Quim. Nova, Vol. 29, No. 6, 1266-1269, 2006
A
rti
g
o
*e-mail: [email protected]
VIBRATIONAL CONTRIBUTION TO DIPOLE POLARIZABILITY AND FIRST HYPERPOLARIZABILITY OF LiH
Marcello F. Costa*
Universidade Estadual do Oeste do Paraná, Campus Toledo, Rua da Faculdade, 2550, 85903-000 Toledo - PR, Brasil Mauro C. C. Ribeiro
Instituto de Química, Universidade de São Paulo, CP 26077, 05513-970 São Paulo - SP, Brasil
Recebido em 6/2/06; aceito em 9/3/06; publicado na web em 11/8/06
The role played by electron correlation and vibrational correction on the polarizability of the LiH molecule is demonstrated. We present results for the dipole moment, polarizability and first hyperpolarizability of the LiH molecule obtained through many-body perturbation-theory, coupled-cluster and quadratic configuration interaction methods. Our best result for the dipole polarizability, obtained using the QCISD(T) scheme, indicates that the vibrational contribution is appreciable, amounting to ca. 10% of the total polarizability. Regarding the first hyperpolarizability, the vibrational contribution is even more important and has opposite sign in comparison with the electronic contribution.
Keywords: ab initio; polarizability; hyperpolarizability.
INTRODUCTION
The important role played by polarizability to understand a large variety of physical phenomena has already been stressed in a number of previous articles1-3. The well-known experimental difficulties to
obtain reliable results of polarizabilities, justifies the need for accurate theoretical results. In some cases, these results are the only source of information regarding polarizabilities. It has been shown that in order to get accurate results for electrical properties, calculations must take into account a careful choice of the function basis set and an appropriate inclusion of electronic correlation effects. In addition to these well-established precautions, another important aspect, which will be considered in this work, is the inclusion of vibrational corrections.
The finite field method4,5 is usually employed to calculate electrical
properties. According to this scheme, the energy of the system is calculated in absence and in the presence of an external electric field, and the properties of interest are obtained by means of numerical differentiations of the energy. A common feature of previous works is the use of the same geometry both in the presence and in the absence of the field. This clearly constitutes a risky procedure when dealing with ionic system, as the positive ions are driven in the direction of the applied field while the negative ions are pulled in the opposite direction. The repositioning of the ions in the presence of the electric field changes the framework and is responsible, as we are going to show in the sequence, for a relevant contribution to the polarizabilities. The effect of nuclear motion in the calculation of electric properties has already been studied using the finite pertubation theory (FTP) by Cohen et al.6
and Bishop and Kirtman7,8 using their perturbation theory method. The
FTP approach has been employed by Papadoupoulos et al.9 in evaluating
vibrational contributions to polarizability and hyperpolarizability of LiH. However, Papadoupoulos et al.9 did not include electronic correlation.
The purpose of this article is to evaluate the importance of simultaneous inclusion of electronic correlation effects and geometry relaxation in the calculation of polarizabilities and hyperpolarizabilities. We choose LiH as a test molecule due to its pronounced ionic character. Furthermore, we obtained new optimized exponents for the basis sets of Li and H based on the convergence of calculated hyperpolarizability.
COMPUTATIONAL DETAILS Methods
Recent calculations10,11 showed that in order to obtain reliable results
for electrical properties it is necessary to use a carefull built basis set and include electronic correlation at high order. In this work, we first performed calculations at Hartree-Fock level, and then we included electronic correlation effects through the following methods: configuration interaction (CI), which is variational but not size consistent12, many-body perturbation theory (MBPT), and
coupled-cluster (CC), which are size consistent but not variational13,14. As these
methods adopt different approaches, the comparison of results will allow useful and interesting analysis of electronic correlation effects. As the size consistency is usually considered more important than provision of an energy upper bound in the calculation of electric properties, the MBPT and CC methods are frequently preferred in this type of calculation. In particular, when compared to MBPT, coupled-cluster has the advantage of including higher order contributions of some substitutions. An interesting scheme is the coupled-cluster with double substitutions (CCD)13,14 which includes all the contributions
within the space of double and quadruple substitutions, up to fourth-order in perturbation theory [DQ-MBPT(4)], and some selected high order contributions. The CCD+ST(CCD) scheme15 includes single
and triple substitutions contributions in the way done by MBPT(4), except that the coefficients of the double substitutions are take from a previous CCD calculation. It has been stressed in the literature that this scheme treats the electronic correlation more properly than MBPT(4)15,16. An alternative approach is the use of the so called
quadratic configuration interaction (QCI) method17,18. This approach
includes some quadratic terms in the CI expansion aiming to restore the size consistency, at the cost of loosing the variational character of the CI method. In this work, we have employed the quadratic configuration interation method, with single and double substituitions (QCISD), adding the contribution of triple substitutions through the QCISD(T) approximation17,18. Both the QCISD(T) and the
1267 Vibrational contribution to dipole polarizability and first hyperpolarizability of LiH
Vol. 29, No. 6
Basis sets
Alkali metal hydrides (LiH, NaH and KH) are the subject of a comprehensive study11-18 and that treats the electronic and vibrational
contributions to dipole moment, polarizability and hyper-polarizabilities. This work is intended to provide a benchmark study for these molecules and we discuss it here to exemplify the kind of results obtainable or the static response functions of small molecule using current state of the art methods.
As important as the correct treatment of the correlation problem, is the care in selecting the basis set11-20. Bad choice of
the basis set does not compensate for the most sophisticated treatment of electronic correlation. Bearing this in mind, we have chosen at the substrate basis set the (6s4p/3s2p) contraction proposed by Sadlej19 for Hand the (10s6p4d/5s3p2d) contraction
from Sadlej and Urban21 for Li. This set has been successfully
applied in the calculation of dipole moment and polarizability of LiH. In our calculations, it has been supplemented by including diffuse and polarization functions in order to ensure a proper behavior of the hyperpolarizability. In the case of LiH there are a number of high level calculations in the literature with which these calculations can be compared and it is of interest at this point to examine how well they have converged on settled values. For the computation of accurate hyperpolarizabilities, one needs a method for the satisfactory calculation of the contribution of the electron correlation and an adequate basis set, which will allow the physically correct polarization of the molecule in the presence of an electric field. It is well documented that the first demand is properly answered, in general, by fourth-order Möller Plesset perturbation theory. As for the design of appropriate basis sets, various rules and strategies have been proposed.
Our option was to use some basis sets for Li and H which we have recently published and which gave good results for the polarizabilities and hyperpolarizabilities of Li + and H+ 9. The
exponents ζi of the polarization and diffuse functions are defined by
ζi = abi
where a and b are parameters optimized to reproduce energy and i takes the values 0, ±1, ±2, ... .In our calculations, it has been supplemented by including diffuse and polarization functions in order to ensure a proper behavior of the hyperpolarizability.
The procedure adopted was a continue adding of new exponents until a modification of no more than 2% was achieved in the first hyperpolarizability at the CISD level. Table I shows the obtained extra exponents. Inclusion of f-type functions has no influence on results. The complete set comprises a total of 86 basis functions, in comparison with ca. 40 of Sadlej and Urban21. Finally, it is worth
mentioning that all orbitals, occupied and virtuals, were employed in our calculations including electronic correlation.
Electric properties
It is well known that the energy of a molecule in an applied field can be expressed as an expansion involving coefficients identified as permanent multipole moments and polarizabilities. Followig McLean and Yoshimine22, when a linear molecule is placed in the
presence of an external electric field, its energy is modified according to the expression
where Ezand Ex represents, respectively, the parallel and perpendi-cular components of the applied field, μ is the permanent dipole moment, αzz and αxx are the parallel and perpendicular components
of the polarizabilities, and βzzz and βxxz are the independent components of the first dipole hyperpolarizability. According to the finite field method, the multipole moments and polarizabilities are obtained as numerical differentiation of the energy calculated in the presence of different values of the applied electric field. In this work, we started by calculating polarizabilities and hyperpolarizabilities of LiH with no change in molecular geometry due to the presence of the field. This is what we call the electronic contribution. The second contribution to the calculated properties, due to the repositioning of the ions, is called here the vibrational contribution.
RESULTS AND DISCUSSION
We first optimized the internuclear distance in the absence of an electrical field at all levels of calculations. Keeping unaltered the optimized geometry of the free molecule, with different magni-tude the energy has been calculated in the presence of applied electric fields of different magnitude. As this procedure does not take into account the nuclear motion, the calculated properties incorporate only the electronic contribution. This has been the procedure usually adopted, which excludes the vibrational contribution. These calculations were performed by using the Gaussian94 package23.
We then optimized the internuclear distance for each value of the applied field. For this purpose, we have performed several independent single-point energy calculations. Following this procedure, we present the molecular reorientation when an electric field is applied perpendicularly to the molecular axis.
In order to calculate the electronic contribution to µ, αzz, αxx
and βzzz we used different values of field: EZ = ± 0.001, ± 0.002, ± 0.003 and Ex = ± 0.002 a.u.. To obtain βxxz, we performed an extra
calculation applying a field with components Ex = ± 0.002 and Ez = ± 0.002 a.u.. These selected values were chosen as a compromise of achieving the desired numerical accuracy, but not disturbing appreciably the electronic density. An eight-degree polynomial was employed to obtain the fitting of the energy as function of the electric field. To obtain αxx we performed calculations applying two different
values of the electric field: Ex = 0.001 and 0.002 a.u.. The fitting of the energy curve was obtained using a four-degree polynomial.
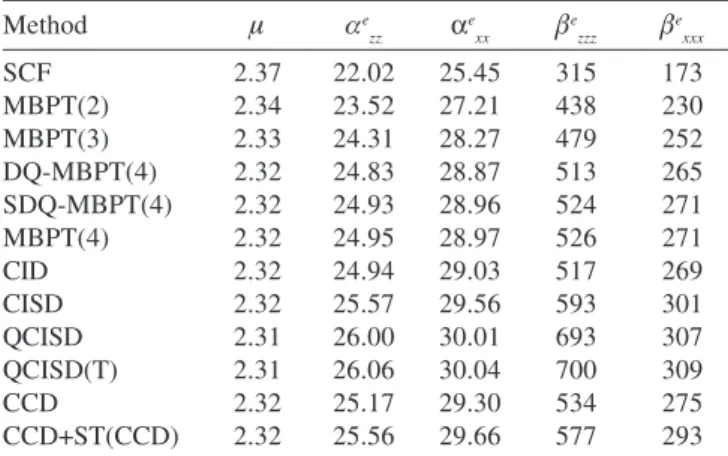
Table 2 shows the calculated electronic contributios for μ, αand β .
Calculated dipole moment at different levels of theory are very similar, indicating that no sophisticated treatment is needed in order to have a reliable estimative of this property. The difference between the SCF result and the best level of correlation, QCISD(T), is only 2%. These results are in very good agreement with those of Roos and Sadlej24, Papadoupoulos et al.9 and Rothstein25.
Concerning parallel and perpendicular components of dipole polarizability, Table 2 shows that electronic correlation effects are Table 1. The extra exponents in the basis sets for H and Li
H Li
s = 0.0102 p = 0.002601
s = 0.0032 d = 4.15340
p = 14.6596 d = 1.16944
p = 4.1216 d = 0.002061
1268 Costa and Ribeiro Quim. Nova
not negligible for this property. The difference between SCF and QCISD(T) results amounts to 15% for both components. Our results are again in good agreement with those of Sadlej and Urban21,
McCullough26, Papadoupoulos et al.9, and Lazzeretti et al.27.
One finds in the literature a number of calculated first hyperpolarizability at SCF level, which are not in agreement between them9,26. Our SCF results for βe
zzz and β e
xxz are in good agreement
with Lazzeretti et al.27 results. The accuracy of our results is
supported by the use of an extensive basis set which has been carefully selected. Correlation effects are of fundamental importance for the calculation of β, as SCF results are ca. half of QCISD(T) results. This finding has been also obtained by Papadoupoulos et al.9, indicating that electronic correlation contribution is increasingly
more important for higher order coefficients in the energy expansion. It is clear that contributions from triple substitutions are negligible by comparing SDQ-MBPT(4) and MBPT(4) or QCISD and QCISD(T) values. Conversely, single substitutions are relevant. As single substitutions are more appropriately included in the QCISD method than in the CCD+ST(CCD) one, we consider that results obtained within the QCISD(T) approximation are our best values for the properties studied here. The small difference between DQ-MBPT(4), CCD, and CID results indicates that contributions beyond fourth-order, arising from double substitutions, do not affect significantly these properties. Finally, it is worth mentioning that contrary to dipole polarizabilities, the perpendicular component of the first hyperpolarizability is smaller than the parallel component. In fact, the βe
xxz value is ca. 50% of the β e
zzz value.
We now turn the discussion to the vibrational contribution on the calculated properties, which is the main purpose of this work. In order to calculate the vibrational contribution, the equilibrium geometry was optimized at all levels of calculation and for each value of the applied electric field. In order to illustrate the changes in the geometry, Fig.1 shows the optimized geometry versus Ez, at SCF, MBPT(2), and QCISD(T) levels of theory. As expected, test calculations indicated that the geometry is not affected by the presence of an electric field in the perpendicular direction. The results for the vibrational contribution, the previously calculated electronic contribution (for comparison), and the total values of αzz,
βzzz and βxxz are listed in Table 3, in which the superscript “e” and “v” stand for the electronic and the vibrational contributions, respectively. The absence of superscripts means the total value.
It is clear from Table 3 that the vibrational contribution for dipole moment and αxx component of the polarizability are null. For the parallel component of dipole polarizability, the vibrational contribution decreases with the inclusion of electronic correlation, whereas the electronic contribution increases. In this way, the relative importance of the vibrational contribution for the total polarizability diminishes with the inclusion of electronic correlation. At the SCF level, this contribution is 14%, whereas it is ca.10% at the QCISD(T) level. Table 3 shows that the vibrational contribution for the first dipole hyperpolarizability is of fundamental importance at all levels of calculation. It should be noted that vibrational contribution have opposite sign to electronic contribution, for both components. Concerning the βzzz component, it can be seen that both contributions increase with the inclusion of electronic correlation, but the rate of increase is less remarkable for the vibrational contribution. For the electronic contribution, the SCF value Table 2. Calculated electronic contribution for dipole moment,
po-larizability and first hyperpopo-larizability of LiH (in a.u.)
Method μ αe
zz α
e
xx β
e
zzz β
e xxx
SCF 2.37 22.02 25.45 315 173
MBPT(2) 2.34 23.52 27.21 438 230
MBPT(3) 2.33 24.31 28.27 479 252
DQ-MBPT(4) 2.32 24.83 28.87 513 265
SDQ-MBPT(4) 2.32 24.93 28.96 524 271
MBPT(4) 2.32 24.95 28.97 526 271
CID 2.32 24.94 29.03 517 269
CISD 2.32 25.57 29.56 593 301
QCISD 2.31 26.00 30.01 693 307
QCISD(T) 2.31 26.06 30.04 700 309
CCD 2.32 25.17 29.30 534 275
CCD+ST(CCD) 2.32 25.56 29.66 577 293
Ref. 21.μ=2.29; Ref. 24.μ=2.31; Ref. 25. αe
xx=21.9; Ref. 27.
αe
zz=26.99; α e
xx=25.29
Table 3. Calculated polarizability and first hyperpolarizability of LiH (in a.u.) together with electronic and vibrational contributions
Method αe
zz α
v
zz αzz β
e
zzz β
v
zzz βzzz β
e
xxz β
v
xxz βxxz
SCF 22.02 3.61 25.63 315 -250 65 173 -55 118
MBPT(2) 23.52 3.08 26.60 438 -346 92 230 -52 178
MBPT(3) 24.31 2.95 27.26 479 -348 131 252 -52 200
DQ-MBPT(4) 24.83 2.89 27.72 513 -353 160 265 -52 213
MBPT(4) 24.95 2.88 27.81 526 -353 173 271 -54 217
CID 24.94 2.87 27.84 517 -354 163 269 -54 215
CISD 25.57 2.77 28.34 593 -358 237 301 -56 245
QCISD 26.00 2.73 28.73 693 -364 329 307 -56 251
QCISD(T) 26.06 2.71 28.77 700 -363 337 309 -56 253
CCD 25.17 2.87 28.04 534 -361 173 275 -52 223
CCD+ST(CCD) 25.56 2.80 28.36 577 -361 216 293 -56 237
1269 Vibrational contribution to dipole polarizability and first hyperpolarizability of LiH
Vol. 29, No. 6
corresponds to only 45% of our best value obtained at the QCISD(T) level, while the MBPT(2) value corresponds to 63%. For the vibrational contribution, the SCF value corresponds to 69% while the MBPT(2) value corresponds to 95% of the QCISD(T) value. For the βxxz component, our results show that the vibrational contribution is pratically independent of the level of approximation.
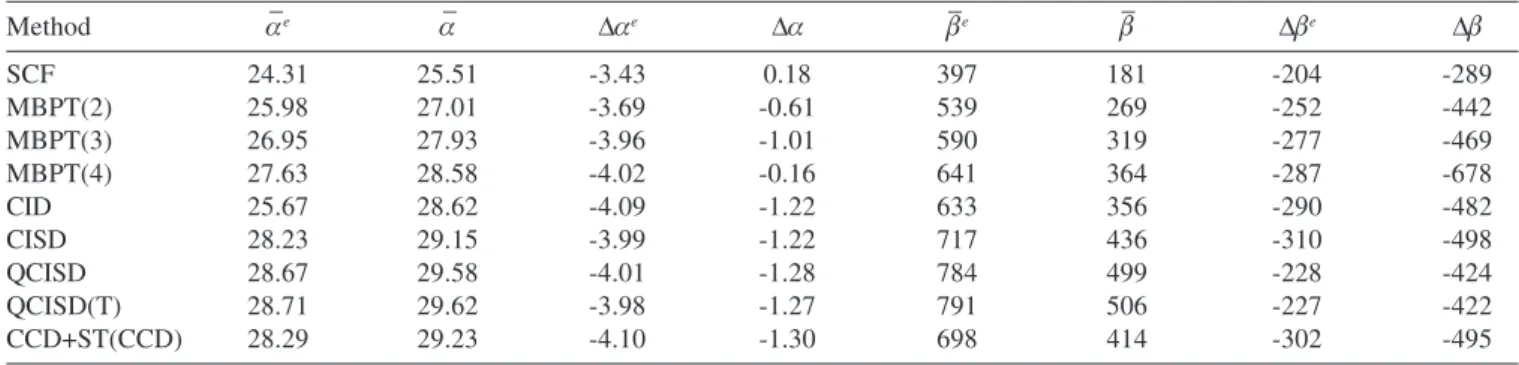
In addition to αzz , αxx , βzzz and βxxz components of polarizabilities,
we show in Table 4 the mean polarizabilities, given by:
and the anisotropies, defined as
Δα = αZZ - αXX Δβ = βZZZ - 3βXXZ
Interestingly, Table 4 shows that inclusion of vibrational contribution decreases the magnitude of anisotropy of the dipole polarizability.
CONCLUSIONS
The results obtained in this work indicate that inclusion of the effect of nuclear repositioning due to the presence of an applied electric field, called in this paper the vibrational contribution, turn to be of fundamental importance to the calculation of dipole polarizability and hyperpolarizability of the LiH molecule. The same conclusion should stand for any molecule which has ionic character. In case of LiH, inclusion of electronic correlation changes significantly calculated values of αν
zz, β
ν
zzz and β
ν
xxz. Convergence of calculated values for vibrational
contributions, with respect to different levels of approximation, is more systematic than for the corresponding electronic values.
The results obtained for the electronic contribution show small differences between the QCISD, QCISD(T) and CCD+ST(CCD) values, which demonstrate that the quadratic configuration interaction (QCI) scheme provides an appropriate description of single and double substitutions for this system and that near-degeneracy effects are negligible. Considering that the contributions of the triple substitutions are relatively small, specially when calculated by means of the QCI scheme, we expect, even in the absence of experimental results for comparison, that the values obtained using the QCISD(T) approach should be in close agreement with the exact figure.
Table 4. Total and the electronic contribution of the mean and anisotropic polarizabilities of LiH (in a.u.)
Method α–e α– Δαe Δα β–e β– Δβe Δβ
SCF 24.31 25.51 -3.43 0.18 397 181 -204 -289
MBPT(2) 25.98 27.01 -3.69 -0.61 539 269 -252 -442
MBPT(3) 26.95 27.93 -3.96 -1.01 590 319 -277 -469
MBPT(4) 27.63 28.58 -4.02 -0.16 641 364 -287 -678
CID 25.67 28.62 -4.09 -1.22 633 356 -290 -482
CISD 28.23 29.15 -3.99 -1.22 717 436 -310 -498
QCISD 28.67 29.58 -4.01 -1.28 784 499 -228 -424
QCISD(T) 28.71 29.62 -3.98 -1.27 791 506 -227 -422
CCD+ST(CCD) 28.29 29.23 -4.10 -1.30 698 414 -302 -495
Finally this paper confirm the statement that an appropriate treatment of the electron correlation is of fundamental importance in order to obtain accurate estimates for the electronic contributions to dipole moments, polarizabilities and hyperpolarizabilities by the molecule ionic character. Our results allow us to state, in addition, that the electron correlation effects are also essential to the evaluation of the vibrational corrections.
ACKNOWLEDGMENTS
This work was supported by CNPq and Fapesp. The computational facilites of LCCA-USP is acknowleged.
REFERENCES
1. Buckingham, A. D.; Adv. Chem. Phys. 1967, 12, 107.
2. Archibong, E. F.; Thakkar , A. J.; Phys. Rev. A:At., Mol., Opt. Phys.1991,
44, 5478.
3. Castro, M. A.; Canuto, S.; J. Phys. B: At., Mol. Opt. Phys.1993, 26, 4301. 4. Urban, M.; Cernusak, I.; Kello, V.; Noga, J. In Methods in Computational
Chemistry; Wilson, S., ed.; Plenum New York, 1987, vol. 1.
5. Dykstra, C. E.; Ab Initio Calculations of the Structure of Properties of
Molecules, Elsevier: Amsterdan, 1988.
6. Cohen, M. J.; Willets, A.; Amos, R. D.; Handy, N. C.; J. Chem. Phys. 1994,
100, 4467.
7. Kirtman, B.; Bishop, D. M.; Chem. Phys. Lett. 1990, 175, 601. 8. Bishop, D. M.; Kirtman, B.; J. Chem. Phys. 1992, 97, 5255.
9. Papadoupoulos, M. G.; Willets, A.; Handy, N. C.; Underhill, A. E.; Mol. Phys. 1996, 88, 1063.
10. Maroulis, G.; J. Phys. Chem. 1996, 100, 13466.
11. Medeiros, R.; Castro, M. A.; Amaral, O. A. V.; Phys. Rev. A: At., Mol., Opt.
Phys.1996, 45, 3661.
12. Szabo, A.; Ostlund, N. S.; Modern Quantum Chemistry, Macmillan: New York, 1984.
13. Bartlett, R. J.; Annu. Rev. Phys. Chem.1981, 32, 359.
14. Jφrgensen, P.; Simons, J., Second Quantization-Based Methods in quantum
Chemistry, Academic Press: New York, 1981.
15. Raghavashari, K.; J. Chem. Phys. 1995, 82, 4607. 16. Raghavashari, K.; J. Chem. Phys. 1995, 82, 4142.
17. Pople, J. A.; Head-Gordon, M.; Raghavashari, K.; J. Chem. Phys. 1990, 87, 5968.
18. Lee, T. J.; Rendell, A. P.; Taylor, P. R.; J. Phys. Chem. 1995, 94, 5463. 19. Sadlej, A. J.; Collect. Czech. Chem. Commun. 1998, 53, 1995. 20. Chong, D. P.; Langhoff, S. R.; J. Phys. Chem. 1990, 93, 570. 21. Sadlej, A. J.; Urban, M.; J. Mol. Struct. 1990, 234, 147. 22. MacLean, A. D.; Yoshimine, M.; J. Chem. Phys. 1967, 47, 1927.
23. Gaussian 94; Frisch, M. J.; et al.; Gaussian, Inc., Pittsburgh PA, 1995.
24. Rothstein, E.; J. Chem Phys. 1969, 50, 1899. 25. Roos, B. O.; Sadlej, A. J.; Chem. Phys.1991, 94, 43. 26. McCullough, E. A.; J. Chem. Phys. 1975, 63, 5050.
27. Lazzeretti, P.; Rossi, E.; Zanasi, R.; J. Phys. B: At., Mol. Opt. Phys. 1982,