Exposure of phosphatidylserine on Leishmania amazonensis isolates is associated with diffuse cutaneous leishmaniasis and parasite infectivity.
Texto
Imagem
Documentos relacionados
Tabela 2 – Efeitos Área (EA) decomposto em Efeito Escala (EE) e Efeito Substituição (ES); Efeito Rendimento (ER), Efeito Localização Geográfica (ELG) segundo
rhoifolium (ZR-EEtOH) and its n-hexane fraction (ZR-FHEX) on infection and infectivity of murine macrophages by promastigote forms of Leishmania amazonensis were investigated..
Valores
Phenotypic characterization of Leishmania spp causing cutaneous leishmaniasis in the lower Amazon Region, western Pará state, Brazil, reveals a putative hybrid parasite,
Recently, a new intermediate form of disease, borderline disseminated cutaneous leishmaniasis (BDCL), was introduced into the clinical spectrum of ACL caused by this parasite, and
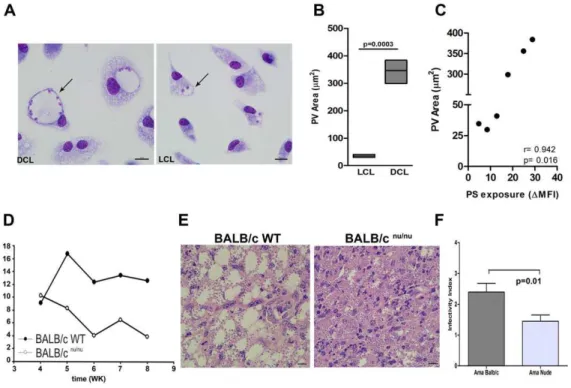
5: histopathological study of the site of infection in hamsters infected in the left hind footpad with Leishmania amazonensis promastigotes and treated with polar
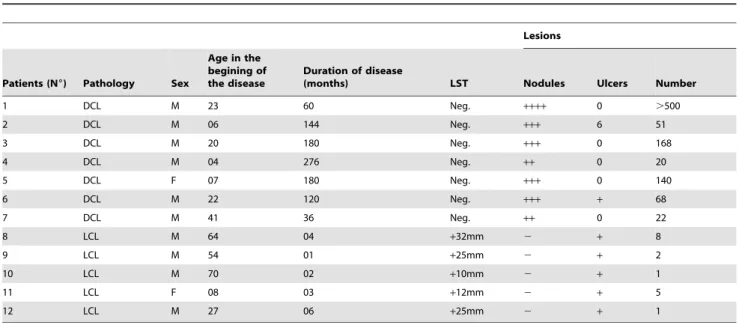
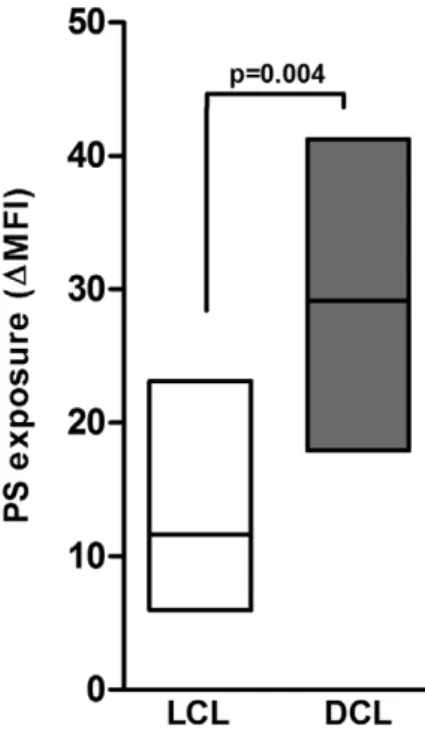
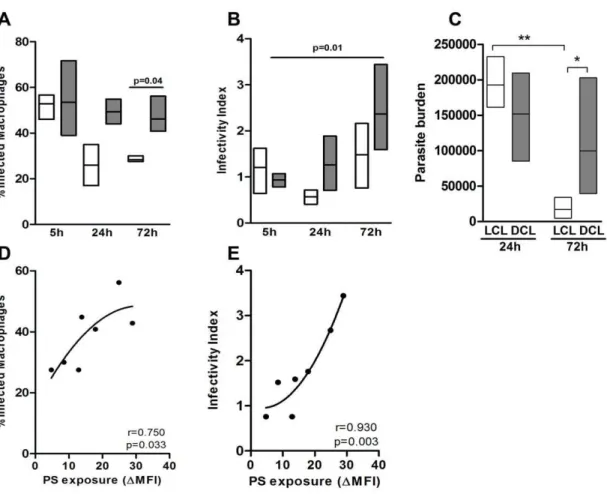
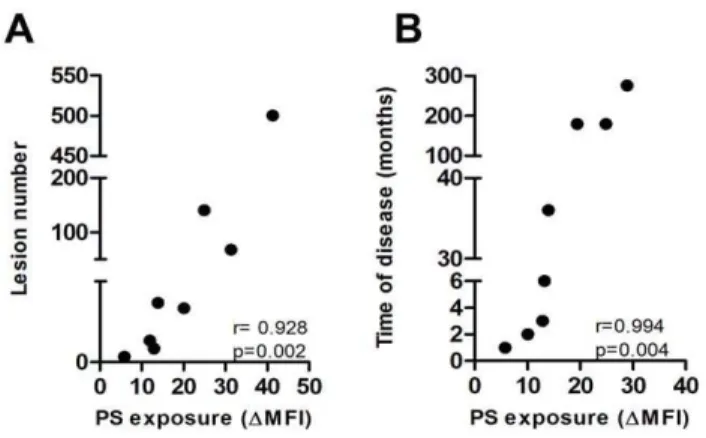
Recently, phosphati- dylserine (PS) exposure and its recognition by a specific receptor (PSR) were implicated in the infectivity of amastigote forms of Leishmania , an
Among the causative species in Brazil, Leishmania ( Leishmania ) amazonensis is an important etiological agent of human cutaneous leishmaniasis that accounts for more than 8%
