w ww.e l s e v i e r . c o m / l o c a t e / b j p
Original
Article
Pharmacognostic
evaluation,
and
development
and
validation
of
a
HPLC-DAD
technique
for
gallocatechin
and
epigallocatechin
in
rhizomes
from
Limonium
brasiliense
Andressa
Blainski
a,
Tânia
M.
Antonelli-Ushirobira
a,
Guilherme
Godoy
b,
Eneri
V.S.
Leite-Mello
c,
João
C.P.
Mello
a,∗aProgramadePós-graduac¸ãoemCiênciasFarmacêuticas,UniversidadeEstadualdeMaringá,Maringá,PR,Brazil bFaculdadedeFarmácia,UniversidadeEstadualdeMaringá,Maringá,PR,Brazil
cDepartamentodeCiênciasMorfológicas,UniversidadeEstadualdeMaringá,Maringá,PR,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory: Received3July2016
Receivedinrevisedform25August2016 Availableonline19October2016
Keywords: Baicuru
Limoniumbrasiliense Pharmacognosticevaluation Qualitycontrol
HPLC-DAD
a
b
s
t
r
a
c
t
Limoniumbrasiliense(Boiss.)Kuntze,Plumbaginaceae,isaplantfromthesoutherncoastofBrazilianthat isusedforthetreatmentofpremenstrualsyndrome,menstrualdisordersandgenito-urinaryinfections. Theaimofthepresentstudywastodeterminethequalitycontrolparametersforrhizomescollected dur-ingdifferentperiodsbypharmacopoeialandnon-pharmacopoeialmethods,andtodevelopandvalidatea HPLC-DADmethodforquantitativecontrolofmarkersubstances.Themeasuredparameterswere: gran-ulometricanalysis(d50=0.21–0.48mm),lossondrying(11.1–12.4%),totalash(4.9–5.7%),dryresidueby
extractionwithacetone:water(7:3,v/v)(30.6–39.5%),totalpolyphenolcontent(8.5–15.8%),and chro-matographicfingerprintbyHPLCandTLC.Besides,theacetone:water(7:3,v/v)extractionsolventin combinationwithaturbo-extractor,yieldedthecrudeextractwithasignificantincreaseintannins (F4,20=37.0,p<0.001).Theantioxidantpotentialofthecrudeacetone:water(7:3,v/v)extract,aswellas
theethylacetateandwaterfractionsobtainedafterthepartitionprocesswasevaluatedbyDPPHandthe resultswere,respectively:IC506.87,5.91,and6.92g/ml.ThevalidationparametersfortheHPLC-DAD
methodshowedadequatespecificity,precisionandaccuracy.Thegallo-andepigallocatechincontents were,respectively,0.8–2.7%and1.2–2.2%.Thesedatacontributetoanalysisofthepharmacognostic qualitycontrolofthecommonlyusedpartfromthisspecies.
©2017SociedadeBrasileiradeFarmacognosia.PublishedbyElsevierEditoraLtda.Thisisanopen accessarticleundertheCCBY-NC-NDlicense(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introduction
Limonium brasiliense (Boiss.) Kuntze, Plumbaginaceae, is a perennialherbthatgrowsinArgentina,Uruguayandthe south-erncoastofBrazil.Itispopularlyknownas“baicuru”or“guaicuru” (Murrayetal.,2004).Preparationsoftheirdriedundergroundparts havebeenusedinthetreatmentofpremenstrualsyndrome, men-strualdisordersandgenito-urinaryinfections(Fenneretal.,2006). Becauseofitstraditionaluse, aclinicalstudywasconductedto testtheassociationofaliquidextractfromL.brasilienseandthe analgesicparacetamol in 36 women withsymptoms related to menstrualdisordersincludingdysmenorrhea,leucorrheaor amen-orrhea.Theimprovementwasexcellentin83.3%andgoodin13.9%
∗ Correspondingauthor.
E-mail:[email protected](J.C.Mello).
ofthewomen,only2.8%ofthemconsideredaregular improve-mentforthistreatment.Thetreatmenttimevariedbetween11 and50days(JanhsandCrescente,1976).Studiesalsoreportthat these extracts have biological activities such as bacteriostatic, anti-inflammatory and antioxidant effects, and a toxicological evaluationoftheextractdemonstratedalowtoxicity( Antonelli-Ushirobira et al., 2015a,b). The presence of hydrolysable and condensed tannins, leucoanthocyanins, flavonoids, -sitosterol, saponins,andcoumarinhasbeenreportedinL.brasiliense(Murray et al.,2004).Arecent morpho-anatomical studyresolved some doubts in the literature as to the nature of the part used for medicinal purposes and defined its structure as the rhizome (Antonelli-Ushirobiraetal.,2015a,b).
Oneofthemaincomplicationsofphytomedicinestudiesisthe complexityoftheprocesstoevaluatenaturalproducts,because theirqualityandcompoundscharacterizationisdifficultand differ-encesmaybeinducedinthepharmacokinetic,pharmacodynamic,
http://dx.doi.org/10.1016/j.bjp.2016.08.009
andsafetyprofiles(NisbetandMoore,1997).Inaddition,the chem-icalconstituentsofmedicinalplantsmayvaryaccordingtogenetic factors,weather,soilqualityandotherexternalfactors( Gobbo-NetoandLopes,2007).Thisway,withoutsuitablequalitycontrol parametersfortheproductionofmedicinalplantsinordertoobtain herbaldrugs,itisimpossibletoensurethereproducibilitybetween differentbatches, or to assayfor the absence of contaminants. Therefore, chemical, physical and physicochemical methods, as wellasmodernanalyticalmethods,shouldbeusedtohelpdefine thefeaturesofnaturaldrugs,inordertorecognizeandunderstand theirpossiblevariations(Gobbo-NetoandLopes,2007;Schulzetal., 2004).Tannins seemtobea featuredchemical classpresent in L.brasiliense,howevertheyareoftenpresentascomplexeswith otherstructuresandtheanalyticaldatadependsonseveral fac-torssuchassamplepreparation,storageandextractiontechnique (Mueller-Harvey,2001;Schofieldetal.,2001).
Inordertoguaranteethesafetyandeffectivenessofthe phy-totherapies,analyticalmethodsforcontentdeterminationarean integral part of thequality control of the sourcematerial. The determinationofqualitycontrolparametersforplantdrugsand extractswithhightannincontentiscomplicatedduetothe struc-turalcomplexityofthisclass.Frequentlythebiological,biochemical andchemicalproceduresmustbeconductedinconcert.Although high-performance liquid chromatography methods are usually conductedaccordingtothemostcommonlaboratorialroutine,its reliabilityrequiresanefficientprocessofdevelopmentand vali-dation(Mueller-Harvey,2001;Lopesetal.,2009).
The purposeof thepresent study wasto evaluate pharma-copoeialandnon-pharmacopoeialparameters,andtodevelopand validateahigh-performance liquidchromatography methodfor thedetermination ofgallocatechin andepigallocatechinfromL. brasilienserhizomes.
Materialsandmethods
Plantmaterial
Rhizomesof Limonium brasiliense (Boiss.) Kuntze, Plumbagi-naceae,werecollectedinRioGrande,inthestateofRioGrandedo Sul,Brazil(S32◦09′22′′/W52◦06′01′′)inMay2006(sampleA),(S 32◦00′00′′/W52◦09′00′′)May2010(sampleB),and(S31◦59′33′′/W 52◦10′43′′)February2013 (sampleC).Voucher specimenswere deposited at theHerbarium of the StateUniversity of Maringá under numbersHUEM-12151, HUEM-21152,and HUEM-27725, respectively.Theplantmaterialwascollectedunderapermitfrom IBAMA-SISBIO,No.11995-2,July5,2007,and11995-3,November 2,2010,authenticationcode46367613,undertheresponsibilityof J.C.P.Mello.Accesstothebotanicalmaterialwasauthorizedand licensedbytheConselhoNacionaldeDesenvolvimentoCientífico eTecnológico(CNPq),registeredunderNo. 010252/2015-0.The plantmaterialswerecleanedwithwatertoremovesoil,driedin acirculating-airoven(37±2◦C),andpowderedinahammermill (TigreASN-5).
Extractpreparation
Thecrudeextract(CE)waspreparedfromthedriedrhizomes (samples:A–0.63kg;B–5.6kg;C–5.0kg)usinganacetone:water solution(7:3,v/v,10%,w/v)andanUltra-Turrax(UTC115KT, Wil-mington,NC,USA;3000rpm,4×5min,t<40◦C).Next,theextract wasfiltered,theorganicphasewasremovedinarotavapor(Büchi R-135)andtheresiduewaslyophilized(ChristAlpha1–4)toyield theCE(A:43.3%;B:28.3%;C:27.6%).TheCEwassolubilizedinwater intheproportionof10%(w/v)andpartitionedwithethylacetate (10timeswiththevolumeofwater).Theethyl-acetate(FAE)and
aqueous(FAQ)fractionswereconcentratedandlyophilizedas pre-viouslydescribedtoyield,respectively,FAE(A:9.2%;B:11.6%;C: 10.4%)andFAQ(A:83%;B:82.4%;C:72.4%).
HPLCmethod
High-performance liquid chromatography (HPLC) was con-ducted on an Agilent Model 1290 Infinity instrument withan AgilentZorbaxC-18(250mm×4.6mm)5mcolumn.Themobile phase was composed of water:concentrated phosphoric acid (100:0.2,v/v,SolventA)andacetonitrile:concentratedphosphoric acid(100:0.2,v/v,SolventB) and usedin thefollowing elution gradient:0min10%B;15min15%B;35min19%B;36min80% B; 44min 80% B; 45min 10% B; 52min 10% B. The oven tem-perature wascontrolledat 24◦C (0–18min), 35◦C (18–45min), and 24◦C (45–52min). The flow rate was 0.4ml/min, and the injectionvolumewas10l.ThedetectionwasbyUVin210nm andDAD(200–400nm)wasemployedfordeterminationofpeak purity.TheFAEsampleswerepreparedin methanol:water(1:1, v/v) at 200.0g/ml with the help of an ultrasonic bath for 10minandfiltered(MilliporeMilex-HV0.45m)priortoinjection. Referencesubstances(gallicacid,gallocatechin,catechin, epigal-locatechin,epicatechin,andellagicacid,Sigma)werepreparedin methanol:water(1:1,v/v)andinjectedintotheHPLCinstrumentto identifythepossiblesubstancespresentinFAEsamplesbythe com-parisonofretentiontimes(RT),absorbancespectrumandincrease topeakareawiththeadditionofreferencesolutionsinsample solu-tion(spikedsample),aswellastheconcentrationsofthesepresent substancesintheFAEforexternstandard.
ValidationoftheHPLCmethod
Forvalidationoftheanalyticalmethod,theguidelines estab-lished bytheInternationalConference ontheHarmonizationof TechnicalRequirementsfortheRegistrationofPharmaceuticalsfor HumanUse(ICH,2005)andbyBrazilianregulationRENo.899/2003 oftheNationalHealthSurveillanceAgency(Anvisa,2003)were employed.TheFAEfromsampleCwasusedtotestthevalidation parameters.
Linearitybetweenthepeakareaand concentrationwas ana-lyzed using three calibration curves obtained from reference solutions of gallocatechin (GC) and epigallocatechin (EGC) at five different concentrations in therange of 3.4–5.1g/ml and 3.2–4.8g/ml,respectively.
Specificitywasdeterminedbypeakpurity(≥0.99)ofGCandEGC peaksfromthesamplesolution.TheDADdetectorwasemployed. Inaddition,thesamplesolutionwasstoredunderrelevantstress conditions(acid/basichydrolysis,heat,oxidationandlight)to pro-motethepartialdegradationofthesampleandtoshowbythepeak purityiftheanalyzedchromatographicpeakcouldbeattributable toonlyonecomponent.Foreachcondition,ca.of2mgofFAEwas weighedintoa10mlvolumetricflask.Foracidstress,1mlof2M HClwasadded,andafter4hatroomtemperatureitwas neutral-izedwith1mlof2MNaOH.Forbasicstress,1mlof0.1MNaOHwas added,andafter3hatroomtemperatureitwasneutralizedwith 1mlof0.1MHCl.Forheatstress,theFAEwasstoredinanoven at60◦Cfor40h.Foroxidationstress,1mlof30%H
2O2wasadded andstoredfor40hatroomtemperature.Forlightstress,two sam-ples,oneinamberflaskandotherintransparentflask,werefor14h inphotostabilitychambers(Weiss,modelPharma500-L).Afterall aboveprocedure,thesamplesweredilutedwithmethanol:water (1:1;v/v)totheconcentrationof200g/mlandsubmittedtoHPLC conditions.
expression10/S,whereisthestandarddeviationoftheresponse
andSistheslopeofthecalibrationcurve.
Precisionwasevaluatedontwolevels:repeatability(intra-day) andintermediate(inter-day).Thetestofrepeatabilitywascarried outusingthreesamplesoftheFAEat80%(ca.1.60mg),at100% (ca.2mg)and120%(ca.2.2mg)ina10mlvolumetricflask.For intermediateprecision,thesameprocedurewasperformedonat leasttwodifferentdaysonanotherHPLCinstrumentofthesame model.Repeatabilityandintermediateprecisionwereexpressed astheresidualstandarddeviation(RSD%)oftheconcentrationsof GCandEGCin thedried FAE.ARSD%over10% wasconsidered unacceptableforthecomplexmatrix.
TheaccuracyofthemethodwasestablishedbasedontheGC andEGCrecoverytestsusingtheadditionofstandard.Thesample solutionswerepreparedfrom1mgofFAEina10mlvolumetric flask(50%ofthenormalpreparation).Theaccuracywasassessed atthree differentlevelsinthreereplicates.Adefinedvolumeof referencesolutionequivalentat12.72gofGCand 12.03gof EGCwasaddedforthelowerconcentration(LC);at21.22gofGC and20.08gofEGCfortheintermediateconcentration(IC),and at29.66gofGCand28.06gofEGCforthehigherconcentration (HC).Theknownconcentrationwasdetermined(basedonassay ofFAEbyprecisionparameter)andthemeasuredvaluewas com-paredwiththetheoreticalvalue.Theaccuracywasassessedasthe recoverypercentageandthemethodwasconsideredaccurateifthe recoverypercentageswerebetween85and115%.
Robustnesswasdeterminedbycomparingsamplesolutionsthat wereanalyzed underthe establishedconditions and by chang-ingthefollowingparameters:fromoventemperaturegradientto aconstanttemperatureat24◦Cand35◦C,andeluentflowfrom 0.4ml/minto0.6ml/min.Inaddition,thestabilityofthesample solutionwasassessedafter30hatroomtemperatureandinthe injectorat10◦C.
Pharmacognosticqualitycontrolassays
Thegranulometryofthepowderwasmeasuredbysievesof mesh sizes 150, 180, 212, 300, 600, and 850m (Bertel AGT-01)accordingtothemethoddescribedbyBrazilianPharmacopeia (2010)andthemediandiameter(d50)couldbedeterminedbythe pointofintersectionofthecurveofthepercentageofresidue frac-tionandpercentageofpassagefraction.Thelevelsoftotalashand lossondryingweredeterminedaccordingtomethodsdescribedby theBrazilianPharmacopeia(2010).Thetotalpolyphenol(TP) con-tentwasdeterminedformilledrhizomesaccordingtothemethod describedbyGlasl(1983).Themilledrhizomes(20g)from sam-plesA,B,andCwereextractedwithacetone:water(7:3,v/v)by turbo-extraction(Ultra-Turrax®UTCIKA-125,4
×5min,t<40◦C) intheproportionof10%(w/v).Theextractionsolutionswere fil-teredandtransferredtoavolumetricflaskanddilutedto200ml. Portionsofthesesolutionswereusedinthedryresiduetest accord-ingtotheGermanPharmacopeia(2015).TheTPcontentexpressed inpyrogallolwasanalyzedintheCE(fromsamplesA,B,andC)by spectrophotometryaccordingtothemethoddescribedbyBlainski etal.(2013).TheFAE(fromsamplesA,B,andC)wereanalyzedby HPLCmethodtocomparetheirchromatographicfingerprintsand determinetheGCandEGCcontents.Forthin-layer chromatogra-phy(TLC),theCE,FAE(samplesA,BandC)andreferencesubstances (gallicacidandgallocathechi–GAandGC,respectively)were pre-paredinmethanolintheconcentrationof40mg/ml,10mg/mland 1mg/ml,respectively;samplesolutionswere5mininultrasonic bathandfollowedtocentrifugation(3min,12,100×g,Eppendorf MiniSpin);40lofeachsampleand10lofreferencesubstances wereappliedinglassplate(TLCSilicagel60F254,20cm×20cm, Merck)byautomaticSampler(Camag);theelutionwaswithethyl acetate:formicacidanhydrous:water(90:5:5,v/v)for15cm;after
drying,theTLCwassprayedwithferricchloride,hexahydrate,1% inmethanol(w/v)andtheimagewascapturedbyinTLCVisualizer (Camag,systemVisionCats).
Comparisonofextractedliquidandselectivefortannins
SampleAextractswerepreparedaccordingtothepreviously describedinextractionpreparationwithaseriesofdifferent solu-tions:hydroethanol50◦GL(ExtractA);hydroethanol70◦GL(Extract B);hydroethanol90◦GL(ExtractC)andmethanol:water(1:1,v/v) (ExtractD), aswellasacetone:water (7:3,v/v)(Extract E).The extractedsolutionswereusedtodeterminedryresidue(German Pharmacopeia,2015),andCEwereobtainedfromremaining solu-tionsandusedtodeterminetheTPandtotaltannin(TT)contents (Glasl,1983).Theseresultswereusedtocomparedifferent extrac-tionsolutionsandtodeterminewhichismoreselectivefortannins inthecontextoftheturbo-extractionprocess.
Antioxidantpotential
SampleBwasusedtodeterminethefree-radical scavenging capacityoftheCE,FAE,andFAQaccordingtothemethoddescribed byAmarowiczetal.(2004).
Statisticalanalysis
Data were analyzed by the Statistica® 8.0 program
(Copy-rightStatSoft,Inc.1984–2007)byaone-wayanalysisofvariance (ANOVA) followed by Tukey’s test considering p<0.05 as the significancelevel,aswellastheT-testfortwogroups consider-ingp<0.05 asthe significance level.Results wereexpressedas mean±standard deviation[residualstandard deviation(RSD%)]. Todeterminethelinearityofthemethod,linecorrelationtestsand residualanalysisweremadebysimplelineregressionconsidering R2valuesequalorgreaterthan0.99andtheresidualsumofsquares wereevaluated.
Resultsanddiscussion
HPLCmethoddevelopment
2000
1000
0
2000
1000
0
2000
1000
0
0 5 10 15 20 25
Minutes
Sample C
30 35 40 45 50
Sample B
mA
U
Sample A
GA GC EGC
Fig.1.HPLCfingerprintoftheethyl-acetatefraction(FAE)fromLimoniumbrasilienserhizomesextractedwithacetone:water(7:3,v/v)forsamplesA,B,andC.GA,gallicacid; GC,gallocathechin;EGC,epigallocatechin.
(RT=17.8min). TheGC andEGC werechosenfor thevalidation procedures.Theothers majorpeaks(RT=18.4;22.9; 25.8;29.4, and32.0min)showedcharacteristicspectraforcondensedtannins withaabsorptionbandatround275nmandashoulderat305nm, commonlyattributed,respectively,totheA-ringandB-ringfrom theflavan-3-olsandflavan-3,4-diolsunits(Vihakas,2014).Fig.1 showstheobtainedthespectraandchromatographicprofiles.The HPLCmethodcarriedoutinthisstudywasaimedatdevelopinga chromatographicsystemcapableofelutingandresolving pheno-liccompoundsinplantmaterials.Thepreliminaryinvestigations weredirectedtowardevaluatingtheeffectofvariousfactorsonthe system.Thefactorsassessedincludesamplepurification,detection wavelength,thecolumntype,andthecompositionofthemobile phase.Theelimination ofhighmolecularweightphenolic com-poundsfromtheplantextractiscriticallyimportant,becauseof theinteractionofthesecompoundswiththestationaryphase.This interactioncanseriouslydamagetheanalyticalcolumn,interfering withthechromatographicprocess(Lopesetal.,2010).
ValidationoftheHPLCmethod
The calibration equation in the linearity test was y=1.44·108x+1.2·107 (n=3, r2=0.9980) for GC and y=1.41·108x+1.0·107 (n=3, r2=0.9950) for EGC. The RSD% of the slopes was 2.3 and 1.8% for GC and EGC, respectively. Table 1 shows the statistical data for the regression equations withanalysisofvarianceandlackoffitcalculationsforcalibration curvesofGCandEGC.Thecalibrationcurvesareadequateforassay determinationandstatisticaldatacomplywithallrequirements for linear regression: calculated F-value is lower than critical
F-value,aswellasthelackoffiterrorthansumofpureerror,and theANOVAtestissignificant.
ThespecificityofthemethodwasshownbycomparingtheUV spectraofGC(13.1min)andEGC(17.8min)inthereference solu-tionwiththosefromthetestsolution.ThepurityoftheGC,EGC, andsixothermainpeakswassuitable(>0.99).Thedegradationof bothpeakswassuitablefortheacid,heatandoxidationandlight stressconditions,becausetheyprovidedthepartialdegradation, bywhichwasallowedthereliableanalysesofthepeakpurityof theremainingGCandEGC(Table2).Ontheotherhand,thesample appearedtobeverysensitivetothebasicconditionandtherewas almostcompletedegradationoftheGCandEGCpeaksevenunder moremildconditions.However,itseemsthatsubstances result-ingfromthedegradationinducedbybasicstressdonotinterfere withtheretentiontimesforGCandEGC.Thus,thechromatography methodisconsideredspecificforassayofGCandEGC.
TheLODisdefinedasthelowestabsoluteconcentrationof ana-lyteinsolutionthatcanbedetectedbutnotnecessarilyquantified under thestated experimentalconditions and was0.161g/ml for GC and 0.238g/ml for EGC. The LOQ is defined as the lowestconcentrationofanalyteinsolutionthatcanbe quantita-tivelydeterminedwithacceptableprecisionandaccuracy.Itwas 0.536g/mlforGCand0.793g/mlforEGC.
Table1
Statisticaldatafortheregressionequationsofthecalibrationcurvesforgallocatechin(GC)andepigallocatechin(EGC)bytheHPLCmethod.
Regressionanalysis CalibrationcurveforGC CalibrationcurveforEGC
Slope(SE) 1.44×108(2.29×1014) 1.41×108(4.78×1014)
Intercept(SE) 1.2×107(7.74×106) 1.0×108(1.1×107)
Regressioncoefficient(R2) 0.9980 0.9950
CalculatedF-value(criticalF-value) 0.30(3.71) 0.30(3.71)
Sumofpureerror 3.51×1016 5.09×1016
Lackoffiterror 3.48×1015 5.04×1015
Analysisofvariance F1,13=6385.7,p<0.001 F1,13=2610.8,p<0.001
95%CLSlope 1.40×108;1.48×108 1.35×108;1.47×108
95%CLIntercept −4.6×106;2.9×107 7.7×107;1.2×108
SS MS SS MS
Regression 1.13×1017 1.13×1017 9.60×1016 9.60×1016
Residual 2.29×1014 1.76×1013 4.78×1014 3.68×1013
Total 1.13×1017 9.64×1016
SE,standarderror;SS,sumofsquares;MS,meansquare;CL,confidencelimits.
Table2
Thepartialdegradationandthereliabilityanalysesofthepuritypeakforthe remaininggallocatechin(GC)andepigallocatechin(EGC)instressconditionsfor specificitytest.
Conditionfordegradation GCcontent (%)/puritypeak
EGCcontent (%)/puritypeak
Normalcondition 2.8/1.0000 2.3/1.0000 Acidstress 2.3/1.0000 1.1/1.0000 Heatstress 2.3/1.0000 1.8/1.0000 Oxidationstress 2.9/1.0000 2.1/1.0000 Basicstress 0.1/0.8771 0.2/0.9153
Lightstress
Amber 2.7/1.0000 2.1/1.0000
Transparent 2.4/1.0000 1.9/1.0000
forthecomplexmatrix,soitisconsidered,thatthemethodis pre-ciseforbothsubstances.
Theaccuracyofthemethodwasestablishedbasedonthe recov-erytestforGCandEGC.Themeanrecoveryratewas102.3%±1.54 [1.5%]forGCand99.7%±1.93[1.9%]forEGC(Table3).Theresultis accordingtotheacceptedcriterion,thusthemethodhasexcellent accuracyforherbaldrugs.
ToensurethattheHPLCmethodisinsensitivetominorchanges in experimental conditions, it tests the robustness parameters. Whentheoventemperaturewaschangedfromagradienttoa con-stanttemperatureof24◦C,theresolutionretentiontime(RT)and peakareaoftheGCandEGChadnosignificantchange.However, thechromatographicprofileafter25minandresolutionofthese peakswaschanged.Ataconstanttemperatureof35◦C,therewere alterationsintheRTofGCandEGC,andtheresolutionbetween EGCandpeakat18.4minwaslostcompletely.Whentheeluent flowwaschangedto0.6ml/min,thischangedtheRTofthemain peaks,includingforGCandEGC.Theresolutionfor somepeaks wasalsolost,buttherewasnosignificantchangeinpeakareafor GCandEGC.Regardingthestabilityofthesamplesolution,no dif-ferencewasobservedforthesamplesleftintheinjectorat10◦C for30h.However,therewasasignificantcontentdecreaseofGC (ca.12%)atroomtemperature.Theseresultsdemonstrated,that itshouldprocedurethemethodunderdeterminateconditionsand somechangecaninducetoproblemswithchromatographicprofile andresolution.
Pharmacognosticqualitycontrolassays
The results of pharmacognostic quality control assays from milledrhizomes(granulometry,totalash,lossondryingandtotal polyphenols),CE(dryresidueandTP),andFAE(GCandEGC)in samplesA,B,andCarepresentedinTable4.
SampleBhasaTPrhizomelowerthanthatoftheother sam-ples,butthisseemstohavenosubstantialdifferencesamongthe samplesinrelationtothedryresidueandTPintheCE.Thed50 determinedfromthecurvesshowsavaluesubstantiallylowerfor sample B(0.21mm) in relationtothe other samples(0.42mm and 0.48mm). However, this lower diameter did not seem to influencetheextractionyield,presumablybecauseitisa turbo-extractionprocess,wherehappensthebreakingofparticles.The granulometricdistributionisanimportantfactorinobtainingplant extracts, mainlybecause the extraction yield is closely related tothesurfaceareaandparticlesizeincontactwiththe extrac-tion liquid. Studies to determinethe existence of positive and negativeinfluencesfromthed50onextractionyieldarepossible throughfactoranalysis(MelloandPetrovick,2000;Delaporteetal., 2001).
Thetotalashresultsrangebetween4.9and5.7%fromthethree differentsamplesshowgoodconsistencyforthetotalash deter-minationforthisspecies.Totalashisameasureofthequantityof non-volatileinorganicimpurities,becauseitisinfluencedby con-taminationoradulterationof plantproducts(Mukherjee,2002). Thetotalashincludesthosederivedfromplanttissue (physiologi-calash)andforeignmaterial,specificallysandanddirtadheringon thesurfaceoftheplant(non-physiologicalash).Thepercentageof totalashmayvarywiththeplantspeciesandmustbedetermined individuallyfor eachspecies.For plantdrugs,thepercentageof totalashisbetween3and15%forsomespeciesdescribedinthe BrazilianPharmacopeia(2010).
Theresultsoflossondryinglesserthan14%showthatthedrying processwasefficient.ForsomespeciesdescribedintheBrazilian Pharmacopeia(2010),thepercentageoflossondryingisbetween8 and14%.Thepercentageoflossondryingisaparameterthatcanbe usedtoestimatetheefficiencyoftheplantdryingprocedure.The excesswaterinplantdrugspromotesthegrowthoffungi, bacte-riaorinsectsandpromotehydrolysisofitsconstituents.Forthis reason,limitsofwatercontentaredescribedforherbs,especially forthosethatreadilyabsorbwaterorthoseforwhichdegradation ispromotedbythepresenceofwater.Thisparameteriscritical todeterminethestability ofthedrugduringthestorageperiod (Mukherjee,2002).
Table3
Accuracyresultsdeterminedbyanalyzinggallocatechin(GC)andepigallocatechin(EGC)insolutionsofknownconcentrationfromLimoniumbrasiliense.
Gallocatechin Epigallocatechin
Theoretical concentration (g/ml)
Measured concentration (g/ml)
%Recovery (¯x±sd)[RSD(%)] Theoretical
concentration (g/ml)
Measured concentration (g/ml)
%Recovered (¯x±sd)[RSD(%)]
LC
3.8266 3.9419 103.0
103.5% ±0.91(0.9%)
3.2580 3.1848 97.8
97.8 ±1.29(1.3%)
3.7543 3.9246 104.5 3.1998 3.1698 99.1
3.6338 3.7400 102.9 3.1029 2.9938 96.5
IC
4.6284 4.7329 102.2
102.4% ±0.88(0.9%)
4.0242 4.0083 99.6
100.2 ±0.87(0.9%)
4.7248 4.7996 101.6 4.1018 4.0888 99.7
4.7007 4.8576 103.3 4.0824 4.1293 101.2
HC
5.5929 5.5731 99.6
100.9% ±1.73(1.7%)
4.9192 4.8997 99.6
101.2 ±1.85(1.8%)
5.5929 5.7554 102.9 4.9192 5.0766 103.2
5.4731 5.4883 100.3 4.9599 4.9921 100.7
Total(x¯±sd)[RSD(%)] 102.3%±1.54(1.5%) 99.7±1.93(1.9%)
¯
x,mean(%);sd,standarddeviation;RSD,residualstandarddeviation.
Table4
ResultsofpharmacognosticqualitycontrolassaysfromLimoniumbrasiliense(samplesA,B,andC).
SampleA SampleB SampleC
Granulometry(d50)–MR 0.42mm 0.21mm 0.48mm
Totalash–MRa 5.6%±0.15[2.7%] 5.7%±0.07[1.4%] 4.9%±0.05[1.0%]
Lossondrying–MRa 11.1%±0.25[2.3%] 12.4%±0.28[2.4%] 11.9%±0.04[0.4%]
PT–MRa 15.8%±0.12[0.8%] 8.5%±0.36[4.3%] 9.4%±0.11[1.2%]
Dryresidue–MRa 39.5%±0.63[1.6%] 36.1%±0.61[1.7%] 30.6%±0.19[0.6%]
TP–CEa 28.9%±0.42[1.5%] 25.0%±1.94[0.8%] 29.1%±0.30[1.1%]
GC–FAEa 2.6%±0.06[2.3%] 0.8%±0.03[3.7%] 2.7%±0.09[3.2%]
EGC–FAEa 1.8%±0.03[1.8%] 1.2%±0.02[1.9%] 2.2%±0.13[5.8%]
ax¯±sd[RSD(%)]: ¯x,mean;sd,standarddeviation;RSD,residualstandarddeviation;MR,milledrhizome;CE,crudeextract;FAE,ethyl-acetatefractions;TP,total
polyphe-nols;GC,gallocatechin;EGC,epigallocatechin.
recommendstheevaluationofa wide numberofsamplesfrom differentyearseasonsandregions.
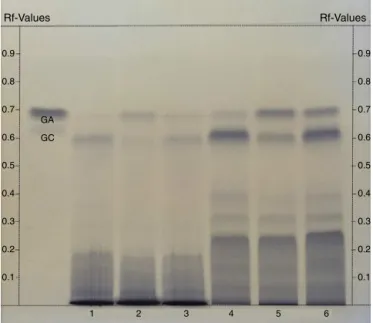
Fig.1showstheHPLCfingerprintsofFAEfromsamplesA,B, andC.Thereisagoodsimilaritybetweenthesamplesregarding theirpeakretentiontime;however,thepeakshavedifferent pro-portions.Fig.2 shows theTLCfingerprintsof CEandFAEfrom samplesA,B,andC.AswellasHPLCfingerprint,itispossibleto noteagoodsimilaritywithdifferenceintensityamongthespots. Forexample,inthesampleB,thespotofGAisstronger,butweak spotofGCisfastabsent,thatitcomplieswithHPLCfingerprint. Forcomplexbotanicals,quantitationofasinglecompoundusually doesnotprovidecompleteinformation.Forthisreason, chromato-graphicfingerprinttechnologyisacceptedbytheWHO(1991)as astrategyforidentificationandevaluationofthequalityofherbal medicines.Thisworkshowsforthefirsttimethechromatographic fingerprintsforL.brasiliense,andtheresultsseemtoprovidea reli-ableandsensitivequalityassessmentmethodwithcharacteristic retentiontime,aswellUVspectraofpeaksforHPLCmethod.The HPLCfingerprintanalysiscanbeusedforqualitativeanalysis, espe-ciallytoidentifyandstandardizeplantmaterialsandpreparations. Thisanalysisrequiresgoodseparationandresolutionofthe com-plexmixtureaswellaspeak puritycontrolin ordertoprevent overlappingofthepeaks(Rehwaldetal.,1994;Dingetal.,2011). HPLCfingerprintinghasbecomeanattractiveoptionforthequality controlofcomplexplantmixturesbecauseofitsfocuson identi-fication,ensuringnoadulterants(Heetal.,2005),andassessing thestabilityoftheplants,seeingthatchangesintheHPLC finger-printcanoccurinsamplesbytheinfluenceofexternalfactorssuch astemperature,humidity,andstressconditions(HeiglandFranz, 2003).SeveralstudieshaveshownthattheHPLCfingerprintcanbe usedtocharacterizeplantdrugsthathavepolyphenolsasprimary
Rf-Values
GA
GC
Rf-Values
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
1 2 3 4 5 6
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
Fig.2. TLCfingerprintofthecrudeextract(CE)andethyl-acetatefractions(FAE) fromLimoniumbrasilienserhizomesextractedwithacetone:water(7:3,v/v)for samplesA,BandC.GA,gallicacid;GC,gallocathechin;1,2and3,CEfromsamples A,BandC,respectively;4,5and6,FAEfromsamplesA,BandC,respectively.
Table5
ResultsforthecomparisonoftheextractionliquidandselectedtanninsinsampleA.
ExtractA ExtractB ExtractC ExtractD ExtractE
TPb 31.0%±0.32[1.0%] 29.3%±0.34[1.2%] 30.2%±1.12[3.7%] 31.3%±0.46[1.5%] 33.1%%±0.64[2.0%]a
TTb 27.6%±0.33[1.2%] 25.3%±0.36[1.4%] 25.9%±1.02[4.0%] 27.7%±0.42[1.5%] 29.5%±0.60[2.0%]a
%TT/TPb 88.9%±0.27[0.3%] 86.6%±0.34[0.4%] 85.7%±0.50[0.6%] 88.7%±0.21[0.2%] 89.2%±0.49[0.6%]a Dryresidueb 36.4%±0.41[1.1%] 32.2%±0.34[1.1%] 29.9%±0.16[0.5%] 36.3%±0.56[1.5%] 39.5%±0.63[1.6%]a
ap<0.001.
b ¯x±sd[RSD(%)]: ¯x,mean;sd,standarddeviation;RSD,residualstandarddeviation;TP,totalpolyphenols;TT,totaltannins.
TLCwasalsoadequatetodetermineacharacteristic fingerprint forthisherbaldrug.DifferencesamongsamplesA,BandCcanbe observed,suggestingthatitisimportanthereaftertocorrelatethe fingerprintswiththeseasonalconditionsandpossibledifferences inpharmacologicalactivities.
Comparisonofextractionliquidandselectivityfortannins
Thepreparationofdifferentextractsfollowedbyanalysisofthe dry residue,TP and TTare shown in Table5. Accordingtothe data,there is a significantincrease indry residue fromExtract E [extraction liquid acetone: water (7:3, v/v)] compared with theotherextracts(F4,20=136.0,p<0.001).Inaddition,ExtractE showssignificantlyhigherlevelsofTP(F4,20=23.7,p<0.001)and TT(F4,20=37.0, p<0.001), and comparatively higherTT/TP ratio (F4,20=86.0,p<0.001).
Thepreparation ofdifferentextracts followedby analysisof dryresidueandchemical compoundcontentmayprovide com-plementarydataforqualitycontrolandcontributetothechoiceof thebestextractionsolvent.Accordingtoourdata(Table5),itwas determinedthatthebestextractionsolventforthepreparationof crudeextractofdryrhizomesoftheL.brasiliensebytubo-extraction processisacetone:water(7:3,v/v).AccordingtoMueller-Harvey (2001),50–70%aqueousacetoneisoftenthebettersolventmore thanwaterormethanoltoextracthydrolysabletannins.However, methanolor watercan extractbetter tanninsof lowmolecular weight.
Consideringthehighquantityoftanninsamongthe polyphe-nolsofdryrhizomesfromL.brasilenseandcomparingwithother plants,itispossibletoaffirmthatitisarepresentativespeciesfor tannins.CommissionE(1994)determinedtannincontentof3%for CrataegusoxyacanthaL.and10%forKrameriatriandraRuiz&Pav.; bothareclassicalplantdrugsfortannins.Stryphnodendron obova-tumBenth.andS.polyphyllumMart.havetannincontentsof19and 12%,respectively(Lopesetal.,2005),andStryphnodendron adstrin-gens(Mart.)Covillehasaminimumof8%oftotaltannins(Brazilian Pharmacopeia,2010).
Antioxidantpotential
TheresultsfortheCE,FAE,andFAQfromsampleBwere, respec-tively,6.87,5.91,and6.92g/ml.TroloxandVitaminCwereused ascontrols,and theirresultswere3.97and4.36g/ml, respec-tively.Theantioxidanteffectofpolyphenolsisknown,however arecentstudysuggestedthatRosacaninaL.fruitextract,whichis richinpolyphenols,mayactnotonlyasanantioxidant,butalsoas aprooxidantinhighconcentrations,showingthatitisnecessary todefineanoptimalconcentrationofpolyphenolsforantioxidant activity(Kilic¸günandAltiner,2010).Althoughitcouldbealready expectedagoodantioxidantpotentialforL.brasiliense,sincethe sampleshowedahighTPandtannicsubstances,fewstudieshave relatedthisactivitytoplantsofthegenusLimonium,andthis inves-tigationproposedtoshowthesepotentialevenwiththesample withlowerphenoliccontents.
Aniyaetal.(2002)showedastrongantiradicalactivity(IC50
7.5g/ml)bytheDPPHmethodforaqueousextractsfromL.wrightii
(Hance)Kuntze.Murrayetal.(2004)showedanIC50of20g/ml for a methanolicextract fromL. brasiliense bythe DPPHassay. Thepresentresultsexhibitedmajorantioxidantpotentialforthe acetone:water(7:3,v/v)extractanditsethyl-acetatefraction,in comparisonwiththeaboveresult.
Conclusions
In the absenceof updated pharmacopoeiadata regarding L. brasiliense,theseresultscanbeusedasqualitycontrolparameters andcontributetothepharmacognosticqualityofsuchextracts.The TLCandHPLCfingerprinthavebeenshowntobeavaluabletoolfor thechemicalanalysisofcomplexmatrices.ThevalidationofaHPLC methodofferschromatographicconditionsfordeterminationand analysisofGCandEGC.Themethodwasfoundtobespecificand suitableforroutineanalysis.Itisalsosimple,sensitive,accurate, andreproducible.
Authors’contributions
AB(Ph.D.student)collectedanddriedtheplantmaterial sam-ples A and C, prepared their voucher specimens, worked on laboratorialtestswithbothsamples,conductedallHPLCandTLC analysis,andwroteandformattedthearticle.TMAUcollectedand driedsampleBoftheplantmaterial,prepareditsvoucher spec-imen,workedinlaboratorialtestswiththissample,andassisted withwriting.GG(undergraduatestudent)assistedin laboratory workwithsample B. EVSLMassisted in theprojectdesign and reviewedthemanuscript.JCPMwasresponsiblefortheproject con-ceptandsupervisionofthestudy,aswellasthewritingandreview ofthemanuscript.
Conflictsofinterest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgments
TheauthorsthankA.Arantesfortechnicalsupportduringthe experiments,AchéLaboratóriosFarmacêuticosS.A.forproviding theUV and HPLCinstrumentation and Finzelberg GmbH& Co. KGforTLCinstrumentationforanalyticalexperiments.Thisstudy receivedsupportfromCNPq,CAPES, FINEP,Fundac¸ãoAraucária, andtheInstitutoNacionaldeCiênciaeTecnologiaparaInovac¸ão Farmacêutica(INCTif).
References
Amarowicz,R.,Pegg,R.B.,Rahimi-Moghaddam,P.,Barl,B.,Weil,J.A.,2004. Free-radicalscavengingcapacityandantioxidantactivityofselectedplantspecies fromtheCanadianprairies.FoodChem.84,551–562.
Antonelli-Ushirobira,T.,Blainski,A.,Gancedo,N.C.,Gaburo,F.,Cardoso,K.A.K.C., Leite-Mello, E.V.S., Mello, J.C.P., Milaneze-Gutierre, M.A., 2015a. Morpho-anatomicalstudyofrhizomeofLimoniumbrasiliense.Rev.Bras.Farmacogn.25, 320–327.
Antonelli-Ushirobira,T.,Blainski,A.,Fernandes,H.G.,Moura-Costa,G.F.,Costa,M.A., Campos-Shimada,L.C.,Salgueiro-Pagadigorria,C.L.,Kaneshima,E.N.,Becker, T.C.A.,Leite-Mello,E.V.S.,Mello,J.C.P.,2015b.Acutetoxicityandlong-termsafety evaluationofthecrudeextractfromrhizomesofLimoniumbrasilienseinmice andrats.J.Ethnopharmacol.174,293–298.
Anvisa,2003.Resoluc¸ãoREn◦899–Guiaparavalidac¸ãodemétodosanalíticose
bioanalíticos.AgênciaNacionaldeVigilânciaSanitária,Brasilia.
Blainski,A.,Lopes,G.C.,Mello,J.C.P.,2013.ApplicationandanalysisoftheFolin Cio-calteumethodforthedeterminationoftotalphenoliccontentfromLimonium brasilienseL.Molecules18,6852–6865.
BrazilianPharmacopeia,2010.FarmacopeiaBrasileira,5thed.ANVISA,Brasília. Chou,G.,Xu,S.J.,Liu,D.,Koh,G.Y.,Zhang,J.,Liu,Z.,2009.Quantitativeandfingerprint
analysesofchinesesweetteaplant(RubussuavissimusS.Lee).J.Agric.Food Chem.57,1076–1083.
Commission, E., 1994. Kommission E, Liste der Monographien. Bundesgesundheitsamt-InstitutfürArzneimittel,Bundesanzeiger.
Delaporte,R.H.,Sánchez,G.M.,Cuellar,A.C.,Mello,J.C.P.,2001.Controldecalidad yactividadantiinflamatoriadelasdrogasvegetalesAltemantberabrasiliana(L.) KuntzeyBoucheaflumtnensis(Vell.)Mold.ActaFarm.Bonaer.20,39–46. Ding,Y.,Wu,E.Q.,Liang,C.,Chen,J.,Tran,M.N.,Hong,C.H.,Jang,Y.,Park,K.L.,Bae,K.,
Kim,Y.h.,Kang,J.S.,2011.Discriminationofcinnamonbarkandcinnamontwig samplessourcedfromvariouscountriesusingHPLC-basedfingerprintanalysis. FoodChem.127,755–760.
Fenner,R.,Betti,A.H.,Mentz,L.A.,Rates,S.M.K.,2006.Plantasutilizadasnamedicina popularbrasileiracompotencialatividadeantifúngica.Rev.Bras.Cienc.Farm. 42,269–394.
GermanPharmacopeia,2015.DeutschesArzneibuch.DeutscherApothekerVerlag, Bonn.
Glasl,H.,1983.ZurPhotometrieinderDrogenstandardisierung–3. Gehaltsbestim-mungvonGerbstoffdrogen.Dtsch.Apoth.Ztg.123,1979–1983.
Gobbo-Neto,L.,Lopes,N.P.,2007.Plantasmedicinais:fatoresdeinfluênciano con-teúdodemetabólitossecundários.Quim.Nova30,374–381.
He,Z.D.,Qiao,C.F.,Han,Q.B.,Cheng,C.L.,Xu,H.X.,Jiang,R.W.,But,P.P.,Shaw,P.C., 2005.Authenticationandquantitativeanalysisonthechemicalprofileofcassia bark(CortexCinnamomi)byhigh-pressureliquidchromatography.J.Agric.Food Chem.53,2424–2428.
Heigl,D.,Franz,G.,2003.Stabilitytestingontypicalflavonoidcontainingherbal drugs.Pharmazie58,881–885.
ICH,2005.TheEuropeanAgencyfortheEvaluationofMedicinalProducts.Topic Q2(R1):ValidationofAnalyticalProcedures:Methodology.
Janhs,R.T.,Crescente,A.S.,1976.Ensaiosfarmacológicoseclínicoscomaassociac¸ão doextratofluidodeLimoniumbrasilienseeN-acetil-p-aminophenol.Trib.Pharm. 44,105–111.
Kilic¸gün,H.,Altiner,D.,2010.Correlationbetweenantioxidanteffectmechanisms andpolyphenolcontentofRosacanina.Pharmacogn.Mag.6,238–241.
Lopes,G.C.,Brainski,A.,Santos,P.V.P.,Diciaula,M.C.,Mello,J.C.P.,2010. Develop-mentandvalidationofanHPLCmethodforthedeterminationofepicatechin inMaytenusilicifolia(Schrad.)Planch.,Celastraceae.Rev.Bras.Farmacogn.20, 789–795.
Lopes,G.C.,Bruschi,M.L.,Mello,J.C.P.,2009.RP-LC–UVdeterminationof proantho-cyanidinsinGuazumaulmifolia.Chromatographia69,175–181.
Lopes,G.C.,Sanches,A.C.C.,Nakamura,C.V.,Dias-Filho,B.P.,Hernandes,L.,Mello, J.C.P.,2005.InfluenceofextractsofStryphnodendronpolyphyllumMart.and StryphnodendronobovatumBenth.onthecicatrisationofcutaneouswoundsin rats.J.Ethnopharmacol.99,265–272.
Marques,L.C.,Pieri,C.,Roman-Junior,W.,Cardoso,M.L.C.,Milaneze-Gutierre,M.A., Mello,J.C.P.,2007.ControlefarmacognósticodasraízesdeHeteropteris aphro-disiacaO.Mach.(Malpighiaceae).Rev.Bras.Farmacogn.17,607–615. Mello,J.C.P.,Petrovick,P.R.,2000.QualitycontrolofBaccharistrimera(Less.)DC
(Asteraceae)hydroalcoholicextracts.ActaFarm.Bonaer.19,211–216. Mueller-Harvey,I.,2001.Analysisofhydrolysabletannins.Anim.FeedSci.Technol.
91,3–20.
Mukherjee,P.K.,2002.QualityControlHerbalDrugs:AnApproachtoEvaluationof Botanicals.BusinessHorizons,NewDelhi,pp.183–245.
Murray,A.P.,Rodriguez,S.,Frontera,M.A.,Tomas,M.A.,Mulet,M.C.,2004. Antioxi-dantmetabolitesfromLimoniumbrasiliense(Boiss.)Kuntze.Z.Naturforsch.59c, 477–480.
Nisbet,L.J.,Moore,M.,1997.Willnaturalproductsremainanimportantsourceof drugresearchforthefuture?Curr.Opin.Biotechnol.8,708–712.
Rehwald,A.,Meier,B.,Sticher,O.,1994.Qualitativeandquantitativereversed-phase high-performanceliquidchromatographyofflavonoidsinCrataegusleavesand flowers.J.Chromatogr.A677,25–33.
Rosenthal,I.,Wolfram,E.,Meier,B.,2014.AnHPLCmethodtodeterminesennoside AandsennosideBinSennaefructusandSennaefolium.Pharmeur.Bio.Sci.Notes 2014,92–102.
Schofield,P.,Mbugua,D.M.,Pell,A.N.,2001.Analysisofcondensedtannins:areview. Anim.FeedSci.Technol.91,21–40.
Schulz,V.,Hänsel,R.,Blumenthal,M.,Tyler,V.E.,2004.Medicinal plants, phy-tomedicines,andphytotherapy.In:RationalPhytotherapy:AReferenceGuide forPhysiciansandPharmacists.Springer-Verlag,Berlin/Heidelberg,pp.1–42. Simirgiotis, M.J., Schmeda-Hirschmann, G.,2010. Directidentification of
phe-nolic constituents in Boldo Folium (Peumus boldus Mol.) infusions by high-performanceliquidchromatographywithdiodearraydetectionand elec-trosprayionizationtandemmassspectrometry.J.Chromatogr.A217,443–449. Soares,L.A.,Oliveira,A.L.,Ortega,G.G.,Petrovick,P.R.,2004.Developmentand validationofaLC-methodfordeterminationofcatechinandepicatechinin aqueousextractivesfromleavesofMaytenusilicifolia.J.Pharm.Biomed.Anal. 36,787–790.
Vihakas,M.,(Doctorthesis)2014.FlavonoidsandOtherPhenolicCompound: Char-acterizationandInteractionwithLepidopteranandSawflyLarvae.Universityof Turku,Turku,135pp.http://www.doria.fi/handle/10024/100939.