DOI: 10.5935/2359-4802.20170077
550 International Journal of Cardiovascular Sciences. 2017;30(6)550-553
CASE REPORT
Mailing Address: Ana Flavia Cassini Cunha
Rua Senador Vergueiro 93 ap 1201. Postal Code: 22230-000. Flamengo, Rio de Janeiro, RJ – Brazil E-mail: [email protected]; [email protected]
Familial Hypercholesterolemia: the Importance of Early Diagnosis and Management
Ana Flavia Cassini Cunha1 and Iliana Ribeiro2
Departamento de Clínica Médica Hospital Geral de Nova Iguaçu;1 Residência médica de clínica médica do Hospital Geral de Nova Iguaçu,2 Rio de Janeiro, RJ – Brazil
Manuscript received November 27, 2016, revised manuscript July 05, 2017, accepted July 11, 2017
Peripheral Arterial Disease; Atherosclerosis; Ankle Brachial Index; Hyperlipoproteinemia Type II.
Keywords
Introduction
Familial Hypercholesterolemia (FH) is an autosomal dominant hereditary disease, a genetic disease of lipoprotein metabolism, mainly due to a defect
in the LDLR gene encoding the LDL receptor.1-4
The diagnosis is established by clinical and laboratory criteria and should always be a diagnostic hypothesis in patients with low-density lipoprotein cholesterol (LDLc) levels greater than 190 mg/dL; and can be confirmed by genetic tests that determine the mutation.1,2
Some diagnostic criteria have been proposed in an attempt to standardize and formalize the diagnosis of FH,
such as the Dutch Lipid Clinic Network (Dutch MEDPED).1
This calculates a patient score based on anamnesis data and physical and laboratory tests such as elevated rates of LDLc; characteristics such as tendinous xanthomas and corneal arch; family history of hypercholesterolemia and/or early coronary artery disease (man < 55 years and woman < 60 years) and identification of genetic
mutations.1,3 The score determines the probability of
an FH diagnosis as possible, probable or definitive
FH.1 The most common mutation related to FH is in
the gene encoding the LDL receptor, resulting in LDL receptors with functional reductions in their ability
to remove LDLc from the circulation.3 There are two
distinct phenotypes: the homozygous form, where two defective genes are inherited and LDL receptors have no functionality; a rare form,1 in 1 million individuals and in this case are observed LDL levels > 650 mg/dL; and the heterozygous form, where a gene defective to the LDL receptor is inherited from one parent and a normal gene
from the other.1 The absence of a functional gene causes
an increase in the plasma level of LDLc; most frequently affects 1 in 500 individuals with LDL levels > 200 mg/dL.1
The homozygous form tends to present cardiovascular
involvement already in childhood.1 The mutation may
also be secondary to defects in the APOB gene encoding apolipoprotein B100, or by functional gain mutations in the pro-protein convertase subutilisin/kexin type 9
(PCSK) gene. -9).1,3,4 In patients with heterozygous FH
the LDL particles circulate longer, being more subject to oxidation and chemical transformations that result in high uptake of LDL modified by macrophages, triggering pro-atherogenic mechanisms, causing atherosclerosis,
coronary artery disease and arterial disease peripheral.1
The nutritional therapies, medication, and regular physical exercises help to control LDL levels and to
prevent cardiovascular disease.1-3 It is recommended to
reduce the intake of foods rich in cholesterol and saturated
fatty acids.1-3 Pharmacological therapy is performed with
high-potency statins, such as Atorvastatin (10-80 mg) and Rosuvastatin (10-40mg), in order to obtain a reduction
of more than 50% of the baseline level.1 In patients with
intolerant to statin therapy, other hypolipidemic agents, such as ezetimibe, niacin or cholestyramine may be used; and may be also combined with each others in patients
who are poorly responding to single statin therapy.1,3
Drug therapy should be individually prescribed and maintained in the long term, with regular medical follow-up always evaluating liver (TGO/TGP) and
muscular (CPK) enzymes.1,2 Lipid profile screening is
recommended in all individuals over 10 years of age and in all first degree relatives of individuals diagnosed as having FH.1 In the presence of risk factors, clinical signs
of FH or atherosclerotic disease, the lipid profile should
be considered from 02 years old on.1
551
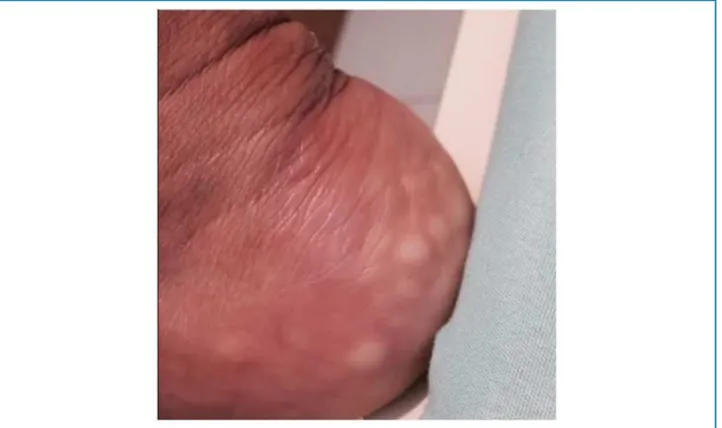
Figure 1 – Xanthoma in the calcaneus. Cunha
Hipercolesterolemia Familiar: diagnóstico e tratamento precoce
Int J Cardiovasc Sci. 2017;30(6)550-553
Case Report
Case Report
A 31-year-old female patient from Rio de Janeiro sought outpatient care in March/ 2015 at the General Hospital of Nova Iguaçu with a history of hypercholesterolemia since the age of 16, but had never taken any medication and only had a diet without laboratory improvement. She used to do regular high-intensity physical activity 05 times/week since the was 18 years old. Family history of hypercholesterolemia (Total Cholesterol > 290 mg/dL) in two first-degree relatives (mother and sibling). Maternal grandfather died of AMI before 50 years. Patient presented a February/2015 laboratory test with lipidogram results: CT: 385 mg/dL; HDLc: 86 mg/dL; LDLc: 252 mg/dL and Triglycerides: 92 mg/dL.
Physical examination at the first consultation: Abdominal Waist: 72 cm, Weight: 66 kg, Height: 1.68 m,
BMI: 23 kg/m2, BP: 110/70 mmHg, HR: 68 bpm. Presence
of tendinous xanthoma (Figure 1). Corneal arch absent. No changes in cardiovascular, respiratory and abdominal examination. Electrocardiogram performed at the 1st consultation without alterations. Laboratory tests were requested to assess dyslipidemia and imaging tests.
According to the criteria of Dutch MEDPED the patient has 13 points (Relative of 1st degree TC > 290 mg/dL - 2 points, tendinous xanthoma - 6 points, LDLc 252 mg/dL - 5 points), being considered as definitive diagnosis of FH.
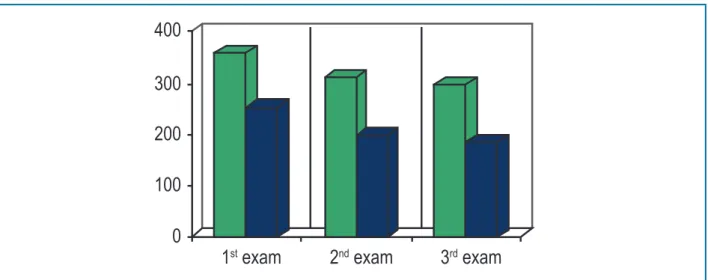
Atorvastatin 20mg was started at the first visit, but the patient irregularly used it and returned to the doctor's office after three months with the results of laboratory and imaging tests: Carotid echocardiography and abdominal USG. These tests didn't show significant alterations. Atorvastatin was prescribed at 40 mg and reassessment scheduled for three months. Patient returned in September/15 with laboratory tests, showing a decrease of Total Cholesterol and LDLc levels (Figure 2) and unchanged hepatic and muscular enzymes (TGP 19U/L, TGO 23U/L, CPK 117U/L); and exercise test without alterations.
552
400
300
200
100
0
1
stexam
2
ndexam
3
rdexam
Figure 2 –Evolution of total cholesterol and LDLc values.
1. Santos RD, Gagliardi ACM, Xavier HZ, Casella Fo A, Araujo DB, et al; Sociedade Brasileira de Cardiologia. I Diretriz brasileira de hipercoleterolemia familar (HF). Arq Bras Cardiol.2012;99(2 Supl 2):1-28. Doi:org/10.5935/1bc.20140097.
2. Xavier HT, Izar MC, Faria Neto JR, Assad MH, Rocha VZ, Sposito C, et al. Sociedade Brasileira de Cardiologia. V Diretriz brasileira de dislipidemias e prevenção da aterosclerose. Arq Bras Cardiol. 2013;101(2 Supl 1):1-20. Doi: org/10.5935/abc.20135010.
3. Genest J, Libby P. Distúrbios das lipoproteínas e doença cardiovascular. In: Libby P, Bonow RO, Mann E, Braunwald E. Tratado de doenças cardiovasculares. Rio de Janeiro: Elsevier; 2013. p.995-1116.
4. Pereira C, Miname M, Makdisse M, Kalil Fo R, Santos RD. Associação da doença arterial periférica e cardiovascular na hipercolesterolmia familiar (HF). Arq Bras Cardiol. 2014;103(2): . doi: org/10.5935/ abc.20140097.
References
Cunha Hipercolesterolemia Familiar: diagnóstico e tratamento precoce Int J Cardiovasc Sci. 2017;30(6)550-553
Case Report
Discussion
The findings described in literature show that FH is a worldwide health problem, with a high risk of early cardiovascular diseases. Early identification of disease carriers contributes to the reduction of morbimortality, through appropriate guidelines and necessary therapeutic measures. The heterozygous form affects one in 500 individuals, corresponding in Brazil to 400,000 people with HF. The described patient was classified as a definitive diagnosis of FH by the Dutch MEDPED criterion, although this has not yet been validated for the Brazilian population. The reported case showed the good result with the use of high-potency statin and the importance of regular medical follow-up. It should be noted that the homozygous form is severe and affects children as early as the first decade of life, so early diagnosis can ensure that these children have the same life expectancy as the general population.
Author contributions
Acquisition of data: Menezes IRR, Cunha AFC. Analysis and interpretation of the data: Menezes IRR, Cunha AFC. Writing of the manuscript: Menezes IRR, Cunha AFC. Critical revision of the manuscript for intellectual content: Menezes IRR, Cunha AFC.
Potential Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Sources of Funding
There were no external funding sources for this study.
Study Association
553
Cunha
Hipercolesterolemia Familiar: diagnóstico e tratamento precoce
Int J Cardiovasc Sci. 2017;30(6)550-553