSÕES E PERSPECTIVAS p. 134
Apêndice A -- Unidades Atômicas p. 136
Apêndice B -- Notação das coordenadas naturais p. 137
CPMD Dinâmica molecular de Car-Parrinello
MBZ Fármaco mebendazol
MBZ-C Polimorfo C do fármaco mebendazol
LDA Aproximação de densidade local
GGA Aproximação generalizada do gradiente
DFT Teoria da funcional da densidade
KH Kohn e Hohenberg
KS Kohn e Sham
3.1 Representação da coordenada interna estiramento. . . p. 40
3.2 Representação da coordenada interna deformação no plano. . . p. 40
3.3 Representação da coordenada interna de torção. . . p. 42
3.4 Representação da coordenada de deformação fora do plano. . . p. 44
3.5 Tabela das coordenadas naturais como apresentada na interface gráfica do
softwarePEDCALC. . . p. 50
3.6 Tabela com a contribuição das coordenadas internas para um modo de
vi-bração. . . p. 54
3.7 Vibração em 606 cm−1 da molécula de benzonidazol. . . p. 56
3.8 Esquema doPEDCALC . . . p. 58
4.1 Fórmula estrutural da molécula de benzonidazol. . . p. 61
4.2 Conformação molecular do benzonidazol com cada átomo identificado. . . p. 62
4.3 Cela unitária do benzonidazol. . . p. 63
4.4 Ligações intermoleculares entre moléculas doBZN. . . p. 64
4.5 Superposição das conformações moleculares teórica (laranja) e
experimen-tal (colorida). . . p. 65
4.6 Funções de ajuste dos espectros Raman calculados. Os gráficos são
mostra-dos para diferentes intervalos de número de onda. . . p. 69
4.7 Funções de ajuste dos espectros infravermelho calculados. Os gráficos são
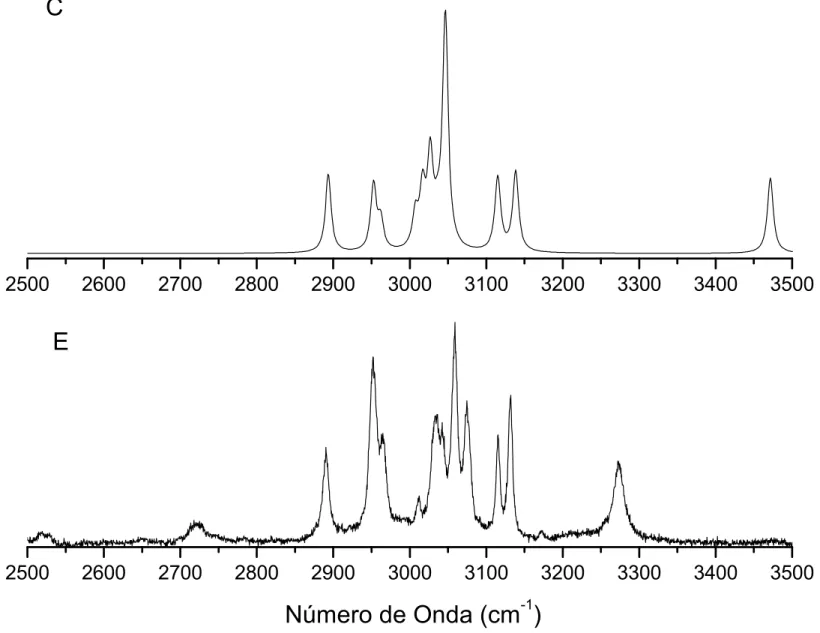
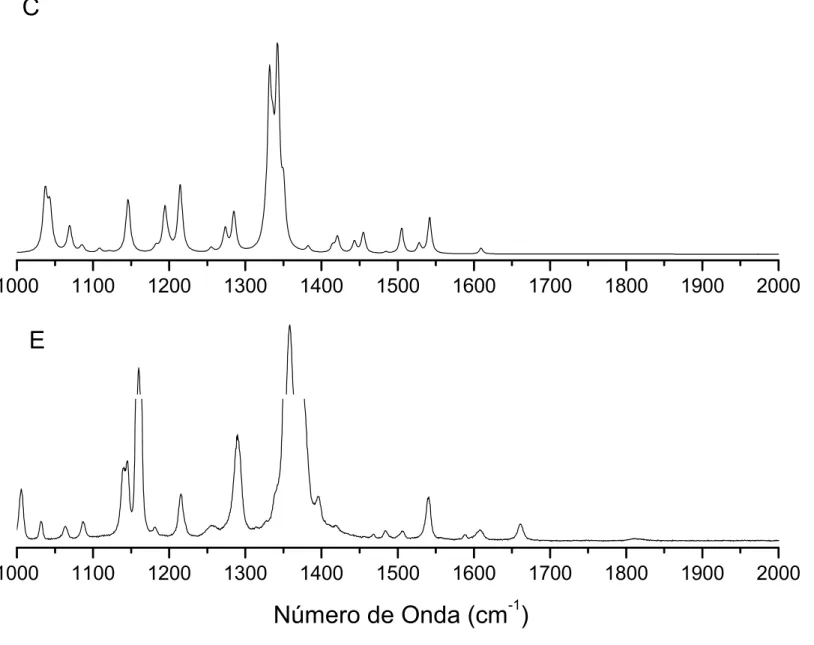
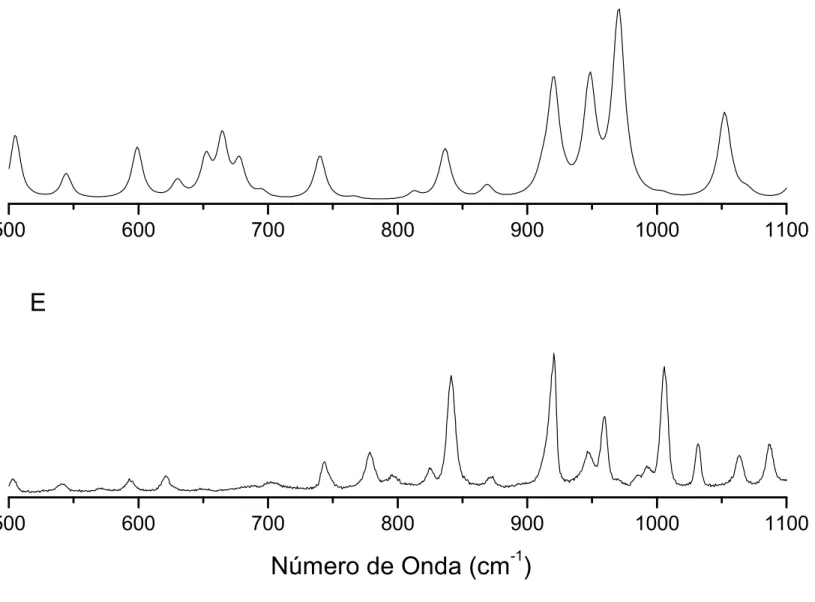
4.9 Espectros Raman calculado (C) e experimental (E) doBZNna região entre
1000 e 2000cm−1. . . p. 72
4.10 Espectros Raman calculado (C) e experimental (E) doBZNna região entre
500 e 1000cm−1. . . p. 73
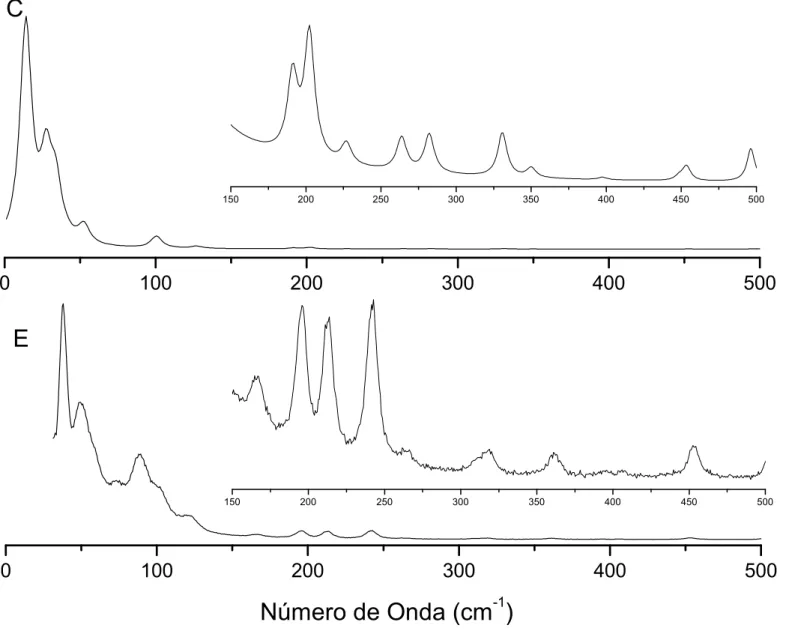
4.11 Espectros Raman calculado (C) e experimental (E) doBZNna região entre
0 e 500cm−1. . . p. 74
4.12 Espectros infravermelho calculado (C) e experimental (E) doBZNna região
entre 2500 e 3600cm−1. . . p. 75
4.13 Espectros infravermelho calculado (C) e experimental (E) doBZNna região
entre 1000 e 2000cm−1. . . p. 76
4.14 Espectros infravermelho calculado (C) e experimental (E) doBZNna região
entre 500 e 1000cm−1. . . p. 77
4.15 Modo vibracional de 3272 cm−1doBZN-G2(ν[N H100]).. . . p. 84
4.16 Modo vibracional de 1661 cm−1doBZN-G2(ν[C N11] +ν[CO72]). . . . . p. 85
4.17 Modo vibracional de 946 cm−1 (R) doBZN- G4(ν[C N12] +δ[ON O45]). . p. 86
4.18 Modo vibracional de 960 cm−1 (R) doBZN- G1 oop[C H99]. . . . p. 86
4.19 Modo vibracional de 38 cm−1 (R) doBZN-G2(
τ[C C36] +τ[C N39]). . . . p. 87
4.20 Modo vibracional de 49 cm−1 (R) doBZN-G2(
τ[C C59]). . . p. 87
5.1 Molécula de mebendazol-C. . . p. 91
5.2 Geometria inicial da molécula de mebendazol-C. . . p. 94
5.3 Variação da energia relativa durante o varredura da torção
C(5)-N(26)-C(1)-O(30). . . p. 95
5.4 Variação da energia relativa durante o varredura da torção
5.6 Tautômeros da molécula deMBZ-C. . . p. 100
5.7 Dímeros tautômeros deMBZ-C. . . p. 101
5.8 Funções de ajuste do dímeroDApara o espectro Raman. . . p. 104
5.9 Funções de ajuste do dímeroDB para o espectro Raman. . . p. 105
5.10 Espectros Raman calculados dos dímeros DA e DB, e experimental (E) do
MBZna região entre 2000 e 3500cm−1. . . p. 106
5.11 Espectros Raman calculados dos dímeros DA e DB, e experimental (E) do
MBZna região entre 1000 e 1800cm−1. . . p. 107
5.12 Espectros Raman calculado dos dímeros DA e DB, e experimental (E) do
MBZna região entre 500 e 1000cm−1. . . p. 108
5.13 Espectros Raman calculados dos dímeros DA e DB, e experimental (E) do
MBZna região entre 0 e 500cm−1. . . p. 109
5.14 Espectros infravermelho calculados dos dímeros DA e DB, e experimental
(E) doMBZna região entre 2000 e 4000cm−1. . . p. 110
5.15 Espectros infravermelho calculados dos dímeros DA e DB, e experimental
(E) doMBZna região entre 1000 e 1800cm−1. . . p. 111
5.16 Espectros infravermelho calculados dos dímeros DA e DB, e experimental
(E) doMBZna região entre 500 e 1000cm−1. . . p. 112
5.17 Modo vibracional de 3404 cm−1doMBZ- G13(
ν[N H31]), G3(ν[N H68]). . p. 114
5.18 Modo vibracional de 2956 cm−1doMBZ- G1-11
νsymmetil[97].. . . p. 115
5.19 Modo vibracional de 1596 cm−1doMBZ-G2-12(ν[CO18]), G5-15(ν[CO30]).p. 116
5.20 Modo vibracional de 608 cm−1 doMBZ-G4
δ6-ring*asym [37]. . . p. 116
5.21 Modo vibracional de 934 cm−1 doMBZ-G4-14 τ[C C11] +oop[C H70].. . p. 117
5.24 Distância dos átomos N63-H48...N59 . . . p. 131
3.1 Principais tipos de coordenadas internas. . . p. 38
3.2 Coordenadas Naturais propostas por Baker. . . p. 51
4.1 Parâmetros geométricos das ligações de hidrogênio. . . p. 64
4.2 Funções de ajuste e regiões do espectro Raman calculado da molécula
iso-lada deBZN. . . p. 67
4.3 Classificação dos modos vibracionais doBZNde acordo com as coordenadas
de simetria . . . p. 78
5.1 Varredura da torção C(5)-N(26)-C(1)-O(30). . . p. 96
5.2 Varredura N(28)-C(5)-N(26)-C(1). . . p. 97
5.3 Ângulos de interesse da primeira otimização de geometria. . . p. 97
5.4 Funções de ajuste e regiões do espectro calculado Raman do dímero DAdo
MBZ-C. . . p. 102
5.5 Funções de ajuste e regiões do espectro calculado Raman do dímero DBdo
MBZ-C. . . p. 103
5.6 Coordenadas Intermoleculares - DímeroDB. . . p. 113
5.7 Classificação dos modos vibracionais do dímero DBdoMBZde acordo com
as coordenadas de simetria. . . p. 117
A.1 Unidades atômicas e fatores de conversão. . . p. 136
B.1 Representação das contribuições das coordenadas naturais para oPED
1
INTRODUÇÃO
A aplicação da Teoria do Funcional da Densidade[1] (DFT) e da dinâmica molecular na física, química[2] e ciência dos materiais é cada vez mais presente. DFT é considerada uma teoria fundamental no cálculo de propriedades de sistemas quânticos de muitos corpos. Enquanto a dinâmica molecular pode nos dar informações valiosas a respeito da dinâmica desses sistemas.
Existem muitas áreas da física e da engenharia onde o progresso científico e tecnológico é limitado somente pelo entendimento das propriedades da matéria a nível atômico. DFT é ferramenta fundamental para obtenção de soluções da equação de Schrödinger, e assim entender o comportamento quântico de átomos e moléculas. DFTé uma teoria de aplicação geral podendo ser aplicado na maioria dos sistemas. Como todos os métodos computaci-onais, DFT é melhor aplicado a um conjunto limitado de problemas: cálculos de reações (termodinâmica, caminho de reações), cálculos envolvendo metais, catálise, etc. A teoria DFTtambém pode ser utilizado para prever geometria e espectros vibracionais de moléculas. Cálculos de otimização de geometria juntamente com cálculos para obtenção de espectros vibracionais são ferramentas padrão na classificação de modos de vibração de moléculas de fármacos.
também são aplicações da dinâmica molecular. A dinâmica molecular aplicada no estudo de biomoléculas investiga propriedades dinâmicas de proteínas, ácidos nucleicos e membranas celulares. Com o auxílio da dinâmica molecular, podemos estudar as propriedades dinâmicas desses sistemas biológicos sem sintetizar essas estruturas, reduzindo custo e tempo. A dinâ-mica molecular é dividida em duas áreas de cálculo: dinâdinâ-mica molecular usando mecânica clássica e aproximações, e dinâmica molecular usando métodos da mecânica quântica. Den-tre os métodos quânticos da dinâmica molecular, destacamos o método Car-Parrinello[3]. A dinâmica molecular de Car-Parrinello é um método eficiente de dinâmica molecular onde o potencial de interação é definido pelo princípio variacional. Ao contrário da dinâmica molecular de Born-Oppenheimer[4] onde os graus de liberdade dos núcleos atômicos são calculados usando forças iônicas que são calculadas a cada iteração com a solução aproxi-mada do problema eletrônico, o método de Car-Parrinello introduz graus de liberdade do problema eletrônico como um conjunto de variáveis dinâmicas e fictícias. Na teoria de Car-Parrinello um sistema de muitos corpos é descrito por um conjunto de equações acopladas, tanto para núcleos atômicos quanto para elétrons.
Na dinâmica molecular de Born-Oppenheimer a minimização do problema eletrônico ao estado fundamental é realizada a cada incremento temporal1. Já na dinâmica molecular de Car-Parrinello a minimização ao estado fundamental é feita uma única vez. A dinâmica fictícia dos elétrons mantem o estado eletrônico fundamental correspondente durante a di-nâmica dos núcleos atômicos. Para que os graus de liberdade dos elétrons não interfiram com os graus de liberdade dos núcleos atômicos, a massa fictícia dos elétrons deve ser suficien-temente pequena, e como consequência a integração das equações de movimento é feita em incrementos temporais muito pequenos2. Na prática, a dinâmica molecular de Car-Parrinello é mais eficiente que outros métodos baseados na aproximação adiabática. Porém, métodos baseados na mecânica clássica ainda são mais eficientes computacionalmente para uma série de problemas que envolvem macro-moléculas, como proteínas. Isso se deve ao fato de que métodos baseados na mecânica clássica somente calculam a dinâmica dos núcleos atômicos.
Ao classificar modos de vibração de uma molécula, calculamos os espectros vibracionais, geralmente usandoDFT. Os espectros calculados são comparados com espectros obtidos
ex-1Um incremento temporal corresponde a um passo na dinâmica molecular.
perimentalmente para que possamos classificar os espectros experimentais em função dos calculados. Geralmente, o processo de classificação dos modos normais consiste em compa-rar os espectros calculados e experimentais usando funções de ajuste, e a partir da visuali-zação dos modos de vibração, determinar quais coordenadas internas contribuem mais para a energia potencial. Esse modelo de classificação dos modos de vibração pode levar a erros difíceis de detectar. Ao visualizar um modo de vibração com o auxílio de umsoftware,
obser-vamos o movimento dos átomos sem quantizar a contribuição de cada coordenada interna à energia potencial. Com esse método de associação, a tendência é que façamos associa-ção da energia potencial com os átomos que mais se movimentam na molécula. Mas em alguns casos os átomos que mais se vibram não são os que mais contribuem para a energia potencial.
Para quantizar a contribuição de cada coordenada interna à energia potencial de um modo de vibração usamos o método da Distribuição da Energia Potencial [5] (Potential Energy Distribution, PED). Esse método calcula a distribuição de probabilidade de cada co-ordenada interna para cada modo de vibração de uma molécula. Sendo assim, podemos associar com segurança quais as coordenadas internas contribuem mais para cada modo de vibração e classificar propriamente os modos de vibração. Nesse Trabalho desenvolvemos um software de cálculo doPEDchamado PEDCALC.
superar as dificuldades encontradas com o uso de GAR2PED e também propor um novo mo-delo de visualização da distribuição da energia potencial baseado em um gradiente de cores no modelo tridimensional da molécula em estudo.
Os métodos de cálculo DFT, Car-Parrinello e PED foram aplicados à classificação dos modos de vibração e dinâmica molecular de dois fármacos: benzonidazol e mebendazol. A geometria otimizada da molécula de benzonidazol e os espectros calculados Raman e infra-vermelho foram obtidos pelo métodoDFTusando o software Gaussian , enquanto a classifi-cação dos modos de vibração foi realizada com o auxílio do softwarePEDCALC. Ao estudar as característica físicas do fármaco mebendazol também utilizamos os softwares Gaussian e PEDCALC para os mesmos propósitos, porém não calculamos otimizações de geometria so-mente para a molécula de mebenzadol, mas também para o dímero de mebenzadol formado a partir de ligações de hidrogênio do tipo N-H...N entre grupos carbamato e benzimidazol de monômeros opostos dos dímeros de mebendazol. Também calculamos dinâmica mole-cular do dímero de mebendazol a fim de mostrar a transferência de prótons nas pontes de hidrogênio N-H...N. A trajetória da dinâmica molecular foi obtida usando o software CPMD (Car-Parrinello Molecular Dynamics) que é uma implementação do método Car-Parrinello.
Os fármacos mebenzadol e benzonidazol são, geralmente, utilizados em regiões onde há precárias condições sanitárias, e existe maior proliferação de doenças parasitárias tropicais. Esses fármacos combatem doenças que podem ser classificadas comodoenças negligenciadas.
Não existem incentivos para o desenvolvimento de pesquisa que levem ao aprimoramento dos fármacos utilizados no tratamento dessas doenças. Os medicamentos utilizados hoje no controle dessas doenças foram, em geral, desenvolvidos há muitos anos. Benzonidazol e me-bendazol são usados para o tratamento de infecções causadas pelos parasitas, Trypanosoma cruzi[13]e nematódeos gastrointestinais, respectivamente.
Trypanosoma cruzi é o parasita que causa à Doença de Chagas[14]. Essa doença foi inicialmente descoberta pelo infectologista Carlos Chagas, em 1909. O principal vetor da Doença de Chagas é o inseto Triatoma Infestans, popularmente conhecido no Brasil como barbeiro. A proliferação do vetor dessa doença se dá em áreas rurais e de florestas. Como
A proliferação da infecção por nematódeos gastrointestinais ocorre em locais onde as condições sanitárias são precárias, incluindo áreas rurais e urbanas. Geralmente, o vetor desses parasitas é o próprio homem. As áreas endêmicas são, em geral, associadas a pobreza e a ausência de políticas governamentais para a saúde pública.
Os capítulos desse trabalho são divididos da seguinte forma: noCapítulo 2discutiremos os métodos de cálculo usados nesse trabalho, incluindo método DFT e dinâmica molecular de Car-Parrinello. Mostraremos os princípios físicos que nos levaram a usar o métodoDFT para calcular otimização de geometria e espectros vibracionais dos fármacos mebendazol e benzonidazol. Também mostraremos as vantagens do método de dinâmica molecular de Car-Parrinello frente os métodos de dinâmica molecular de Born-Oppenheimer. No Capítulo 3 discutiremos a importância da classificação dos espectros vibracionais em termos dos modos normais de vibração. Para isso introduziremos a notação das coordenadas internas, a relação da equação secular com as vibrações moleculares e um método para obter a distribuição da energia potencial em função das coordenadas internas. Também no capítulo 3 mostraremos o software PEDCALC, o método de cálculo usado para detectar redundância no sistema de coordenadas internas, além de um novo método de visualização da distribuição da energia potencial através de uma interface em três dimensões.
No capítulo 4 mostraremos resultados da aplicação do método da análise dos modos normais sobre a molécula do fármaco benzonidazol. Calculamos os espectros vibracionais Raman e infravermelho da molécula de benzonidazol usando a teoriaDFT. Os espectros cal-culados foram ajustados aos espectros experimentais, e a classificação dos modos de vibração foi obtida com o softwarePEDCALC.
2
CÁLCULOS DE PRIMEIROS
PRINCÍPIOS
2.1
DFT
A teoria do funcional da densidade[15–17](Density Funtional Theory,DFT) é uma teoria do estado fundamental da estrutura eletrônica em termos da distribuição da densidade de elétrons. Métodos alternativos aoDFT, como Hartree-Fock[18], são baseados na função de onda de muitos elétrons. A maior vantagem da teoriaDFTfrente a teoria Hartree-Fock é o aumento da precisão nos cálculos computacionais, que depende da escolha do funcional de troca e correlação[19], sem um grande aumento no tempo computacional. Isso se deve ao fato das teorias baseadas na função de onda necessitarem de uma base muito grande para representar as funções de onda, enquanto DFT usa a densidade da nuvem eletrônica para derivar outras grandezas físicas.
2.1.1
Aproximação de Born-Oppenheimer
Dado um sistema de M átomos, uma molécula por exemplo, a propriedade física mais
resolver as equações de movimento dos elétrons usando a Mecânica Quântica. Dada uma configuração dos núcleos atômicos, a solução das equações de movimento dos elétrons nos dá a configuração de menor energia, que chamamos de estado fundamental. A separação
dos núcleos atômicos e dos elétrons em dois problemas distintos é chamada deaproximação de Born-Oppenheimer[20](aproximaçãoBO). Se nosso sistema tem M núcleos atômicos nas
posições R1, ...,RM, então a energia do estado fundamental dos elétrons como função da
configuração dos núcleos atômicos é E R1, ...,RM
. Essa função é conhecida como
superfí-cie adiabática. A superfície adiabática nos diz quanto a energia do estado fundamental dos
elétrons muda de acordo com a configuração dos núcleos atômicos.
Para entender como obter a energia do estado fundamental devemos resolver a equação de Schrödinger.
2.1.2
Equação de Schrödinger
A equação de Schrödinger, independente do tempo e não relativística para um sistema de N elétrons é dada por
Hψ=Eψ (2.1)
onde H é o operador Hamiltoniano, ψ é um conjunto de soluções, ou auto-estados do
sis-tema, e E é um conjunto de números reais que representam a energia do sistema para cada
auto-estado, ou autovalores do sistema. A equação de Schrödinger com o Hamiltoniano expandido, para um sistema de N elétrons interagindo com núcleos atômicos é dada por
h2 2m N X
i=1 ∇2i +
N
X
i=1 V ri
+
N
X
i=1
X
j<i
Uri,rj
ψ=Eψ. (2.2)
O Hamiltoniano é dividido em três termos, em ordem: a energia cinética de cada elétron (m
na equação acima é a massa do elétron), a energia potencial (potencial externo), e interação elétron-elétron. Na equação 2.2 a função de onda é função das coordenadas dos N elétrons,
ψ=ψ r1, ...,rN
, e
Eé a energia do estado fundamental. A função de onda é função das
co-ordenadas dosN elétrons do sistema, mas é possível fazer uma aproximação dessa função de
onda usando o produto de funções de onda de único elétron,ψ=ψ r1
ψ r2,...,
ψ rN.
elé-trons em uma molécula é muito maior que o número de núcleos atômicos, N ≫M. Dessa
forma, o problema quântico demanda maior capacidade de cálculo dos sistemas computaci-onais.
Devido à interação elétron-elétron presente no Hamiltoniano da equação 2.2, os orbitais de único elétron,ψ ri, não podem ser determinados isoladamente sem considerarmos as
interações associadas aos outros elétrons. Com isso, podemos definir a equação de Schrö-dinger para N elétrons como um problema de muitos corpos. Na equação de Schrödinger
as funções de onda não determinam a posição exata dos elétrons, mas sim a probabilidade,
ψ∗i r1, ...,rNψi r1, ...,rN, que os elétrons estejam em um determinado conjunto de
co-ordenadas,r1, ...,rN. Não podemos determinar a posição exata dos elétrons e muito menos
distinguir um elétron do outro. De forma que não é possível mapear a trajetória de elétrons individuais. Então,ψ∗i r1, ...,rN
ψi r1, ...,rN
nos dá a probabilidade de encontrarmos
N
elétrons (independente da ordem) nas coordenadasr1, ...,rN.
As funções de ondaψ ri não são a única maneira de descrevermos um sistema de N
elétrons. Mostraremos que a densidade de carga pode ser usada para determinar unicamente o potencialV rie assim simplificar a descrição do sistema de
N elétrons.
2.1.3
Teorema de Kohn e Hohenberg
A densidade de elétrons em uma posição r é relacionada com a função de onda da seguinte forma
n(r) =2X
i
ψ∗i(r)ψi(r), (2.3)
ondeψ∗i(r)ψi(r)é definido como a probabilidade de que um elétron esteja na posiçãor. A somatória é multiplicada por um fator 2 devido ao princípio de exclusão de Pauli, onde cada função de onda de único elétron pode ser ocupada por dois elétrons de spins diferentes. A densidade de elétrons é uma função de três coordenadas, enquanto a função de onda é uma função de 3N coordenadas. Porém, segundo os dois teoremas desenvolvidos por Kohn e
Hohenberg (KH) a densidade de elétrons tem informação suficiente para resolver a equação de Schrödinger.
primeiro teorema: O estado fundamental de energia da equação de Schrödinger é um funcional única da densidade de elétrons. Dessa forma o primeiro teorema deKH implica que a densi-dade de elétrons do estado fundamental determina todas as propriedensi-dades físicas do estado fundamental: energia, função de onda, etc. Como a densidade de elétrons é uma função de três variáveis, o problema de resolver a equação de Schrödinger passa de um problema de 3N variáveis (no formalismo da função de onda) para um problema de somente três
va-riáveis. Embora o primeiro teorema de KHprove que o funcional da densidade de elétrons existe, não diz como podemos obter esse funcional.
O segundo teorema de Kohn e Hohenberg define uma importante propriedade do funci-onal da densidade de elétrons:o funcional da energia nos dá o estado fundamental de energia do sistema, se e somente se, a densidade de elétrons for a densidade de elétrons do estado
fun-damental. Ou seja, a energia passa a ser a energia do estado fundamental somente se a
densidade de elétrons for a densidade de elétrons do estado fundamental. Se o “verdadeiro” funcional da energia fosse conhecido, bastaria variar a densidade de elétrons até que a ener-gia do sistema fosse mínima. Não existe ainda uma forma analítica do funcional da enerener-gia, mas existem aproximações que podem ser usadas para calcular soluções para a equação de Schrödinger. O funcional da energia pode ser escrito separando os termos conhecidos analiticamente dos termos de troca e correlação:
E[n(r)] =EC[n(r)] +EXC[n(r)] (2.4)
onde EC[n(r)] são os termos cuja forma analítica é conhecida e EXC[n(r)] são todos os outros tipos de interação que não podem ser descritos analiticamente. Os termos de energia conhecidos analiticamente são:
EC[n(r)] = h 2
m
X
i
Z
ψ∗i∇2ψid3r+
Z
V(r)n(r)d3r+e 2
2
Z Z
n(r)n r′ r−r′ d
3r d3r′+E íons,
Kohn e Sham (equaçõesKS). As equaçõesKStêm a forma:
h2
2m∇
2+V(r) +V
H(r) +VXC(r)
ψi(r) =ǫiψi(r) (2.6)
ondeV(r)é o potencial de correlação entre elétrons e núcleos atômicos,VH(r)é o potencial de Hartree eVXC(r)é o potencial de troca e correlação. A solução da equação 2.6 é a função de onda de um único elétron que depende somente de três coordenadas espaciais.
VH(r) =e2
Z
n r′
r−r′ d
3r′. (2.7)
O potencial de Hartree da equação 2.7 descreve a repulsão elétrica entre um elétron (o elétron de índiceiconsiderado na equação 2.6) e todos os elétrons do problema. O potencial
de Hartree inclui termo de “auto-repulsão”, ou seja, termo em que o elétron aplica uma força de repulsão em si mesmo. O termo de auto-repulsão do potencial de Hartree é um efeito não físico desse modelo de cálculo, que é corrigido no termo de troca e correlação. O potencial de troca e correlação pode ser visto como a derivada da energia de troca e correlação:
VXC(r) =
δEXC[r]
δn(r) (2.8)
onde a derivada em questão não é exata, já que não é derivada de uma função simples, mas de um funcional. A solução das equações KS ocorre da seguinte forma: inicialmente um potencial de Hartree deve ser definido, e para definir um potencial de Hartree é necessário conhecer a densidade de elétrons. Para calcular a densidade de elétrons é necessário co-nhecer as funções de onda de único elétron e para calcular essas funções de onda devemos resolver as equaçõesKS. Sendo assim, a solução das equaçõesKSdeve ser obtida de forma iterativa: inicialmente é definida uma densidade de elétrons (não necessariamente a densi-dade de elétrons do estado fundamental), depois as equaçõesKSsão resolvidas tendo como solução as funções de onda de único elétron (ψ(r)), seguido do cálculo da nova densidade de elétrons (n(r) =2X
i
ψ∗i(r)ψi(r)). O processo iterativo termina quando a mudança na
2.1.4
Funcional de Correlação e Troca
Para resolver as equaçõesKSdevemos ter um funcional de troca e correlação,EXC[n(r)]. A teoria de Hohenberg e Kohn garante a existência de um funcional de troca e correlação, mas não diz como obter uma. Existe um caso onde o funcional de troca e correlação é conhecida: o gás de elétrons uniforme. Em um gás de elétrons uniforme a distribuição da densidade de elétrons é uma constante,n(r) =constante. Então, podemos definir o potencial de troca e correlação como:
VXC(r) =VXCgás[n(r)] (2.9)
onde VXCgás[n(r)] é o potencial do gás de elétrons para uma determinada densidade de elé-trons. Essa aproximação usa a densidade local de elétrons para definir o potencial de troca e correlação, e é conhecida comoaproximação de densidade local(local density approximation, LDA). A aproximaçãoLDAnos dá uma solução aproximada da equação de Schrödinger. Para obter a solução exata da equação de Schrödinger seria necessário conhecer o termo de troca e correlação exato.
Existem outros esquemas de aproximação do termo de troca e correlação mais comple-xos. A pesquisa para o desenvolvimento de novas funcionais, que se aproximem mais do “verdadeiro” funcional de troca e correlação, é um campo de pesquisa de grande interesse na física e na química. O conjunto mais conhecido de funcionais é aproximação generalizada do gradiente (generalized gradient approximation, GGA). GGA inclui mais informação do queLDA, já que inclui o gradiente local da densidade de elétrons.
2.1.5
Cálculo de Frequências
Como as vibrações de uma molécula podem ser medidas experimentalmente, é de grande interesse o cálculo das frequências de vibração usando DFT. O espectro vibracional é uma ferramenta importante na classificação e identificação de sólidos e outras substâncias.
Consideremos uma molécula diatômica com átomos fictíciosA1eA2, e a ligação química que mantem esses átomos ligados. A ligação química está sobre o eixo x, logo o estiramento
em função do estiramentos na posição de equilíbrios0 é dada por:
E=E0+ s−s0 d E
ds
s−s0
+1 2 s−s0
2
d2E ds2
s−s0
+... (2.10)
A derivada primeira na equação 2.10 é zero já que a expansão de Taylor é em torno de um ponto de mínima energia. Para pequenos deslocamentos, negligenciamos termos de ordem maior que dois. Essa aproximação é chamada deaproximação harmônica. A expressão
reduzida da energia em função do comprimento do estiramento da ligação química entre os átomosA1e A2 é:
E=E0+
α
2 s−s0
2, (2.11)
ondeα=
d2E ds2
s−s0 .
A equação de movimento do estiramento é:
d2s(t)
d t2 =α
m
2+m1 m2m1
s−s0 (2.12)
onde m1 e m2 são as massas dos átomos A1 e A2, respectivamente. A solução da equação
2.12 é conhecidas=s0+acosωt, ondeaé uma constante arbitrária eω=
r
α
m
2+m1 m2m1
. A frequência de vibração é dada por
ν= ω 2π=
1 2π
r
α
m
2+m1 m2m1
. (2.13)
Para calcular a frequência de vibração da molécula diatômica usandoDFTduas informa-ções são necessárias: o comprimento da ligação química que minimiza a energia da molécula e a segunda derivada da energia. Em cálculosDFTnão dispomos de uma expressão analítica para a segunda derivada da energia, mas podemos aproximar a derivada segunda usando a seguinte expressão:
d2E ds2
s0 ∼
= E s0+δs
−2E s0+E s0−δs
δs02
(2.14)
Além da aproximação dada ao cálculo da frequência na teoriaDFT, a equação de Schrö-dinger não tem solução exata nesse método. Dessa forma, os valores das frequências ob-servados não são iguais aos valores calculados usando o métodoDFT, mas suficientemente próximos para uma boa descrição dos modos de vibração.
Uma solução mais geral para o cálculo das frequências de uma molécula pode ser obtido se considerarmos uma molécula comN átomos, com 3N coordenadas cartesianas associadas
aosN átomos escritas em um vetor,q= q1,...,q3N
. Se o vetor de coordenadas cartesianas
no qual a molécula se encontra em um mínimo local de energia éq0, podemos definir uma nova coordenada como sendox=q−q0. A expansão da série de Taylor em torno da posição de mínimo de energia para uma molécula de N átomos é
E=E0+1 2
3N
X
i=1
3N
X
j=1
∂2E ∂xi∂xj
x=0
xjxj. (2.15)
As derivadas parciais da equação 2.15 podem ser substituídas por uma matriz com 3N
linhas e 3N colunas conhecida como matriz Hessiana, cujos elementos são
Hi j=
∂2E ∂xi∂xj
x=0
. (2.16)
Se considerarmos as forças agindo sobre o sistema deN átomos, temos:
d2x
d t2 =Ax, (2.17)
onde A é uma matriz de 3N linhas e 3N colunas cujos elementos são Ai j=
Hi j
mi
. A solução da equação 2.17 é dada como uma combinação dos modos normais de vibração
x(t) = 3N
X
i=0
aicos(ωt) +bisen(ωt)
ei, (2.18)
onde ai e bi são constantes. Os modos normais de vibração podem ser vistos como uma
descrição completa dos átomos de uma molécula.
deve ser calculada usando a seguinte aproximação:
Hi j=
∂2E ∂xi∂xj
x=0
∼ = E
δxi,δxj
−2E0+E−δxi,−δxj
δxiδxj
, (2.19)
onde Eδxi,δxj
é a energia quando somente as coordenadas xi e xj não são nulas. A
partir dos elementos da matriz Hessiano, a matrizApode ser calculada e por consequência os
autovalores,νi=
p
λi
2π , e os autovetores que por sua vez definem o movimento da molécula
para as 3N−6 frequências.
2.1.6
Intensidade dos Espectros Raman e Infravermelho
Além das frequências dos modos de vibração, para obter o espectro completo pela teoria DFT, geralmente são calculadas as intensidades de espectros Raman e infravermelho.
A espectroscopia Raman é uma técnica baseada no espalhamento inelástico da luz. Como o espalhamento é feito de forma inelástica, a luz monocromática incidente (geralmente laser monocromático) muda após interação com a amostra. Fótons da luz incidente são absorvidos pela amostra e depois emitidos com uma frequência diferente da luz absorvida. A mudança da frequência de luz emitida em relação a frequência da luz absorvida é chamado de efeito Raman.
O efeito Raman é baseado na deformação da distribuição eletrônica determinada pela polarizabilidade α. A luz incidente pode ser considerada como uma onda eletromagnética
com vetor campo elétricoEoscilante, que após interagir com a amostra induz uma polariza-çãoP=αEque é capaz de deformar a distruibuição eletrônica. A luz monocromática, de um laser por exemplo, transforma as moléculas da amostra em dipolos oscilantes. As moléculas excitadas pela luz monocromática podem emitir luz com três frequências diferentes:
Espalhamento Rayleigh: onde a luz incidente de frequência ν0 não é absorvida e é
emitida sem deslocamento de frequência.
Espalhamento Anti-Stokes: esse tipo de espalhamento também é um espalhamento Raman, onde um fóton de frequênciaν0 é absorvido por uma molécula Raman ativa que já
se encontra em um estado excitado de energia. Nesse caso, a molécula emite a energia em excesso em forma de luz com frequênciaν0+νm.
A maior parte do espalhamento de luz, mais de 99,9%, é do tipo espalhamento Rayleigh, enquanto menos de 1% é espalhamento inelástico (Raman ativo).
A intensidade do espalhamento[21–23]é dada por
I=Nl
64π2
3c2 ν0±∆ν
4 Pl m
2
, (2.20)
ondeNl é o número de moléculas no estado iniciall,ν0é a frequência de radiação incidente,
∆ν é o deslocamento da frequência, e Pl m é a probabilidade de transição de um estado
quântico. Quando l=mnão temos o espalhamento elástico de Rayleigh.
Nos cálculos do espectro vibracional usandoDFTa intensidade de espalhamento Raman pode ser estimada com a derivada da polarizabilidade em relação a uma coordenada normal. A intensidade Raman calculada usando a aproximação harmônica é dada por
IRaman= (2π)4
45 ·
h
8π2cωi
· ω0−ωi
4
1−e(−hcωi/kBT)·
Si, (2.21)
ondeSi é o fator de espalhamento Raman calculado em função das derivadas da
polarizabi-lidade.
Moléculas absorvem a radiação infravermelha em frequências características de sua es-trutura. As absorções ocorrem emfrequências ressonantesonde a frequência de vibração da
molécula é a mesma da radiação incidente, sendo que sua intensidade é calculada como a derivada do momento de dipolo em relação a uma coordenada normal.
2.2
Dinâmica Molecular
movi-mento dos núcleos atômicos é geralmente modelado usando-se as leis da Mecânica Clássica. Existem aplicações da Mecânica Quântica na Dinâmica Molecular, mas muitos problemas po-dem ser resolvidos com boa aproximação aos resultados experimentais usando-se somente a Mecânica Clássica.
O método da Dinâmica Molecular foi primeiro introduzido por Alder e Wainwright[24], no final da década de 50, ao estudar a interação de esferas rígidas. Muitos estudos impor-tantes surgiram a partir da contribuição de Alder e Wainwright, principalmente no estudo de líquidos simples. Outra grande contribuição para a Dinâmica Molecular ocorreu em 1964 quando Rahman[25] conduziu a primeira simulação usando um potencial não fictício para argônio líquido. A primeira simulação de uma molécula foi realizada por Rahman e Stillin-ger[26] para água em estado líquido. A primeira simulação de proteína ocorreu em 1977 com a simulação de um inibidor da tripsina pancreática bovina[27]. Hoje, encontramos na literatura uma grande variedade de métodos e aplicações em Dinâmica Molecular.
2.2.1
Teoria de Car-Parrinello
A teoria de Car-Parrinello combina a descrição quântica da dinâmica dos elétrons com a descrição clássica da dinâmica dos núcleos atômicos em uma única estrutura. Devido a grande diferença nas massas e escalas de tempo entre elétrons e núcleos atômicos, podemos separar a dinâmica dos núcleos atômicos e a dinâmica dos elétrons por meio daaproximação adiabática. O movimento dos núcleos atômicos pode ser considerado clássico. Então a
dinâmica dessas partículas é regida pelas equações de Newton, enquanto a dinâmica dos elétrons recebe um tratamento quântico.
cada nova configuração dos núcleos atômicos. Para que a condição adiabática se mantenha durante a dinâmica molecular é necessário que a massa fictícia dos elétrons seja pequena o suficiente para que não haja grande transferência de energia entre os graus de liberdade ele-trônico e iônico. Além disso, as equações de movimento devem ser integradas em pequenos tempos discretos da ordem defs(10−15 segundos).
Car e Parrinello[3]postularam o Lagrangiano dado pela equação abaixo
L=X
i
1 2µ
Z
Ω
d3rψ˙i 2 +X I 1
2MIR˙2I +
X
v
1
2µvα˙2v−E
{ψi},{RI},{αv}, (2.22)
onde os orbitaisψi devem obedecer à condição de ortonormalidade
Z
Ω
d3rψ∗i(r,t)ψj(r,t) =δi j. (2.23)
Os termos a direita da equação 2.22 são, em ordem: a energia cinética dos orbitais (parte eletrônica do problema), a energia cinética dos núcleos atômicos, energia cinética das restrições ao sistema (volume, por exemplo) e energia potencial. OndeMI é a massa de
cada núcleo atômico,µeµv são parâmetros arbitrários.
O Lagrangiano da equação 2.22 gera as seguintes equações de movimento:
µψ¨i(r,t) =−
δE δψ∗i(r,t)+
X
k
Λikψk(r,t), (2.24)
MIR¨i(t) =−∇RiE, (2.25)
µvα¨v=− ∂E
∂ αv
. (2.26)
ondeΛik são os multiplicadores de Lagrange que são introduzidos para satisfazer a condição de ortonormalidade da equação 2.23. A dinâmica dos núcleos atômicos da equação 2.25 tem significado físico, enquanto as dinâmicas dos orbitais,
ψi , e das restrições ao sistema
(como o volume, por exemplo),
αv , são fictícias.
Dadas as seguintes condições: (i) se a massa fictícia µ for suficientemente pequena, e
(ii) se as equações de movimento forem integradas em intervalos de tempo discretos da ordem de f s, na condição de mínimo de energia a Lagrangiana da equação 2.22 descreve
Em termos gerais, uma dinâmica molecular de Car-Parrinello evolui seguindo os seguin-tes passos: (i) a solução das equações KS garante que o sistema esteja no estado funda-mental (os orbitais são minimizados), (ii) forças são aplicadas sobre os núcleos atômicos, atualizando o tempo, ∆t, para os núcleos, ao mesmo tempo, as forças fictícias são
aplica-das sobre os orbitais, atualizando o tempo para os orbitais. O sistema é levado a uma nova configuração sobre a superfície adiabática. (iii) repete o processo a partir do passo (ii).
Cada passo na dinâmica nuclear é acompanhado de um passo na dinâmica eletrônica. A completa minimização dos orbitais ocorre somente no início da simulação. Seµfor pequeno
o suficiente, a nova configuração eletrônica permanece próxima do estado fundamental du-rante a evolução da dinâmica dos núcleos atômicos. Por outro lado, µ deve ser grande o
suficiente para que os incrementos de tempo,∆t, não sejam muito pequenos, resultando em
um menor custo computacional da integração das equações de movimento.
3
MODOS NORMAIS DE VIBRAÇÃO
3.1
Introdução
Os espectros Raman e infravermelho são ferramentas importantes no estudo da maté-ria sólida. Os espectros obtidos de um matematé-rial são função de características estruturais e geométricas das moléculas constituintes da matéria. Sendo assim, entender a estrutura e a geometria das moléculas constituintes da matéria sólida é de grande importância na classifi-cação dos espectros Raman e infravermelho. Podemos, sem exageros, nos referir ao espectro vibracional como “impressão digital” da estrutura molecular da matéria.
Apesar dos avanços tecnológicos da espectroscopia Raman e infravermelho, somente al-gumas moléculas de poucos átomos podem ter seus espectros vibracionais classificados de forma analítica. Existem dificuldades experimentais na classificação dos espectros vibracio-nais, como a sobreposição de linhas espectrais ou a pouca simetria de moléculas. Como con-sequência das dificuldades experimentais, existe um grande interesse no desenvolvimento de técnicas de cálculo que possam determinar as frequências e intensidades de vibração. A
análise das coordenadas normais é uma ferramenta de cálculo das frequências de vibração
através de cálculos de primeiros princípios como cálculosab initiousando a técnicaDFT.
No cálculo do PED usamos o método da mecânica clássica onde as vibrações são cal-culadas usando a aproximação harmônica e as frequências são determinadas pela solução da equação secular que relaciona energia cinética e potencial. Na nossa análise, usamos coordenadas internas que representam os graus de liberdade de uma molécula, menos as translações e rotações da molécula como um todo, ou seja, as coordenadas internas repre-sentam unicamente as vibrações de uma molécula. PEDquantifica a contribuição de cada coordenada interna[29] aos modos vibracionais. Dessa forma, o problema de classificação dos modos vibracionais passa a ser um procedimento simples, principalmente se o cálculo é conduzido com o auxílio do softwarePEDCALC que desenvolvemos durante esse trabalho.
3.2
Equação Secular
Uma molécula não linear tem 3N−6 modos normais de vibração, onde N é o número
de átomos, enquanto uma molécula linear tem 3N−5 modos normais de vibração. Não incluímos nesses números os movimentos de translação e rotação da molécula como um todo. Cada modo normal de vibração pode ser descrito por uma série de coordenadas, podendo ser uma combinação de comprimentos das ligações químicas e ângulos entre essas ligações. Nesse caso, os 3N−6 graus de liberdade do sistema podem ser representados por coordenadas internas ou combinação dessas.
Dentre as vantagens do uso das coordenadas internas destacamos: (i) capacidade de representar com maior clareza os movimentos internos de uma molécula, (ii) facilidade de tranferência, já que não necessitam de um referencial específico.
Os deslocamentos dos átomos em uma molécula a partir da posição de equilíbrio po-dem ser representados pelo conjunto de coordenadas cartesianas X1,Y1,Z1 para o átomo 1, X2,Y2,Z2 para o átomo 2, XN,YN,ZN para o átomo N, ou de maneira mais geral,q1,q2,q3,
..., q3N. Usamos 3N coordenadas porque essas representam os graus de liberdade do
sis-tema. Os átomos podem se movimentar nos três eixos cartesianos. Por conveniência1,
pressamos as coordenadasqi na forma
q1=pm1X1, q2=pm1Y1, q3=pm1Z1, q4=pm2X2, etc. (3.1)
ondem1,m2, ...,mN são as massas dos átomos.
As 3N coordenadasqi, ondei=1, 2, 3, ... 3N, são usadas para expressar a energia
ciné-tica T em termos da derivada temporal
2T= 3N
X
i=1 ˙
q2i onde ˙qi=
dqi
d t . (3.2)
A energia potencial é uma função do deslocamento. Se os deslocamentos forem peque-nos, podemos expandir a energia potencial V em função das coordenadasq em uma série
de potências em torno da configuração de equilíbrio:
2V = 2V0+2 3N
X
i=1
∂V
∂qi
0 qi+
3N
X
i,j=1
∂2V ∂qi∂qj
0
qiqi+...
= 2V0+2 3N
X
i=1 fiqi+
3N
X
i,j=1
fi jqiqi+... . (3.3)
Como temos liberdade para escolher o referencial de energia, para simplificar a expres-são da energia potencial fazemos V =0 na origem. Aplicando a derivada na origem, onde temos um mínimo de energia, teremos:
∂V
∂qi
=0 ⇐⇒ fi=0 i=1, 2, 3, ... 3N.
Para amplitudes de vibração suficientemente pequenas podemos negligenciar os termos maiores que o quadrado deqi, então
2V = 3N
X
i,j=1
fi jqiqj, (3.4)
onde as constantes de força são dadas por
fi j=fji=
∂2V ∂qi∂qj
0
As equações de Euler-Lagrange podem ser escritas como:
d d t
∂T ∂˙qi
+∂V
∂qi
=0 i=1, 2, 3, ... 3N. (3.6)
Podemos simplificar a equação 3.6 com uma substituição simples da energia cinética e energia potencial
¨
qj+
3N
X
i=1
fi jqi=0 j=1, 2, 3, ... 3N. (3.7)
Essa é a equação de um oscilador harmônico de ordem 3N, cuja solução é conhecida
qi=Aicos
λ12t+ε, (3.8)
onde λ, Ai e ε são constantes. Substituindo o valor de qi da equação 3.8 na equação 3.7
obtemos
3N
X
i=1
fi j−δi jλ
Ai=0 j=1, 2, 3, ... 3N, (3.9)
onde δi j=0 quando i6= j, e δi j=1 quando i= j. A equação 3.9 pode ser vista na forma
matricial como
f11−λ f12 ... f1,3N
f21 f22−λ ... f2,3N
... ... ... ...
f3N,1 f3N,2 ... f3N,3N−λ
=0. (3.10)
Na equação 3.10 temos a equação secular na forma matricial em coordenadas cartesia-nas. Com isso podemos aplicar propriedades de determinantes para obter soluções. Devemos observar que devido à maneira que escolhemos as coordenadas
fi j=
Fi j
p
mimj
, (3.11)
onde Fi j é uma constante de força conhecida. Os Fi j são constantes de força geralmente
obtidas através de cálculos ab initio, já os fi j são constantes de força ajustadas para o
con-junto de coordenadasqi de forma que a equação secular não apresente termos explicitos das
molécu-las não lineares seis2 soluções não representam vibrações. Essas soluções em queλ=0 são translações e rotações da molécula como um todo.
3.3
Coordenadas Internas
Os tipos de coordenadas internas mais comuns são mudanças nos comprimentos das li-gações químicas e os ângulos entre essas lili-gações a partir de uma posição de equilíbrio. O uso dessas coordenadas é apropriado, pois a ideia de forças químicas mostra que essas forças produzem uma resistência às mudanças dos comprimentos das ligações químicas e aos ân-gulos entre essas ligações de suas posições de equilíbrio. Os principais tipos de coordenadas internas são apresentadas na tabela 3.1.
Tabela 3.1: Principais tipos de coordenadas internas.
Coordenada Descrição
estiramento mudança no comprimento de uma ligação química deformação no plano mudança no ângulo entre ligações químicas
torção mudança em um ângulo diedral formado pelas ligações
químicas
deformação fora do plano mudança no ângulo entre ligação química e o plano da
molécula
A relação entre coordenadas internas e coordenadas cartesianas é dada pela seguinte transformação de coordenadas
St=
3N
X
i=1
Bt iqi t=1, 2, 3, ..., 3N−6, (3.12)
e de forma simplificada
S=BX, (3.13)
que podemos expandir como S1 S2 ... =
B11 B12 ... B21 B22 ... ... ... ... · q1 q2 ... . (3.14)
Como mostra a equação 3.12, as coordenadas internas são uma combinação das coor-denadas cartesianas, onde Bt i são constantes que dependem da geometria da molécula. A
matrizBé a matriz de transformação do sistema de coordenadas cartesiano para o sistema de coordenadas internas. A matriz B tem 3N−6 linhas e 3N colunas para moléculas não
lineares, ou 3N−5 linhas e 3N colunas para moléculas lineares. Cada linha da matriz B representa a mudança nas coordenadas cartesianas para uma coordenada interna, sendo as coordenadas cartesianas de cada átomo indexadas de três em três em cada linha. Ou seja, em uma linha as três primeiras colunas são coordenadas cartesianas do primeiro átomo, e as três colunas seguintes são coordenadas cartesianas do segundo átomo e assim por diante.
Para tornar a notação mais compacta, usamos um vetor ̺α que representa o
desloca-mento em coordenadas cartesianas do átomoα, substituindo as coordenadas cartesianasqi,
qi+1 e qi+2. Assim obtemos uma notação mais simples para a definição das coordenadas internas:
St= N
X
α=1
stα·̺α, (3.15)
ondestαe ̺α são vetores. O significado físico destα é a direção em que o átomoαdeve ser
deslocado para produzir a maior mudança na coordenada internaSt.
Abaixo listamos as quatro principais coordenadas internas e os respectivos vetoresstα
que compõem a matrizB.
3.3.1
Estiramento
O estiramento é uma coordenada interna muito simples, mostrada na figura 3.1, que
representa a mudança na distância entre os átomos. Os vetores st1 e st2 representam a
1 2 r12
st1 st2
Figura 3.1: Representação da coordenada interna estiramento.
Nessa coordenada, os átomos 1 e 2 se deslocam de acordo com os vetores st1 e st2,
respectivamente.
st1=e21=−e12 st2=e12. (3.16)
A notação de vetores unitários foi escolhida de tal forma que ex y aponta do átomo x
para o átomo y.
3.3.2
Deformação no Plano
A coordenada de deformação no plano, ou simplesmente deformação, é dada pelo ân-gulo formado pela interseção de duas ligações químicas que têm um átomo em comum. Essas ligações químicas são representadas pelos segmentos de retar31 er32, como mostrado na figura 3.2. Três átomos estão envolvidos nesse tipo de coordenada, um átomo central, aqui representado pelo átomo 3, e outros dois átomos, 1 e 2, sendo os três átomos não coli-neares. Os vetoresst1,st2 e st3 mostram a direção do movimento desses átomos que induz
a maior variação da coordenadaSt= ∆φ
r32 r31
3
2
1
φ
st3
st2
st1
Figura 3.2: Representação da coordenada interna deformação no plano.
respectivamente. Como a deformação no plano é uma coordenada interna, o centro de massa da molécula não deve ser afetado, logo a soma de todos os vetoresst i deve ser nula3.
st3=−st1−st2, (3.17)
Os vetores st1 e st2 são perpendiculares a r31 e r32, respectivamente. O comprimento do
vetorst1 é de
1
r31, já que um deslocamento de uma unidade na direção de st1 aumenta o ânguloφde 1
r31. De maneira análoga,st2tem comprimento 1
r32, e de acordo com a equação 3.17st3 tem comprimento|st1+st2|.
Os vetores unitáriose31 e e32 têm a mesma direção que os vetoresst1e st2,
respectiva-mente. Podemos definir os deslocamentos dos átomos na coordenadadeformaçãoem função
desses vetores unitários.
st1 = e31×e31×e32 r31sinφ
= e31 e31·e32
−e32 e31·e31
r31sinφ
= e31cosφ−e32
r31sinφ . (3.18)
Realizando um cálculo similar para o caso do vetorst2 e com o auxílio da equação 3.17
para o vetorst3, podemos obter as seguintes relações:
st1=
cosφe31−e32
r31sinφ , (3.19)
st2=
cosφe32−e31
r32sinφ , (3.20)
st3=
r31−r32cosφe31+ r32−r31cosφe32
r31r32sinφ . (3.21)
3.3.3
Torção
A coordenada interna de torção é dada pela mudança do ângulo diedral, τ, formado
pelos planosΠ1,2,3e Π2,3,4, respectivamente, onde 1, 2, 3 e 4 são átomos representados na figura 3.3. 1 φ2 φ3 2 3 4 r12 r23 r34
Figura 3.3: Representação da coordenada interna de torção.
A definição da coordenada interna torção,τ, é dada pela seguinte relação:
cosτ= e12×e12
· e23×e34
sinφ2sinφ3 , (3.22)
onde o ângulo τ está compreendido no intervalo −π < τ≤π, de forma que τ é positivo
quando o átomo 2 estiver mais perto do observador do que o átomo 3, quando visto ao longo da ligação química 2, 3. O ânguloτé visto como a projeção de 2, 1 e 3, 4 na direção
horária.
st1=−
e12×e23 r12sin2φ2
, (3.23)
st2=r23−r12cosφ2
r23r12sinφ2
e12×e23 sinφ2
+ cosφ3
r23sinφ3
e43×e32
sinφ3 , (3.24)
st3=r32−r43cosφ3
r32r43sinφ3
e43×e32 sinφ3
+ cosφ2
r32sinφ2
e12×e23
sinφ2 , (3.25)
st4=−
e43×e32 r43sin2φ3
3.3.4
Deformação Fora do Plano
É a coordenada formada pelo ângulo entre uma ligação química e o plano de três átomos. A figura 3.4 mostra que o átomo 1 está fora do plano Πformado pelos átomos 2, 3 e 4. A ligação química, representada por r41, forma um ângulo θ com o plano Π. O ângulo θ é a coordenada de interesse, sendo st1, st2, st3 e st4 os vetores que mostram a direção preferencial de deslocamento dos átomos 1, 2, 3 e 4, respectivamente.
Como definimos anteriormente, as vibrações em estudo são muito pequenas. No caso da coordenada deformação fora do plano, a vibração é tão pequena que podemos definir o
comprimento do vetorst1como sendo o mesmo que o comprimento do arco formado quando
o átomo 1 deixa o planoΠ,st1=
1
r41.
st1=
1
r41, (3.27)
st2=
sinφ2
r42sinφ1, (3.28)
st3=
sinφ3
r43sinφ1, (3.29)
st3=−
1
r41−
sinφ2 r42sinφ1−
sinφ3
r43sinφ1. (3.30)
3.4
Equação Secular em Coordenadas Internas
A energia cinética e a energia potencial em coordenadas internas são dadas pelas duas equações abaixo
2T=˙S′G−1˙S, (3.31)
2V =S′FS (3.32)
φ1
φ2
φ3
r43 3
2 1
4
r42 r41
Figura 3.4: Representação da coordenada de deformação fora do plano.
como
Gt t′=
3N
X
i=1
1
mi
Bt iBt′i t,t′=1, 2, ..., 3N−6, (3.33)
ou na forma matricial
G=BM−1B′, (3.34)
ondeMé um vetor coluna de 3N elementos representado a massa dos átomos da molécula,
sendo os elementos da forma mi=mi+1=mi+2 para o átomo de índice i de um total de N
átomos.
As coordenadas internas são definidas em função das coordenadas normais através da transformação de coordenadas
S=LQ, (3.35)
ondeSsão as coordenadas internas,Qsão coordenadas normais eL é a matriz de transfor-mação das coordenadas normais em coordenadas internas. Para cada coordenada normal
Qj todos os átomos de uma molécula vibram com mesma frequência e passam
simultanea-mente pela posição de equilíbrio. Cada modo normal de vibração é caracterizado por uma única coordenada normal e autovetorLj que determina a direção de vibração de cada átomo