FACULDADE DE FARMÁCIA
Targeting MicroRNAs for Modulation of the Anti-Tumor
Human γδ T Cell Differentiation
Master Thesis Dissertation
Gisela Silva Gordino
Master in Biopharmaceutical Sciences - FFUL
Supervisor
Dra. Julie C. Ribot Molecular Medicine Institute
Targeting MicroRNAs for Modulation of the Anti-Tumor Human γδ T Cell Differentiation
UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIATargeting MicroRNAs for Modulation of the Anti-Tumor
Human γδ T Cell Differentiation
Master Thesis Dissertation
Gisela Silva Gordino
Master in Biopharmaceutical Sciences - FFUL
Supervisor
Dra. Julie C. Ribot Molecular Medicine Institute
Abstract
Recent developments in the immunology field have validated immunotherapy as being capable of extending cancer patients survival, highlighting its role as a promising anti-tumor treatment. However, a wider and more successful application of adoptive cell therapy requires a better understanding of the molecular determinants, in effector lymphocytes, of functional differentiation, recognition and elimination of tumor cells.
γδ T cells have been proposed as the first line of immune defense, acting in response to a variety of stress-inducible or pathogen-associated metabolites. Their activation includes a potent cytolytic and inflammatory activity against a wide range of malignant cells as one of its most important features. Although clinical trials have aimed at modulating their activity, they were only able to achieve objective responses in the range of 10-33%. Therefore, it has become evident that understanding the mechanisms involved in regulating γδ T cell activation and functional differentiation is critical for their manipulation in clinical settings.
Importantly, data from the host laboratory has recently shown that the anti-tumor effector properties of γδ T cells were selectively acquired upon stimulation with interleukin (IL)-2, but not with IL-7. The effects caused by IL-2 depended on the Mitogen-Activated Protein Kinase/Extracellular Signal-Related Kinase (MAPK/ERK) signaling and induced de novo expression of the transcription factors (TFs) T-bet and eomesodermin, as well as the cytolytic enzyme perforin. Based on this background, this project proposes to explore an additional layer in the regulation of γδ T cell differentiation, by characterizing the post-transcriptional mechanisms mediated by microRNAs (miRs).
In mammals, more than 700 miRs have already been identified and shown to regulate many developmental and differentiation processes. For instance, with regard to the immune system, overexpressing miR-491 decreases the interferon (IFN)-γ production in cluster of differentiation (CD)8+ T cells, miR-583 inhibits natural killer (NK) cells differentiation process, and miR-181a expression regulates activation of memory T helper (Th)17 cells through modulation of ERK phosphorylation. This notwithstanding, no miRs have yet been implicated in human γδ T cell differentiation into anti-tumor effectors.
By performing a ribonucleic acid-sequencing (RNA-seq) analysis, we have been able to identify a discrete repertoire of miRs possibly implied in regulating γδ T cell type 1 functional differentiation. Selected miRs candidates have been validated by reverse transcriptase-polymerase chain reaction (RT-PCR): miR-135b, 10a and 20b were upregulated in mature γδ T cells while; miR-181a and 196b were downregulated in mature γδ T cells.
We next manipulated the expression of these miRs following two different gain-of-function strategies (electroporation with mimics and retroviral transduction). We observed that miR-181a and 196b overexpression decreases γδ T cell differentiation into type 1 effectors, while enhancing their proliferation. On the other hand, our preliminary results indicate that miR-135b, 10a and 20b impair γδ T cell proliferation without affecting their anti-tumor functions. If confirmed, these findings could have major implications for the manipulation of γδ T cells in cancer immunotherapy.
Resumo
Desenvolvimentos recentes na área da imunologia validaram a imunoterapia como sendo capaz de aumentar a sobrevivência de pacientes com cancro, evidenciando o seu papel promissor no tratamento desta doença. No entanto, uma aplicação mais abrangente e bem-sucedida da terapia celular adoptiva requer um conhecimento profundo dos determinantes moleculares, em linfócitos T efectores, de diferenciação funcional, reconhecimento e eliminação de células tumorais.
Os linfócitos T γδ têm sido propostos como a primeira linha de defesa imunológica, em resposta a uma variedade de metabolitos induzidos pelo stress ou associados a organismos patogénicos. A activação destas células inclui uma actividade citolítica e inflamatória potente, contra uma vasta gama de células malignas, como uma das suas características principais. Embora os ensaios clínicos tenham tentado modular a actividade destes linfócitos, estes só alcançaram respostas objectivas na ordem dos 10-33%. Tornou-se assim evidente que a compreensão dos mecanismos envolvidos na regulação da activação e da diferenciação funcional dos linfócitos T γδ é crítica para a sua manipulação em contextos clínicos.
De forma importante, resultados obtidos pelo laboratório de acolhimento demonstraram que as propriedades anti-tumor efectoras dos linfócitos T γδ eram adquiridas, selectivamente, mediante estimulação com IL-2, mas não quando estimulados com IL-7. Os efeitos mediados por IL-2 eram dependentes da via de sinalização de MAPK/ERK e induziam a expressão de novo dos factores de transcrição T-bet e eomesodermina, assim como da enzima citolítica perforina. Com base nestes dados, este projecto propõe explorar um outro nível de regulação na diferenciação de linfócitos T γδ, caracterizando os mecanismos pós-transcrição mediados por miRs.
Em mamíferos, mais de 700 miRs foram já identificados e demonstrados como sendo capazes de regular vários processos de desenvolvimento e diferenciação. Em relação ao sistema imunitário, os padrões de expressão destas pequenas espécies de RNA variam consoante o tipo de linfócitos e também de acordo com o estadio de desenvolvimento. Curiosamente, alguns estudos demonstraram que esses perfis de expressão são únicos em cada etapa do desenvolvimento das células T tímicas. De acordo com esta informação, a remoção dos miRs maduros na fase inicial do desenvolvimento de timócitos resultou num bloqueio do seu desenvolvimento e na consequente redução de linfócitos T αβ periféricos assim como reduziu a pool de células iNKT. Adicionalmente, vários miRs têm sido também correlacionados com a desregulação das capacidades efectoras de diversos tipos de linfócitos. Alguns exemplos incluem: a sobreexpressão do miR-491 em linfócitos T CD8+ que demonstrou ser capaz de diminuir a produção de IFN-γ; a expressão do miR-583 foi correlacionada com a inibição da diferenciação de células NK e; a expressão do miR-181a demonstrou regular a activação de linfócitos de memória Th17 ao modular a fosforilação da ERK. Apesar disso, não surgiram ainda miRs implicados na diferenciação de linfócitos T γδ humanos em efectores anti-tumorais.
Neste trabalho fomos capazes de identificar um repertório discreto de miRs possivelmente implicados na regulação da diferenciação funcional do tipo 1 em linfócitos T γδ. Para tal, recorremos à análise dos dados obtidos em ensaios de sequenciação de RNA em linfócitos T
γδ imaturos versus maduros, isolados a partir de amostras de timo humano. Utilizando a metodologia de RT-PCR validámos esses dados, reduzindo a lista inicial de treze para cinco potenciais candidatos: miR-135b, 10a e 20b que se encontravam sobreexpressos em linfócitos T γδ maduros e; miR-181a e 196b que tinham expressão reduzida em linfócitos T γδ maduros. Surpreendentemente, quando analisada a presença destes miRs por RT-PCR em linfócitos T γδ isolados da periferia (i.e. amostras de sangue de dadores saudáveis), o padrão de expressão para os cinco candidatos era mais semelhante à sua expressão em timócitos γδ recém-isolados – ou seja, imaturos – do que à expressão observada em timócitos γδ em cultura com IL-2 – ou seja, maduros/diferenciados. Tais resultados parecem sugerir que o padrão de expressão destes miRs candidatos poderá estar associado a um processo transiente, capaz de restaurar os seus níveis basais assim que as células se encontrem completamente diferenciadas e entrem num processo de repouso. De acordo com esta hipótese, quando os linfócitos T γδ periféricos foram colocados em cultura com as citosinas IL-7 ou IL-2 – que atribui às células um estado imaturo ou maduro, respectivamente – o estímulo induzido pelas citosinas demonstrou ser capaz de romper com o estado de repouso das células, conforme comprovado pela alteração nos padrões de expressão destes miRs. Mais ainda, em linfócitos T γδ periféricos cultivados com IL-7, os miRs-135b, 10a e 20b aparentaram ter um comportamento diferente do observado em timócitos γδ em cultura também com IL-7, visto que a sua expressão surge aumentada nestas amostras periféricas.
Importantemente, estas experiências demonstraram também que as subpopulações Vδ1 and Vδ2 isoladas a partir do sangue de controlos saudáveis obtiveram valores semelhantes para a expressão dos miRs. Estes resultados estão então de acordo com o facto de se terem registado padrões semelhantes na expressão dos miRs candidatos, quando comparados os resultados entre as amostras de linfócitos T γδ isoladas a partir do timo com as amostras recém-isoladas a partir de sangue humano.
De seguida procedeu-se à manipulação da expressão destes miRs utilizando duas estratégias de ganho-de-função diferentes: electroporação com mimetizadores ou transdução retroviral. Ao sobreexpressar os miRs candidatos, usando o sistema Neon, fomos capazes de observar que nos linfócitos T γδ periféricos em cultura com IL-7+IL-2 a proliferação aparenta estar comprometida nas amostras que sobreexpressaram os miRs-135b, 10a e 20b, sobretudo quando delimitados à subpopulação Vδ2. Tal resultado é consistente com os dados obtidos durante as experiências de RT-PCR, que sugerem um papel para estes três miRs na regulação das respostas funcionais em linfócitos T γδ que já se encontrem completamente diferenciados. De forma importante, apesar de só se ter conseguido explorar a manipulação destes linfócitos numa amostra de timo, quando utilizando o sistema Neon, os resultados obtidos foram também indicativos de um papel na regulação do desenvolvimento e diferenciação de linfócitos T γδ por parte dos miRs-181a e 196b, visto que estes foram capazes de reduzir a expressão de TNF-α em cerca de 10% dentro da subpopulação Vδ1, ao mesmo tempo que parecem induzir a sua proliferação.
De acordo com estes dados, a transdução retroviral do miR-181a em linfócitos T γδ periféricos demonstrou ser capaz de aumentar a proliferação destas células enquanto, ao mesmo tempo, diminuiu a expressão de IFN-γ e TNF-α. No que concerne os resultados do processo de diferenciação, embora tenham ocorrido pequenas diferenças nas duas
subpopulações, tais alterações na subpopulação Vδ2 foram demasiado reduzidas para serem consideradas. Pelo contrário, a expressão de IFN-γ e TNF-α em células Vδ1+ foi significativamente comprometida pela presença do miR-181a, reduzindo a produção destas citosinas em cerca de 12%. Interessa salientar que esta não é a primeira vez que a sobreexpressão do miR-181a é correlacionada com a redução da produção de IFN-γ e TNF-α, visto que tal foi já observado recentemente em linfócitos humanos CD4+ ou na linha celular HEK293K, respectivamente.
Estes resultados, em conjunto com outros relatórios semelhantes, sustentam um papel regulatório do miR-181a na diferenciação funcional de células T, que deve ser aprofundado no que diz respeito aos linfócitos T γδ.
No seu conjunto, estas experiências de ganho-de-função permitiram-nos observar que a sobreexpressão dos miRs-181a e 196b diminui a diferenciação dos linfócitos T γδ em efectores do tipo 1, ao mesmo tempo que aumenta a sua proliferação. Por outro lado, estes resultados preliminares indicam que o miR-135b, 10a e 20b comprometem a proliferação destes linfócitos sem afectar as suas propriedades anti-tumorais. Interessantemente, em ambas as experiências de ganho-de-função, as células Vδ1+ surgiram com sendo a subpopulação que registou as alterações mais significativas. Deste modo, seria importante sobreexpressar estes miRs em mais amostras de timo, uma vez que os timócitos γδ são enriquecidos em células Vδ1. Se validados, estes resultados poderão ter grandes implicações na manipulação dos linfócitos T γδ para a imunoterapia em cancro.
Como seguimento lógico deste projecto, pretende-se identificar as redes de mRNAs controladas pelos nossos miRs candidatos, recorrendo a uma combinação de ferramentas bioinformáticas e bioquímicas. Esta visão integrada dos efeitos de um determinado miR no transcriptoma humano será fundamental para compreender o seu papel na diferenciação funcional dos linfócitos T γδ.
Adicionalmente, pretende-se tentar definir um perfil patogénico para estes miRs na diferenciação funcional dos linfócitos T γδ, tendo como base amostras de pacientes com cancro. Interessantemente, a presença e as capacidades efectoras dos linfócitos T γδ periféricos foram anteriormente relatadas como estando comprometidas em amostras de pacientes com melanoma, glioblastoma e cancro do estômago. Como tal, cremos ser possível observar uma expressão anormal dos miRs envolvidos no processo de diferenciação funcional dos linfócitos T γδ em amostras obtidas a partir de sangue periférico deste género de pacientes. Para verificar essa hipótese, contamos com a parceria estabelecida com o Departamento de Oncologia do Hospital de Santa Maria, através do Dr. Luís Costa, que nos permitirá ter acesso a amostras de sangue obtidas a partir de um grupo definido de pacientes, aquando a altura do diagnóstico.
Acknowledgements
I wish to take this opportunity to thank to everyone who has been involved in this journey, which allowed me to grow, not only professionally but also personally.
To begin, I would like to thank Professor Dr. Bruno Silva-Santos for giving me the opportunity to integrate this already large “family”, known as the BSS lab. Thanks for always believing in me and in this project, and to always try to push me further. Your constructive criticism not only allowed me to grow scientifically, but thanks to you I have always tried to improve my work every step of the way.
I also wish to give thanks to all of my colleagues in the BSS lab, who made me feel welcomed since the first day, but in particular to those who have also endowed me with some of their skills. In special, I want to thank Nina for the enormous sympathy and the amazing help she gave me by teaching me how to work with the Neon system, and also with the retrovirus. I could always come to you with a million questions that you always answered with kindness and a smile on your face. Thank you for that! To Tiago, my main “retrovirus teacher”, who taught me almost everything I know about culturing bacteria and producing virus. Thank you for the many hours you have spent unveiling the mysteries of this “small world” to me and for making these experiments look more “user friendly”. To Anita, thanks for always trying to help me with my microRNA doubts and for teaching me how to do the sequencing step. To Natacha, thanks for teaching me how to do an RT-PCR with those amazing 384 well plates, and for helping me to do it three times!
There are other several colleagues, who have somehow made my life in the lab easier, and to whom I wish to thank: to Karine for always trying to help when I was having some doubts in my experiments and for listening to my first oral presentation when I was so nervous; to Sérgio, for showing me where (and how) to pick up the thymus and blood samples, and for processing the thymus for me sometimes, when I was too busy; to André, Biagio, Cláudia, Paula and Pedro, for their selfless help, their sympathy and their kind word during rough times; to Helena, our more recent teammate, in whom I have found an unexpected friend, thank you for that special hug when I needed the most; and to Sofia, your constant help is priceless, you have literally saved the final step of my retrovirus experiment, so if I have results to show here it is thanks to you!
I also wish to give thanks to some of my friends and family. To my dad and my grandparents, for supporting me in this step of my education, for never judging and for taking care of me the best you knew and you were able to. Thank you for your enormous heart!
To my closest friends Rita and Andreia, for listening and for giving me support, and for never complaining about me not having the time to visit you, even if just to drink a coffee. Thank you for being there for me, always!
Finally, there are two special persons to whom I owe and to whom I want to thank the most. To Julie: my mentor, my teacher and my friend. You have always trusted in me and in my skills from the beginning and you always believed I would thrive not only in the lab, but also in other many aspects of my life. Thank you for being my role model: an excellent hardworking scientist and also an excellent person, as I have only met few during my whole
life. I could not have asked for a better supervisor. In you I could always find a word of encouragement, a sparkle of optimism and a solution for every problem. Even when I saw no light at the end of the tunnel, you did, and you always made me believe that we were able to pull this through, as we did! So thank you for this marvelous journey, for teaching me, for letting me try to do things my own way, for having my back every time and for making me go beyond my limits, but most of all thank you for choosing me.
To my boyfriend: I could write a whole page just to thank you and still there would be a lot more to say! Thanks for being there for me every step of the way, for helping picking me up when I was down, for believing in me and for listening to me talking about my work, even when I had the same conversation over and over again, hopping that you were able to provide me with the answers. Thank you for waking up earlier than you had to just so you could make me company in those hard days when I had to deal with my “ghosts”, I would never forget that in a million years! Thank you for the sleepy breakfasts, the long conversations at late (and earlier) hours, and for your unselfish and unconditional love. You always said that everything was going to be just fine, and you were right, it really did!
I cannot finish without thanking to my guardian angel, my mom, who raised me to be the women I am today, and to whom I turn to in my darkest hours. I know you are always looking out for me and that somehow you had always given the strength to achieve my goals. Wherever you are, I hope I am making you proud.
Table of Contents
Abstract ... 3 Resumo ... 4 Acknowledgements ... 7 List of Figures ... 11 List of Abbreviations ... 13 1. Introduction ... 171.1 γδ T Cells as Key-Players in Immunity ... 17
1.1.1 Adaptive vs Innate Immune System ... 17
1.1.2 γδ T Cell Development and Differentiation ... 18
1.1.3 γδ T Cell Target Recognition and Response ... 21
1.2 Cancer and γδ T Cells ... 23
1.2.1 The Immune System and Cancer ... 23
1.2.2 γδ T Cells in Cancer ... 24
1.3 MiR Role in Gene Regulation ... 27
1.3.1 MiR Biogenesis and Maturation ... 27
1.3.2 MiR Silencing Mechanisms ... 28
1.3.3 MiR-Mediated Regulation of T Cell Biology ... 29
2. Aims of the Thesis ... 33
3. Materials and Methods ... 35
3.1 Ethics ... 35
3.2 Lymphocyte Preparations ... 35
3.3 Cell Culture ... 35
3.4. Quantitative RT-PCR ... 36
3.5 γδ T cell Transfection using Neon System for Mimics Delivery ... 36
3.6 MiR-Overexpressing Recombinant Retrovirus Production ... 37
3.7 γδ T cell Transduction with the Retroviral Constructs ... 38
3.8 FACS Analysis ... 39
3.9 Statistical Analysis ... 40
4. Results ... 42
4.1 MiR Signature Associated with γδ T Cell Type 1 Differentiation ... 42
4.2 RT-PCR Validation of the miR Candidates ... 43
4.2.2 MiR Candidates Expression Profile in Peripheral γδ T Cells is more Similar to
Freshly-isolated than to IL-2 Cultured γδ Thymocytes ... 44
4.2.3 Comparison of miR Candidates Expression on Vδ1 vs Vδ2 Subpopulations Shows No Significant Differences in their Expression Levels ... 46
4.3 Setup of the Conditions for an Efficient miR Transfection using the Neon System ... 47
4.4 Analysis of the Impact of Mimics Delivery on γδ T Cell Differentiation and Proliferation ... 50
4.4.1 Peripheral Vδ2 γδ T Cells Transfected with miR-135b, 10a and 20b Mimics tend to Proliferate Less when Cultured with IL-7 plus IL-2 ... 51
4.4.2 Peripheral γδ T Cells Transfected with the miR Mimics show No Significant Differences in IFN-γ and TNF-α Production ... 52
4.4.3 MiR-181a and miR-196b Overexpression Decreases TNF-α Production in Vδ1 Electroporated γδ Thymocytes ... 53
4.5 Overexpression of Candidate miRs using Retroviral Constructs ... 54
4.5.1 MiR Overexpression was Not Efficient for All of the miR Constructs ... 54
4.5.2 MiR-181a Overexpression in Peripheral γδ T Cells Increases Cell Proliferation ... 55
4.5.3 MiR-181a Overexpression in Peripheral γδ T Cells Increases Vδ1 T Cell Activation and Programmed Cell Death ... 56
4.5.4 Overexpression of miR-181a in Peripheral γδ T Cells did not Influence their Effector/Memory Phenotype ... 58
4.5.5 Overexpression of miR-181a in Peripheral γδ T Cells Reduces IFN-γ and TNF-α Production in the Vδ1 Subpopulation ... 60
5. Discussion ... 63
6. Future Plans ... 72
List of Figures
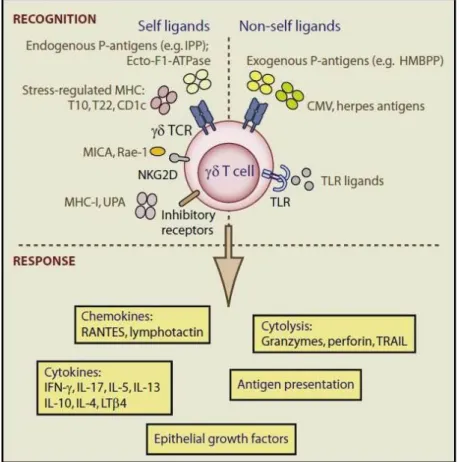
Figure 1 - γδ T cell recognition and response. ... 22
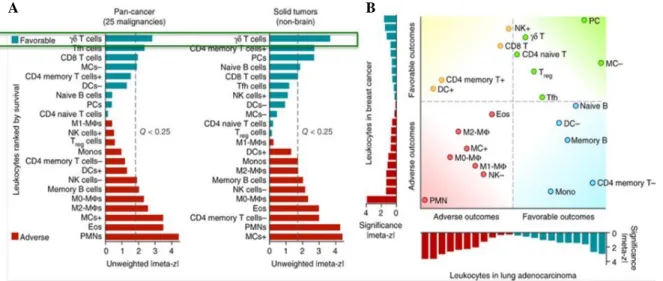
Figure 2 – Inferred leukocyte frequencies and prognostic associations in 25 human cancers. ... 24
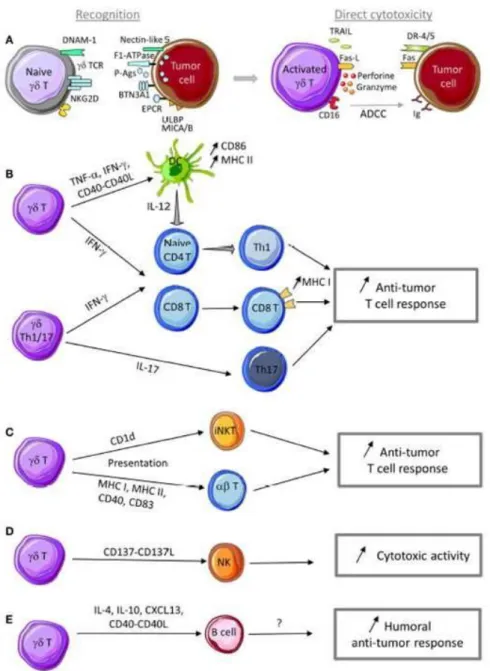
Figure 3 – Anti-tumor functions of γδ T cells. ... 26
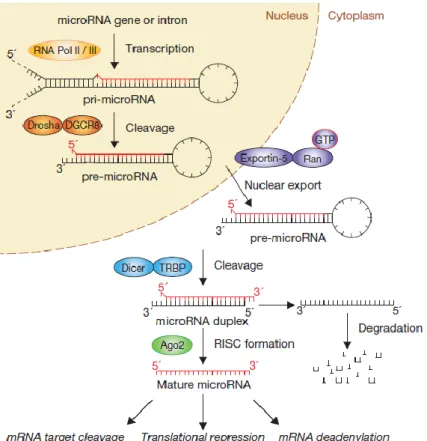
Figure 4 – The “linear” canonical pathway of miR processing. ... 29
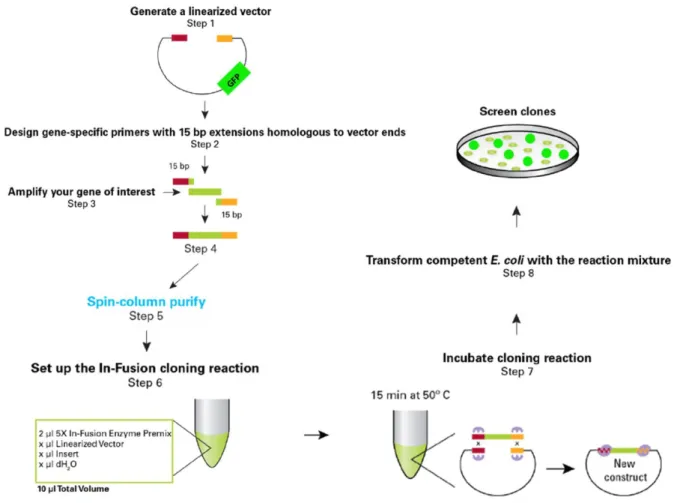
Figure 5 – Schematic representation of the procedure to develop a retroviral construct with the DNA of interest. ... 38
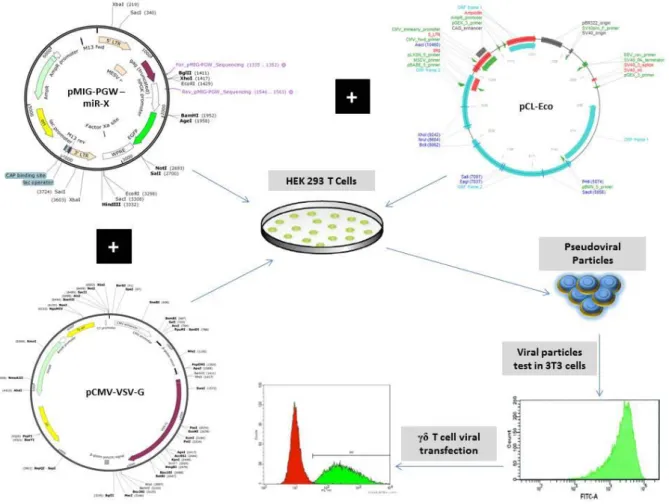
Figure 6 – Schematic representation of the transfection process. ... 39
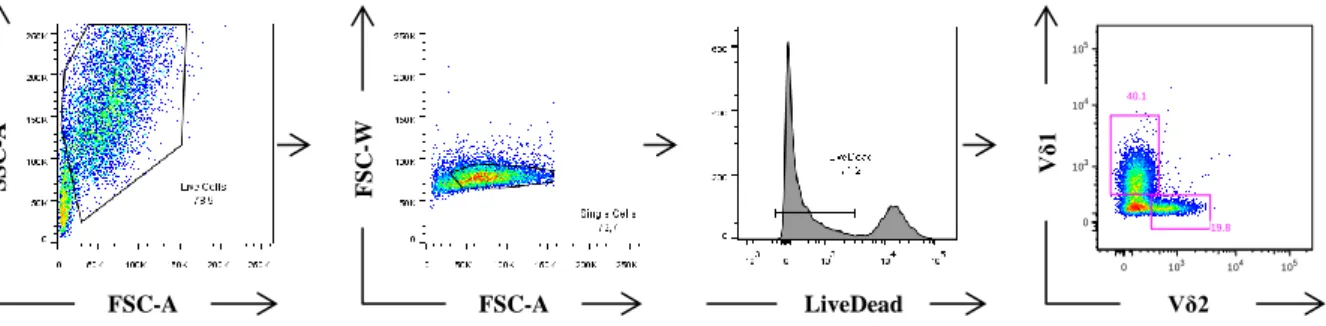
Figure 7 – FACS gating strategy. ... 40
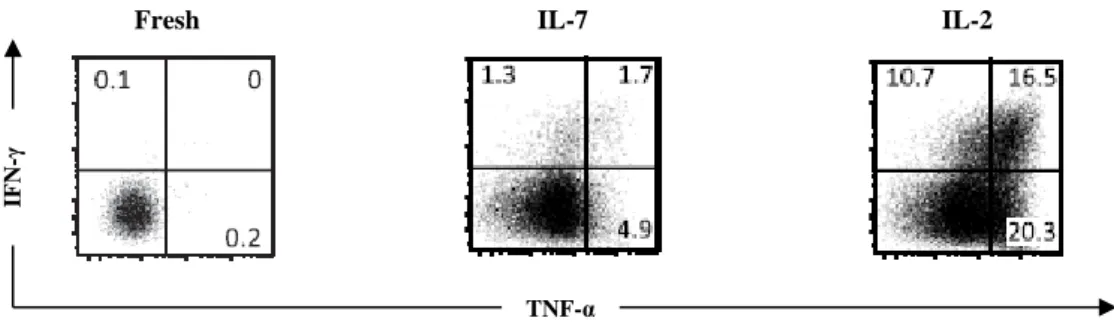
Figure 8 – Human γδ thymocytes are devoid of IFN-γ production and cytotoxic functions but IL-2 signal differentiates them into cytotoxic type 1 effector T cells. ... 42
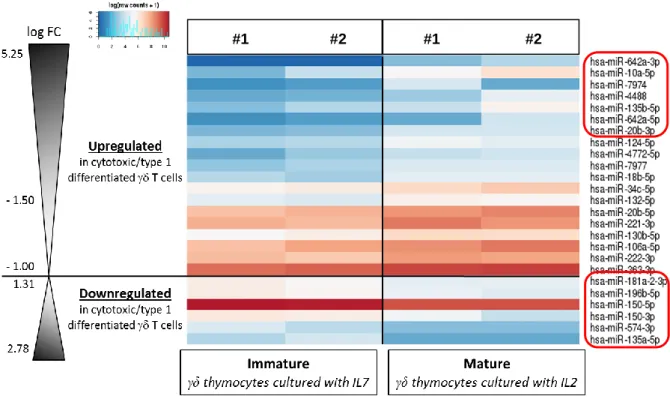
Figure 9 – MiR signature associated with γδ T cell type 1 differentiation. ... 43
Figure 10 - RT-PCR in γδ thymocytes validates top five miR candidates. ... 44
Figure 11 - MiR candidates expression profile in peripheral γδ T cells is more similar to freshly-isolated than to IL-2 cultured γδ thymocytes. ... 45
Figure 12 – MiR-135b, 10a and 20b expression is upregulated in IL-7 or IL-2 cultured peripheral γδ T cells indicating a rupture in their resting status. ... 46
Figure 13 - Comparison of miR candidates expression on Vδ1 vs Vδ2 subpopulations shows no significant differences in their expression levels. ... 47
Figure 14 – Transfection of the siRNACD45 using the Neon system proves to be efficient on silencing CD45 in γδ T cells. ... 48
Figure 15 – Transfection of the siRNACD45 using the Neon system does not compromise γδ T cell survival. ... 49
Figure 16 – Neon protocol timeline for mimics delivery. ... 50
Figure 17 – Mimics delivery to PBMC-isolated γδ T cells using the Neon system occurred efficiently. ... 50
Figure 18 – Peripheral Vδ2 γδ T cells transfected with miR-135b, 10a and 20b mimics tend to proliferate less when cultured with IL-7 plus IL-2. ... 51
Figure 19 – Peripheral γδ T cells transfected with the miR mimics show no significant differences in IFN-γ and TNF-α production. ... 52
Figure 20 – MiR-181a and miR-196b overexpression decreases TNF-α production in Vδ1 electroporated γδ thymocytes. ... 53
Figure 21 –MiR overexpression was not efficient for all of the miR constructs: 3T3 cells. ... 54
Figure 22 – MiR overexpression was not efficient for all of the miR constructs: γδ T cells. ... 55
Figure 23 – MiR-181a overexpression in peripheral γδ T cells increases cell proliferation. ... 56
Figure 25 – MiR-181a overexpression in peripheral γδ T cells increases Vδ1 T cell programmed cell death. ... 58 Figure 26 – Overexpression of miR-181a in peripheral γδ T cells did not influence their effector/memory phenotype. ... 59 Figure 27 – Overexpression of miR-181a in peripheral γδ T cells reduces IFN-γ and TNF-α production in the Vδ1 subpopulation... 60 Figure 28 – Summary of the results obtained upon overexpression of miR-181a in peripheral γδ T cells... 61
List of Abbreviations
Ab: antibodyADCC: Ab-dependent cell cytotoxicity Ag: antigen
AGO: Argonaute
AICD: activation-induced cell death APC: antigen-presenting cells ATPase: F1-Adenylpyrophosphatase B: bone marrow-derived
BFA: brefeldin
BTN: butyrophilin family
C. elegans: Caenorhabditis elegans CD: cluster of differentiation
cDNA: complementary deoxyribonucleic acid CLL: chronic lymphocytic leukemia
CM: central memory CMV: cytomegalovirus D: diversity
DC: dendritic cell
DETC: dendritic epidermal γδ T cells
DGCR8: DiGeorge syndrome critical region 8 DN: double-negative
DOT: delta one T DP: double-positive
EA-1: early activation antigen EM: effector memory
ERK: extracellular signal-related kinase Exp5: exportin-5
FACS: fluorescence-activated cell sorting FISH: fluorescent in situ hybridization γδ NKT cells: NK1.1+
γδ cells
GFP: green fluorescent protein GLUT1: glucose transporter 1
HEK293T TAT: human embryonic kidney 293 cells with large T antigen and a transcription transactivator
HITS-CLIP: high-throughput sequencing of RNA isolated by cross-linking immunoprecipitation
HIV: human immunodeficiency virus
HMBPP: (E)-4-hydroxy-3-methyl-but-2-enylpyrophosphate IFN-γ: interferon-γ
Ig: immunoglobulin IL: interleukin
iNKT: invariant natural killer T IPP: isopentenyl pyrophosphate J: junctional
LNA: locked nucleic acid mAb: monoclonal Ab
MACS: magnetic cell sorting
MAPK: mitogen-activated protein kinase MDSCs: myeloid derived suppressor cells MFI: mean-fluorescence intensity
MHC: major histocompatibility complex MICA: MHC Class-I chain-related A MICB: MHC Class-I chain-related B miR: microRNA
mRNA: messenger RNA
NEAA: minimum essential amino acids NKG2D: natural killer group 2 member D NK: natural killer
NKR: NK cell receptor NKT: natural killer T P-Ag: phosphoantigen
PBL: peripheral blood lymphocyte PBMC: peripheral blood monocyte cells PCR: polymerase chain reaction
Pen/Strep: penicillin and streptomycin
pMIG-PGW: MSCV-IRES-GFP-PGW plasmid pre-miR: precursor miR
pri-miR: primary miR
PRKC: protein kinase C epsilon PRR: pattern recognition receptor qPCR: quantitative PCR
RISC: RNA-induced silencing complex RNA: ribonucleic acid
RNase: ribonuclease
RNA-seq: RNA sequencing RT: reverse transcriptase siRNA: small interfering RNA
SNORD44: small nucleolar RNA C/D Box 44 SP: single-positive
T: thymic-derived
T-ALL: T cell acute lymphoblastic leukemia TAL: tumor-associated leukocytes
TCR: T-cell receptor TF: transcription factor Th: T helper
TIL: tumor infiltrating lymphocytes TLR: toll-like receptor
TME: tumor microenvironment TNF-α: tumor necrosis factor α UTR: untranslated region V: variable
1. Introduction
1.1 γδ T Cells as Key-Players in Immunity
1.1.1 Adaptive vs Innate Immune SystemDefense against microbial assaults is an essential necessity for all living organisms. Consequently, all life forms have evolved strategies that are designed to limit the invasion of the host by microorganisms (Schenten and Medzhitov, 2011). Immunity, as it is known, refers to the global ability of the host to resist the predation of microbes that would otherwise destroy it. It has many facets, but the greatest dichotomy separates adaptive immunity – usually known as “acquired immunity” – from innate immunity – also termed as “natural immunity” or “innate resistance” (Hoebe et al., 2004).
Traditionally, innate immunity is assumed to be rapid, “non-specific”, and identical qualitatively and quantitatively each time the same pathogen is encountered. Many of the innate immune cells are considered to be short-lived, making “memory” a moot concept. On the other hand, adaptive immunity features are considered to include the generation of long-lived, antigen (Ag)-specific cells after initial exposure to an Ag or pathogen. Thus, these cells respond faster and more robustly to subsequent encounters with the same Ag or pathogen, which consists the basis of vaccination (Clem, 2011; Lanier and Sun, 2009).
The adaptive immunity is usually attributed to two classes of specialized cells: thymic-derived (T) lymphocytes and bone marrow-derived (B) lymphocytes, which further differentiate into plasma cells in order to secrete antibodies (Ab) (Lanier and Sun, 2009; Medzhitov and Janeway, 2000).
Deletion of self-reactive T and B cell clones during lymphocyte development forms the basis for the discrimination between self and non-self by the adaptive immune system (Schenten and Medzhitov, 2011). Lymphocytes able to overcome this clonal deletion will display a single kind of structurally unique receptor, creating a very large and diverse repertoire of antigen receptors in the entire lymphocyte population, in a process called clonal selection, thus increasing the probability of an individual lymphocyte to encounter an Ag that binds to its receptor. Upon infection these lymphocytes are activated and start to proliferate, by a process known as clonal expansion, a mechanism which is essential for the generation of an efficient immune response (Medzhitov and Janeway, 2000; Mogensen et al., 2009).
Adaptive immunity comprises many of the same effector mechanisms that are used in the innate immune system, but is able to target them with greater precision. However, it takes three to five days for sufficient numbers of clones to be produced and to differentiate into effector cells, creating a time gap that would allow for most pathogens to damage the host. In contrast, the effector mechanisms of innate immunity are activated immediately after infection and rapidly control infecting pathogen replication. For this reason, containing the infection until the lymphocytes can begin to deal with it is considered the main function of the innate immunity (Janeway et al., 2001; Medzhitov and Janeway, 2000).
Granulocytes, monocytes, macrophages, dendritic cells (DCs), and NK cells have been delegated to the innate immune system, which also comprises epithelial cell barriers, proteins
of the complement system and anti-microbial peptides, and also other soluble factors (Hoebe et al., 2004; Lanier and Sun, 2009).
The innate immune response relies on recognition of evolutionarily conserved structures on pathogens, termed pathogen-associated molecular patterns (PAMPs), through a limited number of germ line-encoded pattern recognition receptors (PRRs), of which the family of Toll-like receptors (TLRs) is the most well-known (Akira et al., 2006; Medzhitov and Janeway, 2000; Mogensen, 2009). In specific cases, PRRs also recognize host factors as “danger” signals, as for instance when they are present in aberrant locations or abnormal molecular complexes as a consequence of inflammation, infection or in other types of cellular stress (Beg, 2002; Mogensen, 2009).
PAMPs are characterized by being invariant among entire classes of pathogens, essential for pathogen survival, and distinguishable from “self” (Janeway, 1989). Detection of PAMPs by PRRs leads to the induction of inflammatory responses and innate host defenses. PRR-induced signal transduction pathways ultimately result in the activation of gene expression and synthesis of a broad range of molecules, which include: cytokines, chemokines, cell adhesion molecules, and immunoreceptors (Akira et al., 2006; Iwasaki and Medzhitov, 2015; Mogensen, 2009). In addition, the sensing of microbes by PRRs expressed on antigen-presenting cells (APCs), particularly DCs, will lead to the activation of adaptive immune responses (Iwasaki and Medzhitov, 2015).
It is now universally recognized that innate instruction of adaptive immunity is a critical step that controls the activation, types, and duration of the adaptive immune response. Innate instruction occurs initially during the interaction between APCs and T cells. While this interaction is critical for the generation of an adaptive immune response, it is clear that innate control of the adaptive immunity is a process that occurs at multiple stages throughout the immune response and involves all cell types contributing to a particular response (Hoebe et al., 2004; Schenten and Medzhitov, 2011). Importantly, the distinctions between innate and adaptive immunity have recently become blurred: certain subsets of B and T cells, such as B1 cells, γδ T cells, and invariant natural killer T (iNKT) cells, are often referred to as innate-like lymphocytes (Murphy et al., 2008); while some innate immune cells, as for instance the NK cells, seem not to fit the conventions, by showing features normally attributed exclusively to cells of the adaptive immune system (Lanier and Sun, 2009; O’Leary et al., 2006; Sun et al., 2009). In this project we will be focusing our attention to the less well-known subset of T lymphocytes, which seems to comprise features from both the adaptive and the innate immune system: the γδ T cells.
1.1.2 γδ T Cell Development and Differentiation
T lymphocytes develop from a common lymphoid progenitor in the bone marrow that also gives rise to B lymphocytes, but those progeny destined to give rise to T cells leave the bone marrow and migrate to the thymus (Janeway et al., 2001). T cell differentiation in the thymus can be divided into discrete stages based on CD4 and CD8 expression: CD4 and CD8 double-negative (DN) early thymic progenitors; more differentiated CD4 and CD8 double-positive
(DP) thymocytes and; differentiated CD4 or CD8 single-positive (SP) thymocytes. The DN stage is heterogeneous and can be subdivided into four distinct subsets in mice, based on the expression of CD44 and CD25 (Germain, 2002). In humans, three distinct DN stages can be recognized: a CD34+CD38-CD1a- stage that represents the most immature thymic subset and the consecutive CD34+CD38+CD1a- and CD34+CD38+CD1a+ stages, with CD1a expression correlating with T lineage commitment (Dik et al., 2005; Joachims et al., 2006).
T-cell development has to accommodate the production of two distinct lineages of T cells with different types of T-cell receptor (TCR): αβ and γδ (Janeway et al., 2001). Although most T cells express αβ TCR and either of the TCR-associated molecules CD4 or CD8 on their surface, a small minority (1–10%) demonstrates a γδ TCR, predominantly CD4–CD8– (i.e. DN) phenotype, an observation that can be explained by the fact that γδ TCR directly binds to an Ag superstructure in an manner that is independent of the Major Histocompatibility Complex (MHC)/peptide complexes (Deniger et al., 2014; Girardi, 2006; Kabelitz et al., 2014; Lafont et al., 2014). Expression of a γδ TCR heterodimer at the surface of a T cell in the thymus inhibits recombination of βTCR-chain locus during the CD4–CD8– stage, leading to commitment of the T cell to the γδ lineage (Deniger et al., 2014; Xiong and Raulet, 2007). When exiting the thymus, this DN status is typically maintained probably because co-receptors are dispensable for functional γδ TCR binding to Ags (Hayday, 2009; Prinz et al., 2013). Importantly, commitment to the γδ T cell lineage has consistently been shown to be dependent on the Notch signaling pathway, with the Notch-activators NOTCH1 and NOTCH3 genes instructing the earliest human intrathymic precursors to adopt a γδ T-cell fate (Ciofani et al., 2006; García-Peydró et al., 2003; Van de Walle et al., 2013).
γδ T cells are defined by the expression of γ and δ heterodimer of TCR chains (γTCR/δTCR) (Deniger et al., 2014; Xiang et al., 2014). For these two TCR loci, recombination of variable (V), diversity (D, for δ chain), and junctional (J) region sequence elements generates a TCR diversity, similar to the generation of B-cell Ab diversity via recombination of heavy and light chains (Girardi, 2006).
The human γδ TCR complex is composed by the γδ TCR itself and various CD3 chains following the stoichiometry: TCRγδCD32γδζ2 (Siegers et al., 2007). The assembly of a γδ TCR complex in thymic progenitors has immediate consequences for γδ T-cell development as the “strong” signals stemming from this kind of TCR – when compared to the “weaker” signals from the pre-TCR – drive γδ/αβ common precursors into the γδ lineage (Haks et al., 2005; Hayes et al., 2005; Zarin et al., 2015).
Functional responses by γδ T cells can be stratified by the V region of the TCRδ chain, also usually termed as Vδ (Deniger et al., 2014; Kabelitz et al., 2014). In humans, γδ T cells are limited to a small repertoire of V gene segments when undergoing chain rearrangement in comparison with those available for Vα, Vβ, Immunoglobulin (Ig) light or Ig heavy chain rearrangements. Three main Vδ gene segments – Vδ1, Vδ2 and Vδ3 – are most frequently used in the rearrangement of this chain, although there are five less used Vδ segments that have both a Vδ and a Vα designation, due to the location of the δ locus within the α locus on the 14th human chromosome (Adams et al., 2015; Boneville et al., 2010; Thedrez et al., 2007). Seven functional Vγ gene segments - Vγ2, Vγ3, Vγ4, Vγ5, Vγ8, Vγ9 and Vγ11 -
located within the γ locus on chromosome 7 in humans are used for rearrangement of the γ chain, but several Vγ pseudogenes who are also found in this locus are not used in productively arranged γδ TCRs (Adams et al., 2015). This restricted repertoire of Vδ and Vγ gene segments available for rearrangement has led to speculate that these TCRs recognized only conserved self-proteins of low variability. However, even though there are much more V elements that may be used for α and β gene rearrangements, γδ TCRs have a greater potential diversity than αβ T cells because of their ability to incorporate multiple tandem copies of their D elements (Adams et al., 2015; Elliott et al., 1988; Girardi, 2006).
Productive recombination and pairing of matching the γ and δ chains facilitates the transition of a “γδ quality control checkpoint”, selecting γδ T-cell precursors for their competence to transduce signals via their TCR (Livak et al., 1995; Passoni et al., 1997; Prinz et al., 2006; Prinz et al., 2013). In mice, naïve γδ T cells – as defined by a CD44loCD27+CD62L+ phenotype – can leave the thymus at this point and populate secondary lymphoid organs and blood (Prinz et al., 2013; Turchinovich and Pennington, 2011). However, γδ T cells that do not exit the thymus at this stage can undergo further intrathymic differentiation before they are exported to the periphery. This differentiation process will result in the development of multiple murine γδ T-cell subsets, starting with the dendritic epidermal γδ cells (DETCs), followed by the IL-17A-producing γδ cells (γδ17 cells), and ultimately the NK1.1+ γδ cells (γδ NKT cells) (Azuara et al., 1997; Haas et al., 2009; Prinz et al., 2013; Vicari et al., 1996; Zarin et al., 2015). Also, γδ T cells can be further differentiated into IFN-γ producers (Schmolka et al., 2013; Shibata et al., 2012; Shibata et al., 2014; Zarin et al., 2015).
Mouse and human γδ T cells share many developmental and functional properties (Bonneville et al., 2010; Vantourout and Hayday, 2013), such as the fact that γδ T cells are highly effective at killing tumor cells and providing IFN-γ-mediated protective responses against cancer. Moreover, the main determinants of tumor cell recognition — namely the γδ TCR and NK cell receptors (NKRs), such as the natural killer group 2 member D (NKG2D) — are shared by both species (Correia et al., 2013; Silva-Santos et al., 2015).
However, in contrast to what has been observed in mice, where functional properties of γδ T cells can be acquired during their development in the thymus by a process known as “developmental pre-programming” (Ribot et al., 2009), human γδ thymocytes are immature (Ribot et al., 2014). In fact, their Th1/cytotoxic functions are selectively acquired only upon ex vivo stimulation with IL-2 or IL-15 – but not when using IL-4 or IL-7 – through a process dependent on the MAPK/ERK signaling (Ribot et al., 2014). This stimulus induced the de novo expression of the TFs T-bet and eomesodermin, as well as the cytolytic enzyme perforin, required for the cytotoxic type 1 program (Ribot et al., 2014).
In humans, γδ T cells can usually be found in the human mucosa, tongue, vagina, intestine, lung, liver, and skin and can comprise up to 50% of the T cell population in intestinal epithelial lymphocytes. Interestingly, in adult thymus, Vγ3, Vγ4 and Vγ1 rearrangements are suppressed by the programmed rearrangement process, favoring production of Vγ2+ and other adult-type γδ T cells. This indicates that a maturation process might confer these cells with their specific homing properties which enables them to exit the thymus and home to secondary lymphoid organs (Xiong and Raulet, 2007).
Additionally, circulating γδ T cells can be found in the blood and lymphoid organs and represent up to 0,5–16% (on average: 4%) of all CD3+ cells found in adult peripheral blood (Carding and Egan, 2002; Deniger et al., 2014; Lafont et al., 2014). Specifically, the Vδ2 isotype is expressed by 50–95% of γδ T cells from human peripheral blood, whereas TCRs including the Vδ1 and/or Vδ3 isotypes are predominantly found in γδ T cells from tissues (skin, intestine, thymus) (Bonneville et al., 2010; Deniger et al., 2014; Lafont et al., 2014, Ribot et al., 2014).
1.1.3 γδ T Cell Target Recognition and Response
Usually considered innate-like T cells (deBarros et al., 2011; Lafont et al., 2014), γδ T cells possess a combination of innate and adaptive immune cell qualities. In fact, they can rearrange TCR genes to produce J diversity and develop a memory phenotype, which is a feature of the adaptive immune system, but they can also use a restricted TCR as a pattern recognition receptor, which is a feature of the innate immune system (Girardi, 2006).
Some of their main features include being involved in the stress response to injured epithelia and in tissue homeostasis by limiting the dissemination of malignant or infected cells and by regulating the nature of the subsequent adaptive immune response. Additionally, γδ T cells have potent MHC-unrestricted cytotoxicity, a high potential for cytokine release and a broad-spectrum recognition of cancer cells (Hannani et al., 2012). Furthermore, ligands that interact with γδ TCRs can be MHC molecules, MHC-like molecules or MHC-unrelated molecules (figure 1), and receptor recognition varies accordingly with the γδ T cell subtype (Bonneville et al., 2010; Hayday, 2009).
Most of the known human γδ T cell ligands are specific for the Vδ1 or the Vδ2 isotype (Deniger et al., 2014). Vδ1+ γδ T cells are capable of recognizing several members of the MHC superfamily family, all of which deemed MHC-like ligands, including members of the CD1 family presenting lipids (Adams et al., 2015; Bonneville et al., 2010; Cardigan and Egan, 2002). For instance, both Vγ1Vδ1 and Vγ2Vδ1 recognize the non-polymorphic MHC molecule CD1c; Vγ5Vδ1 is a receptor for a galactosylceramide-CD1d complexes commonly described in the activation of natural killer T (NKT) cells (Deniger et al., 2014; Spada et al., 2000) and; recently CD1d has been shown to be a ligand for at least a subset of both Vδ1+ and Vδ3+
γδ T cells (Adams et al., 2015). Moreover, Vδ1+ cells have specificity for MHC Class-I chain-related A and B (MICA and MICB, respectively), molecules that participate in evasion of immune surveillance following viral infection and are usually present on tumor cells in response to cellular stress (Bonneville et al., 2010; Gomes et al., 2010; Spada et al., 2010; Xu et al., 2011).
Vδ2+
γδ T cells have preferred pairing with Vγ9, originating Vγ9Vδ2 cells, and this is the most extensively studied sub-group of human γδ T cells. Several of its ligands have already been identified, which include: the phosphoantigen (P-Ag) isopentenyl pyrophosphate (IPP) and its isomer (E)-4-hydroxy-3-methyl-but-2-enylpyrophosphate (HMBPP) (Alexander et al., 2008); members of the butyrophilin family (BTN), such as BTN3A1 (Harly et al., 2012); the F1-Adenylpyrophosphatase (ATPase) (Scotet et al., 2005); the apolipoprotein A-I (Scotet et
al., 2005) and; Mycobacterium tuberculosis and Mycobacterium leprae (Constant et al., 1994; Lafont et al., 2001).
In contrast to the Vδ2 subpopulation, much less is known about the Vδ1 subset and even less is known about the cells expressing the remainder Vγ chains. However, similarly to the observed in Vγ9Vδ2 T cells, there is indirect evidence of a Vγ3 role in immunity against cytomegalovirus (CMV) (Déchanet et al., 1999; Vermijlen et al., 2010) and human immunodeficiency virus (HIV) (Wesch et al., 1998).
Figure 1 - γδ T cell recognition and response. For γδ T cells to be suitable for stress surveillance they recognize a spectrum of molecules signifying dysregulation. These molecules may be self-encoded TCR and NKG2D ligands (T10 and T22, P-Ags, MICA, etc.), or non-self-encoded, e.g., common products of multiple pathogens (HMBPP), unique products of very common pathogens (e.g., putative CMV or Herpes ligands), or TLR ligands. The cells can then deploy several types of function appropriate to different types of stress, directed against non-self or self-targets. The responses are negatively regulated via inhibitory receptors for MHC-I, UPA, and probably other ligands. [Hayday, 2009]
Besides activation through the TCR- ligand mechanisms, γδ T cells can also be indirectly activated by pro-inflammatory cytokines released by TLR-induced DCs and NK cells (Bonneville et al., 2010; Hannani et al., 2012; Hao et al., 2010). Human γδ T cells can also recognize and be activated by Ab-opsonized cells or microorganisms through binding of IgGs, which mediates Ab-dependent cell cytotoxicity (ADCC) (Chen and Freedman, 2008; Seidel et al., 2014).
Upon activation, these cells become capable of producing inflammatory cytokines, chemokines, directly lyse infected or malignant cells, and establish a memory response to attack pathogens upon re-exposure (Deniger et al., 2014; Hayday, 2009; Wesch et al., 2014), as described below.
1.2 Cancer and γδ T Cells
1.2.1 The Immune System and Cancer
Bearing several differences when compared to normal cells, cancer cells have two unique main characteristics: uncontrolled growth and metastasis (Zhou, 2014). Additionally, recent studies have shown that cancer cells have eight hallmarks, which include sustained proliferative signaling, evading growth suppressors, cell death resistance, replicative immortality, angiogenesis induction, metastasis and invasion activity, energy metabolism reprogramming, and evading immune destruction (Hanahan and Weinberg, 2011; Zhou, 2014). Currently, cancer can be treated by using several methodologies, which include surgery (Recht and Houlihan, 1995), radiotherapy (Bijker et al., 2006), chemotherapy (Goffin et al., 2010), biological therapy (Manganoni et al., 2000), hormone therapy (Prezioso et al., 2004), and targeted therapies (Urruticoechea et al., 2010; Vanneman and Dranoff, 2012). However, since an occurrence of 20-30 million new cases of cancer is predicted to occur all over the world by 2030 and, of those, it is expected that 13-17 million people will die from the disease, no cancer therapy presently available seems to be entirely satisfactory (Katikireddi and Setty, 2013; Siegel et al., 2012). Thus, cancer treatment remains a challenging issue for both scientists and clinicians.
Recent developments in immunology, as for instance the successes of sipuleucel‑T and ipilimumab in Phase III clinical trials, have validated the principle that immunotherapy can also extend cancer patient survival, stressing out this type of therapy as a new anti-tumor promising candidate (Vanneman and Dranoff, 2012; Zhou, 2014). Interestingly, research published over the past decade in tumor immunology has validated the concept of cancer immune surveillance which predicts that the immune system can recognize precursors of cancer and, in most cases, destroy these precursors before they become clinically apparent (Dunn et al., 2002; Galon et al., 2013; Hamaï et al., 2010). In fact, several studies have identified specific patterns of immune activation associated with patient survival, proving that the immune system can recognize and eliminate aberrant cancer cells arising within the human body (Zhou, 2014). Additionally, twenty two types of leukocytes have been recently associated with twenty five different types of cancer, whose presence or absence can indicate a favorable or an adverse prognostic depending on the tumor type (figure 2) (Gentles et al., 2015), with a special emphasis for the favorable outcome attributed to γδ T cell presence. Thus, it has become clear that the immune system not only protects the host against tumor development but also sculpts the immunogenic phenotype of a developing tumor and can favor the emergence of resistant tumor cell variants (Galon et al., 2013; Hamaï et al., 2010; Zhou, 2014).
In cancer patients, the immune system is apparently not proficient in eliminating cancer cells, suggesting a suppression of its anti-tumor function. Indeed, it has been shown that several factors may contribute to this immunosuppression such as a low frequency of high-avidity anti-tumor T cells or the presence of CD4+CD25+ regulatory T cells (Dugué et al., 2013; Frey and Monu, 2006; Zhou, 2014). Therefore, using immunological methods capable of removing this anti-tumor immunosuppression and/or able to increase the anti-tumor immunity can be very useful for cancer treatment (Zhou, 2014).
Figure 2 – Inferred leukocyte frequencies and prognostic associations in 25 human cancers. A) Global prognostic associations for 22 leukocyte types across 25 cancers (n = 5,782 tumors; left) and 14 solid non-brain tumors (n = 3,238 tumors; right), ranked by unweighted meta-z score, with a false discovery rate (FDR) threshold of 25% indicated for each plot. B) Concordance and differences in tumor-associated leukocytes (TAL) prognostic associations between breast cancers and lung adenocarcinoma. Resting and activated subsets in are indicated by “–” and “+”, respectively. Red and blue bars in indicate adverse and favorable prognostic associations, respectively. [Adapted from Gentles et al., 2015]
It is by now well established that the immune context of the tumor microenvironment (TME) can influence cancer progression and outcome. Within the TME, several sub-populations of effector cells can participate in cancer cell control and removal. All subsets of immune cells can be found within tumors, but their presence differs accordingly with the tumor type and stage of the disease and also between individuals (Hanahan and Weinberg, 2011; Lafont et al., 2014).
In jawed vertebrates, B cells, αβ T cells and γδ T cells use genetically recombined receptors to survey the environment and mediate the host defenses against disease. Among these populations, the best studied T cell lineage is the one expressing αβ TCRs: within this group the T cells often described as ‘‘conventional’’ are the most fully understood; less well understood are the αβ T cell specialized populations that recognize non-peptide presenting MHC molecules, who are often present at high frequencies in particular tissues or organs (Adams et al., 2015; Deniger et al., 2014). Even more enigmatic are cells that express a γδ TCR since this cell lineage remains the most poorly understood in terms of Ag recognition and also to what concerns differentiation into effector cell subsets (Adams et al., 2015; Hayday, 2009; Ribot et al., 2014). Albeit this fact, γδ T cells are already being targeted for cancer immunotherapy due to multiple promising preclinical studies (Gomes et al., 2010; Liu et al., 2008; Silva-Santos et al., 2015).
1.2.2 γδ T Cells in Cancer
γδ T cells have been proposed as the first line of immune defense that responds to a variety of stress-inducible or pathogen-associated proteins or metabolites (Hayday, 2009). However,
contrary to mouse γδ thymocytes, human γδ thymocytes are functionally immature, but nonetheless are highly poised to become type 1 cytotoxic effector cells. In fact, exogenous IL ‑2 or IL‑15 signals alone, in the absence of TCR stimulation, can upregulate type 1 TFs and endow γδ thymocytes with IFN-γ-producing and tumor-killing functions (Ribot et al., 2014; Silva-Santos et al., 2015).
Therefore, it is not surprising that γδ T cells actively contribute to the immune response against many tumors, which include lymphoma, myeloma, melanoma, breast, colon, lung, ovary, and prostate cancer (Bouet-Toussaint et al., 2008; Cordova et al., 2012; Dieli et al., 2007; Kang et al., 2009; Lafont et al., 2014; Meraviglia et al., 2010). Of note, when expanded in vitro in the presence of IL‑2, γδ T cells isolated from patients with melanoma, glioblastoma, neuroblastoma or renal, breast, lung, ovarian, colon, pancreatic or blood cancers efficiently killed tumor cell lines and/or primary cancer samples (Lo Presti et al., 2014; Silva-Santos et al., 2015). This anti-tumor role can be accomplished directly through their cytotoxic activity against tumors (Lafont et al., 2014). Moreover, γδ T cells can indirectly regulate the biological functions of other cells, such as DCs, NK cells, NKT cells, CD4+CD8+ T cells and IFN-γ-producing CD8+ T cells by producing the pro-inflammatory cytokines IFN-γ and tumor necrosis factor (TNF)-α, by Ag presenting or by producing signaling agents, such as IL-17, IL-10 and IL-4 (figure 3) (Bonneville et al., 2010; Hao et al., 2010; Lafont et al., 2014; Rei et al., 2015; Thedrez et al., 2007).
Owing to these potent effector functions, γδ T cells are currently attractive mediators for immunotherapy, namely against cancer. However, clinical trials completed to date have shown objective responses of only 10-33% (Gomes et al., 2010). This lack of response to treatment could be explained by a deficient expansion and/or functions of effector γδ T cells. Of note, Vδ1+
T cell lines have generally outperformed their Vδ2+ counterparts (Correia et al., 2011; Lo Presti et al., 2014) which makes it somewhat paradoxical that almost all of the clinical applications of γδ T cells have thus far concentrated on Vγ9Vδ2+
T cells (Silva-Santos et al., 2015).
Research on γδ T cell activation molecular mechanisms has demonstrated that TCR co-stimulation plays a central role in this process, thus creating a possibility for positive (in case of infection or cancer) or negative (in chronic inflammation or autoimmunity) modulation of γδ T cell responses in the clinic (Ribot and Silva-Santos, 2013). Namely, CD27 expression endows Vγ9Vδ2 peripheral blood lymphocytes (PBLs) with enhanced proliferative capacity, expanding the γδ T cell group capable of producing IFN-γ, which clearly could be useful for cancer immunotherapy (deBarros et al., 2011).
Earlier this year, recent advances in γδ T cell-based immunotherapy gave rise to the first clinical application based on the Vδ1 subpopulation reported thus far: Delta One T (DOT) cells, described as highly reproducible cells for selective, large scale expansion and differentiation of cytotoxic Vδ1+
T cells, have showed a high potential in pre-clinical models of chronic lymphocytic leukemia (CLL). Importantly, development of these cells did not require any genetic manipulation and was able to specifically targeted leukemic, but not healthy cells, in vitro. Their application prevented wide-scale tumor dissemination to peripheral organs in vivo, without any signs of healthy tissue damage, providing
proof-of-principle for clinical application of DOT cells in adoptive immunotherapy of CLL (Almeida et al., 2016).
Figure 3 – Anti-tumor functions of γδ T cells. A) γδ T cells can recognize tumor cells through interaction with (i) TCR ligands, such as P-Ags, F1-ATPase, BTN3A1, EPCR,…, and (ii) innate receptor ligands, such as ULBP, MICA/B, and nectin-like 5. Following sensing of tumor antigens or stress signals, γδ T cells are activated and can kill tumor cells through cytotoxic mechanisms that rely on the perforin/granzyme pathway, the death receptor pathway in response to TRAIL or Fas-L expression, and ADCC in the presence of tumor-specific antibodies. B) γδ T cell activation leads to TNF-α and IFN-γ production and CD40-L expression that promote DC maturation and T cell differentiation into Th1 cells. IL-17-producing γδ Th17 cells favor Th17 effector cell development. Th1 and Th17 effector T cells display anti-tumor functions to control tumor development. C) Through a trogocytosis mechanism, activated γδ T cells can capture and express CD1d molecules and then promote iNKT cell activation. Activated γδ T cells can also display antigen-presenting cell functions (MHCI and II, CD40, CD83, and CD86 expression) and activate both naïve and effector T cells with cytotoxic activity against tumor cells. D) Activated γδ T cells can provide a co-stimulatory signal to NK cells through CD137L expression to promote their anti-tumor activity. E) In the presence of specific signals, activated γδ T cells can display a Tfh profile (i.e., IL-4, IL-10, and CXCL13 production and CD40-L expression) to help B cell antibody production. Although not yet demonstrated, production of antibodies against specific tumor antigens could be involved in the humoral anti-tumor response. [Lafont et al., 2014]
However, some reports have emerged that suggest a pro-tumor role of γδ T cells in cancer. Despite the well-established concept of γδ T cells as potent anti-tumor tumor infiltrating lymphocytes (TIL), a study in 2007 on human breast cancer surprisingly revealed a potential pro-tumor function of these cells (Peng et al., 2007; Rei et al., 2015), with the γδ TILs being the most significant predictor of relapse and poor survival in these cancer patients (Ma et al., 2012; Silva-Santos et al., 2015). Moreover, recent reports have revealed an unexpected series of pro-tumor functions of γδ T cells in mouse models and human patients (Rei et al., 2014, Rei et al., 2015). In particular, IL-17-producing (Vδ1+) γδ T cells have been reported, for the first time in human, to promote the chronic inflammation associated with colorectal cancer, through recruitment of myeloid derived suppressor cells (MDSCs) (Wu et al., 2014).
Additionally, γδ T cells have been shown to promote cancer progression by enhancing angiogenesis (Wakita et al., 2010). Once more, the common mediator for all of these functions appears to be the cytokine IL-17, whose pathogenic effects seem to be able to override the anti-tumor immune response orchestrated by IFN-γ production (Rei et al., 2015). Given this apparent dual role of γδ T cells in the context of cancer, depending on their cytokine profile, it becomes fundamental to understand the molecular mechanisms associated with the regulation of their functional differentiation.
1.3 MiR Role in Gene Regulation
1.3.1 MiR Biogenesis and MaturationMiRs are evolutionarily conserved, short (20–23-nucleotide), single-stranded, non-coding RNAs that regulate target genes at the post-transcriptional level by antisense binding to their target 3’-untranslated regions (UTRs) (Sheppard et al., 2014; Winter et al., 2009; Zhou et al., 2011), which results in translational repression and/or degradation of the targeted transcript (Carthew and Sontheimer, 2009). MiRs were first discovered in Caenorhabditis elegans (C. elegans) in 1993 (Lee et al., 1993; Zhou et al., 2011), and are estimated to regulate about 90% of human protein-coding genes, which clearly indicates the powerful role of miRs in regulating human genes (Friedman et al., 2008; Mogilyansky and Rigoutsos, 2013). In mammalian cells, more than 700 miRs have been already identified and shown to regulate many developmental and differentiation processes (Friedman et al., 2008).
Most of the human miRs reside in intergenic regions and use their own gene promoter for expression (Lau et al., 2001;Lee et al., 2004; Zhou et al., 2011). The remainders are located mostly in the introns of coding genes and are generally transcribed coincidentally with their host genes (Saini et al., 2007).
MiR genes are very similar to protein coding genes since the majority of them are transcribed by RNA Polymerase II, resulting in primary miR (pri-miR) transcripts that are then capped and polyadenylated (Cai et al., 2004; Lee et al., 2004). Moreover, miR promoters are regulated by the same epigenetic marks as those used in protein coding genes (Barski et al., 2009). In the nucleus, the double-stranded RNA hairpin structure in a pri-miR transcript is processed by a type III ribonuclease (RNase III) known as Drosha and a non-catalytic protein
named DiGeorge syndrome critical region 8 (DGCR8). By forming a complex with the RNase III enzyme, DGCR8 orients the catalytic domain of Drosha that releases hairpins from pri-miRs, resulting into 60–80 nucleotide stem-loop structures, called precursor miRs (pre-miRs) (Gregory et al., 2006; Zhou et al., 2011). However, not all miRs are processed in this way. A special subset, known as mirtrons are not cleaved by Drosha, as the spliced intron already corresponds to a specific processed miR precursor (Okamura et al., 2007).
After being formed, pre-miRs fold into small helical structures, allowing for their recognition by Exportin-5 (Exp5). Then, Exp5 in complex with Ran-GTP exports the pre-miR from the nucleus to the cytoplasm, where a second type III RNase, Dicer, cleaves the pre-miR into 18– 24 base pair duplexes, generating mature miRs (Winter et al., 2009). These mature species contain a guide strand, which targets specific messenger RNAs (mRNAs) through the seed sequence, and an antisense strand, also known as passenger strand or miR* strand (Meijer et al., 2014).During miR maturation in the cytoplasm, the Argonaute protein (AGO) – a critical component of RNA-induced silencing complexes (RISC) – is thought to stabilize the guide strand, which is crucial for miR function (Kai and Pasquinelli, 2010; Zhou et al., 2011).
1.3.2 MiR Silencing Mechanisms
In order to elicit their silencing mechanisms, mature miRs are incorporated into the RISC by loading their guide strand into the AGO protein. This protein normally uses the seed sequence of the 5’ terminus – which is the thermodynamically less-stable end of the miR duplex – to recognize complementary mRNA transcripts and then proceed with their degradation or translational silencing (figure 4) (Bartel, 2004; Hutvágner and Zamore, 2002; Podshivalova and Salomon, 2013; Winter et al., 2009).
There are four AGO proteins in human cells (AGO1-AGO4). AGO2 possesses catalytic activity and can cleave bound mRNAs. The function of AGO1 is less well-defined but it can affect miR-mediated inhibition of translation, splicing, and transcription (Matsui et al., 2015). Although all AGO proteins have the ability to interact with small RNAs, it has been shown that passenger strand cleavage and RNA chaperone activities that are intrinsic to both AGO1 and AGO2 are sufficient to load these small RNAs into the RISC complex (Wang et al., 2009; Wang et al., 2012), highlighting the importance of these two proteins in the miR silencing mechanisms.
Assembly of the miR into RISC is regulated by thermodynamic properties but it can also be subject to additional regulation as the ratio miR:miR* varies dramatically depending on the miR duplex properties, on the tissue where they are being processed and on the developmental stages (Ro et al., 2007). Importantly, although direct cleavage of the targeted mRNA will cause a reduction of the mRNA levels, inhibition of protein translation will not affect the mRNA levels of the targeted protein (Zhou et al., 2011).
Figure 4 – The “linear” canonical pathway of miR processing. Canonical maturation includes production of the pri-miR by RNA polymerase II (or III) and cleavage of the pri-miR by the Drosha–DGCR8 complex in the nucleus. The resulting pre-miR, is exported by Exp5–Ran-GTP. Once in the cytoplasm, Dicer cleaves the pre-miR hairpin to its mature length. The functional (guide) strand of the mature miR is loaded together with AGO proteins into the RISC, where it guides RISC to silence target mRNAs, whereas the passenger strand (black) is degraded [Winter et al., 2009]
1.3.3 MiR-Mediated Regulation of T Cell Biology
MiR expression patterns vary among lymphocyte subsets and stages of development, indicating that these small RNAs may contribute to lymphocyte identity or functional state (Jeker and Bluestone, 2013). By instance, miR expression profiles at each thymic T cell developmental stage have been shown to be unique, with some miRs undergoing expression changes up to three orders of magnitude during maturation (Kirigin et al., 2012). Accordingly, removal of all mature miRs at early stages of thymocyte development via Dicer or Drosha knockouts has resulted in developmental blockage and consequent reduction of the peripheral mature αβ T and iNKT cell pool (Cobb et al., 2005; Podshivalova and Salomon, 2013; Seo et al., 2010; Zhou et al., 2011).
Furthermore, it has been shown that expression of miR-181 promotes NK cell development, through the suppression of NLK – a Notch inhibitor – providing an important link between miRs and this signaling pathway (Cichocki et al., 2011). In agreement with this, deletion of miR-181a-1/b-1 inhibited the development of Notch1 oncogene-induced T cell acute lymphoblastic leukemia (T-ALL). Importantly, Notch oncogenes use normal thymic progenitor cell genetic programs for tumor transformation. However, comparative analyses of miR-181a-1/b-1 function in normal thymocyte and tumor development demonstrated that