Human Vav1 expression in hematopoietic and cancer cell lines is regulated by c-Myb and by CpG methylation.
Texto
Imagem


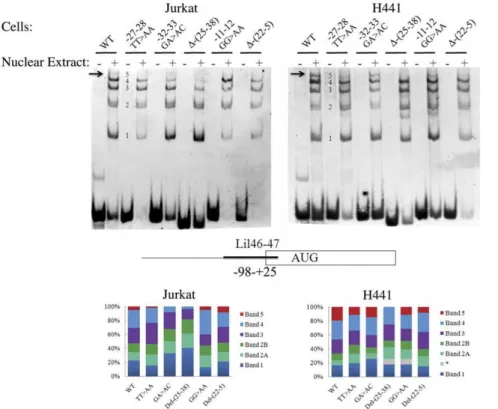

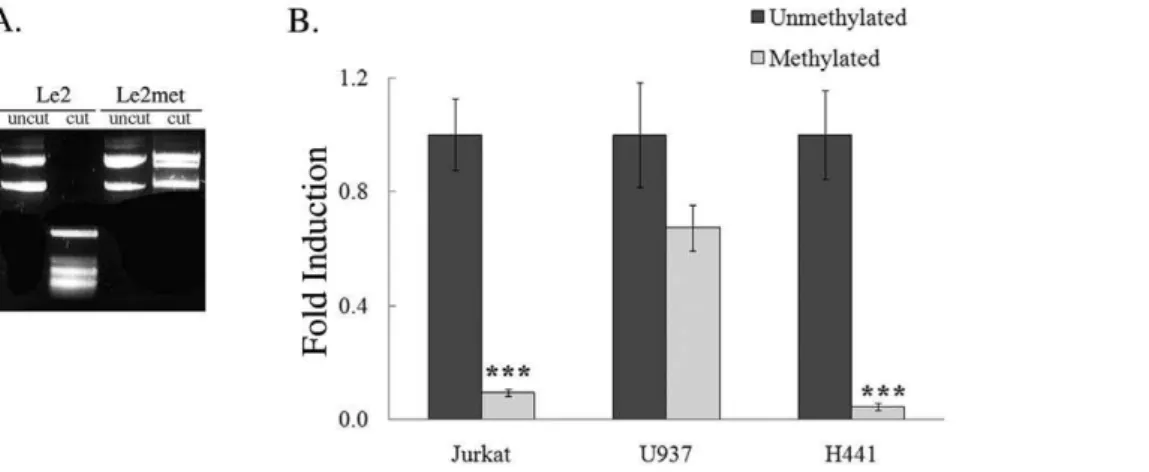
Documentos relacionados
mRNA expression level of ABCB1 , ABCC1 and SLC47A1 was evaluated by qPCR and protein expression levels MDR-1 was performed by western blotting in malignant ascites cells
No decorrer de apenas quatro anos de atividades da ITL, alguns resultados podem ser visualizados: oportunidades de novas alternativas de desenvolvimento econômico e tecnológico para
Proliferation and secretion of cytokines (IL10 and TNF a ) was determined in primary immune cells and the human cell lines (THP1, Jurkat E6-1 and Raji cell lines) stimulated
To overcome this hurdle for studying the functional role of Vav1 in human breast cancer, we overexpressed Vav1 in two breast cancer cell lines, AU565 and MCF-7, achieving Vav1
High PD-L1 expressing basal breast cancer cell lines (N = 12) demonstrate higher levels of STAT1 expression and lower levels of IRF2BP2 compared to low PD-L1 expressing cell lines (N
In fact, based on the high constitutive RANK expression in breast cancer specimens and cell lines, recent data suggest that RANK expression status of cancer cells determines
recombinant FVIII products (expressed in rodent cell lines),. the authors use a human cell line in their
Durante todo este período de ins- talação do modo de produção manufactu- reira, os ((trabalhadores da fábrica)) irão ocupar o lugar, de maneira progressiva e
