Universidade de Aveiro 2006 Departamento de Biologia
PEDRO MIGUEL
GUEDES DOS
SANTOS
ANÁLISE “IN SILICO” DE TRANSPOSASES DA
FAMÍLIA ISL3
Universidade de Aveiro 2006 Departamento de Biologia
PEDRO MIGUEL
GUEDES DOS
SANTOS
ANÁLISE “IN SILICO” DE TRANSPOSASES DA
FAMÍLIA ISL3
dissertação apresentada à Universidade de Aveiro para cumprimento dos requisitos necessários à obtenção do grau de Mestre em Microbiologia Molecular, realizada sob a orientação científica do Dr. António Correia, Professor Associado do Departamento de Biologia da Universidade de Aveiro
agradecimentos Gostaria de deixar expressos os agradecimentos a todas as pessoas que directa ou indirectamente contribuíram para a realização deste trabalho e em particular: Ao Prof. Doutor António Correia (Orientador da Dissertação) pela solidariedade, orientação, e tempo despendido, sem o qual não teria sido possível a sua concretização.
À Prof. Doutora Sónia Mendo (Coordenadora de Mestrado) o apoio e
disponibilidade na resolução dos problemas inerentes à realização da dissertação. Às Caves Transmontanas Lda., ao Engº Celso Pereira e ao colega António Pinheiro a voz de incentivo durante a realização deste trabalho.
À Sónia, minha namorada, aos meus pais e à minha irmã pelo apoio incondicional demonstrado nas diversas etapas deste trabalho.
palavras-chave Transposase, ISL3, relações evolutivas.
resumo Sequências de inserção são elementos genéticos móveis que codificam genes responsáveis por eventos de inserção. Apresentam na sua constituição enzimas denominadas transposases, que apresentam elevada variabilidade intraespecífica. De acordo com essa variabilidade, as sequências de inserção estão classificadas em grupos de afinidade, designados de famílias. Uma dessas famílias de
transposases é a ISL3.
Procedeu-se à exploração de bases de dados de proteínas e sequências de aminoácidos para extrair a informação existente sobre sequências de transposases da família ISL3. Utilizando as aplicações informáticas ClustalW e TreeView, efectuou-se a análise comparativa de trinta e quatro sequências de transposases da família ISL3 e determinação das relações evolutivas das respectivas sequências com base nos alinhamentos e cladogramas gerados pelos programas acima indicados.
As trinta e quatro transposases analisadas são agrupadas em três grupos evolutivos distintos, sendo a transposase da IS1489v1 (de Pseudomonas putida ML2), a transposase mais próxima da raiz do cladograma.
Das trinta e quatro transposases seleccionaram-se seis que se encontram presentes nos genomas de bactérias Gram-positivas de elevado contudo percentual em G+C e procedeu-se ao seu alinhamento. Verificou-se que as transposases das IS31831 e IS13869 apresentam as sequências de aminoácidos com maior grau de homologia (79,1%) entre si.
keywords Transposase, ISL3, evolutionary relationships
abstract Insertion sequences are defined as mobile genetic elements that are known to encode functions involved in insertion events. They usually encode a transposase protein, that have a very high intraspecific variability The specific variability allows to organize them in to affinity groups, called families. ISL3 is one of those families. Conduction of amino acids and proteins data bases searches to obtain information about ISL3 family transposases. Comparative sequence analysis of thirty four ISL3 family transposase sequences and determination of the evolutionary relationships based on the alignments and cladograms generated using ClustalW and TreeView applications.
The analysed transposases evolved into three different families. The IS1489v1 transposase (of Pseudomonas putida ML2) is the transposase near the root of the cladogram.
Selection and alignment of six transposases presents in Gram-positive bacteria with high G+C content. The results indicate that IS31831 and IS13869 transposases, presented the amino acid sequences with the higher (79,1%) homology degree.
INDICE GERAL
1. INTRODUÇÃO...1
1.1. SEQUÊNCIAS DE INSERÇÃO...2
1.2. TRANSPOSASES...4
1.2.1. DOMÍNIOS ESTRUTURAIS...4
1.2.1.1. DOMÍNIO DE LIGAÇÃO AO DNA (DBD)...5
1.2.1.2. DOMÍNIO CATALÍTICO...6
1.2.1.3. DOMÍNIO DE MULTIMERIZAÇÃO...7
1.2.2. PROTEÍNAS ASSOCIADAS...8
1.3. ACTIVIDADE E EXPRESSÃO...9
1.4. FAMÍLIA ISL3...10
2. OBJECTIVOS DO PRESENTE TRABALHO...13
3. MATERIAL E MÉTODOS...14
3.1. OBTENÇÃO DAS SEQUÊNCIAS DAS TRANSPOSASES...14
3.2. ALINHAMENTO DAS SEQUÊNCIAS DE AMINOÁCIDOS DAS TRANSPOSASES...15
3.3. CONSTRUÇÃO DE CLADOGRAMAS...16
3.4. ANÁLISE DE DOMÍNIOS CONSERVADOS...17
3.4.1. ALINHAMENTO DE SEQUÊNCIAS DE AMINOÁCIDOS...17
3.4.2. CONSULTA DA BASE DE DADOS Pfam...18
3.5. DETECÇÃO DE MOTIVOS CONSERVADOS EM GENOMAS PROCARIÓTICOS...19
3.5.2. DETERMINAÇÃO DO NÚMERO DE CÓPIAS...20
4. RESULTADOS E DISCUSSÃO...21
4.1. SEQUÊNCIAS OBTIDAS DO GENBANK...21
4.2. ALINHAMENTO DAS SEQUÊNCIAS DE AMINOÁCIDOS DAS TRANSPOSASES...23
4.3. CONSTRUÇÃO DE CLADOGRAMAS...24
4.4. RELAÇÃO TAXONÓMICA...28
4.5. TRANSPOSASES DE IS PRESENTES NOS GENOMAS DE ORGANISMOS PERTENCENTES AO RAMO DAS BACTÉRIAS GRAM-POSITIVAS DE ELEVADO CONTEÚDO PERCENTUAL EM G+C...30
4.6. DOMÍNIO CONSERVADO TRANSPOSASE_12...35
4.6.1. ORGANIZAÇÃO ESTRUTURAL...35
4.6.2. ANÁLISE FILOGENÉTICA...36
4.7. ESPÉCIES REPRESENTATIVAS DAS ACTINOBACTERIAEA...37
4.7.1. ALINHAMENTO...38
4.7.2. ORGANIZAÇÃO ESTRUTURAL...39
4.7.3. NÚMERO DE CÓPIAS EM GENOMAS COMPLETOS...40
5. CONCLUSÕES...42
6. REFERÊNCIAS BIBLIOGRÁFICAS...45
INDICE DE TABELAS
Tabela 1. Sequências de Inserção pertencentes à família ISL3. ...11 Tabela 2. Sequências de inserção pertencentes à família ISL3 e não utilizadas no
presente estudo...21
Tabela 3. Transposases codificadas em IS pertencentes à família ISL3 e
utilizadas no presente estudo. ...22
Tabela 4. Caracterização de transposases de IS presentes em genomas de
bactérias Gram-positivas de elevado conteúdo percentual em G+C. ...30
Tabela 5. Matriz de identidade do alinhamento das sequências das transposases
de IS presentes em genomas de bactérias Gram-positivas de elevado
conteúdo percentual em G+C. ...32
Tabela 6. Cópias da região transposase_12 nos genomas de Corynebacterium glutamicum ATCC 13032 e Corynebacterium efficiens YS-314. ...41 Tabela 7. Aminoácidos conservados no alinhamento 2 e respectivas posições nas
INDICE DE FIGURAS
Figura 1. Organização típica de uma IS. ...3 Figura 2. Organização conhecida da transposase (ORF AB) da IS911. ...7 Figura 3. Cladograma que descreve as relações evolutivas entre as transposases
pertencentes à família ISL3. ...25
Figura 4. Ramo superior do cladograma que descreve as relações evolutivas de
transposases pertencentes à família ISL3. ...26
Figura 5. Ramo intermédio do cladograma que descreve as relações evolutivas
de transposases pertencentes à família ISL3. ...27
Figura 6. Ramo inferior do cladograma que descreve as relações evolutivas de
transposases pertencentes à família ISL3. ...28
Figura 7. Distribuição taxonómica dos organismos contendo transposases
pertencentes à família de sequências de inserção ISL3. ...29
Figura 8. Resultado do alinhamento das sequências das transposases de IS
presentes em genomas de bactérias Gram-positivas de elevado conteúdo percentual em G+C. ...32
Figura 9. Cladograma que descreve as relações evolutivas entre as transposases
das sequências de inserção que se encontram presentes nos genomas de bactérias Gram-positivas de elevado conteúdo percentual em G+C. ...33
Figura 10. Análise estrutural de uma transposase integrada numa sequência de
inserção da família IS2. ...35
Figura 11. Distribuição por espécies do domínio conservado Transposase_12,
Figura 12. Resultado do alinhamento das sequências de aminoácidos do domínio
conservado presentes em bactérias representativas das Actinobacteriaea. .38
Figura 13. Cladograma que descreve as relações evolutivas entre as sequências
de aminoácidos do domínio conservado transposase_12 presentes em
bactérias representativas das Actinobacteriaea. ...39
Figura 14. Representação gráfica da organização estrutural do domínio
conservado transposase_12 presente nas espécies A) Brevibacterium linens, B) Mycobacterium smegmatis C) Corynebacterium efficiens. ...40
LISTA DE ABREVIATURAS [a.a.] – aminoácido(s)
[A+T] – Adenina e Timina
[DBD] – Domínio de ligação ao DNA [DDE] – Motivo DDE
[DNA] – Ácido desoxirribonucleico [DR] – Repetição directa Direct repeat [ET] – Elementos transponíveis
[G+C] – Guanina e Citosina
[GI] – GenInfo Identifier sequence identification number [HTH] – Motivo hélice-volta-hélice
[IR] – Repetição invertida Inverted repeats
[IRL] – Inverted repeat a montante da transposase [IRR] – Inverted repeat a jusante da tranposase [IS] – Sequência de inserção
[LZ] – Motivo fecho-de-leucina [Mg2+] – Magnésio bivalente [ORF] – Fase de leitura aberta [pb] – Pares de bases
[Tpase] – Transposase
[3`-OH] – Grupo hidroxilo no terminal 3` [5`-P] – Grupo fosfato no terminal 5`
[16S rRNA] – RNA presente na sub-unidade ribossomal, cuja velocidade de sedimentação é de 16 Svedberg (S).
Aminoácido Código de três letras Código de uma letra Alanina ala A Arginina arg R Asparagina asn N
Ácido aspártico asp D
Asparagina ou Ácido aspártico asx B
Cisteína cys C
Ácido glutâmico glu E
Glutamina gln Q
Glutamina ou Ácido glutâmico glx Z
Glicina gly G Histidina his H Isoleucina ile I Leucina leu L Lisina lys K Metionina met M Fenilalanina phe F Prolina pro P Serina ser S Treonina thr T Triptofano try W Tirosina tyr Y Valina val V
1. INTRODUÇÃO
Todos os organismos têm a capacidade de rearranjar o seu genoma, de forma a modificar ou diversificar o seu pool de genes e/ou o seu nível de expressão. Esses mecanismos de recombinação genética, essenciais à evolução, podem ser classificados em três categorias: i) recombinação homóloga, ii) recombinação específica de local, e iii) recombinação transposicional. A transposição é o mecanismo capaz de gerar maior diversidade, sendo os rearranjos provocados, resultantes de um forte carácter aleatório [31].
Os elementos transponíveis (ET) foram identificados pela primeira vez nos anos 50 por Barbara McClintock e o novo conceito de elementos genéticos móveis, então introduzido, estava totalmente à margem das ideias dominantes sobre a estrutura dos cromossomas. Este conceito apenas foi reconhecido em 1960, após a descoberta de sequências de DNA móveis em células bacterianas, sendo identificadas três famílias de elementos transponíveis (i.e. capazes de ser transferidos de um local dador para um local receptor, por acção de uma enzima de recombinação especializada): as sequências de inserção (IS), os transposões e os fagos mutagénicos. Ao longo dos 30 anos seguintes, o número de ET bacterianos e eucariotas identificados não parou de crescer, revelando uma grande diversidade de estruturas e de mecanismos que asseguram a sua mobilidade [24].
Uma classe de elementos transponíveis (transposões cut and paste) move-se de uma molécula de DNA para outra (ou de um locus para uma molécula de DNA), por um processo que envolve a excisão do transposão de DNA do local original e a sua integração no local alvo. Este processo de excisão e integração de DNA é catalizado por uma proteína denominada transposase (Tpase) [29].
Os ET são ubíquos e dada a sua capacidade de provocar inserções, delecções, inversões e fusão de cromossomas, desempenham funções importantes de manutenção da diversidade genética [15]. Em 1994, Kato et tal., observou a
transferência lateral de sequências de inserção (IS6100) em Mycobacterium fortuitum e Flavobacterium (Arthrobacter). Acontecimentos genéticos como este ocorrem na natureza [23], porque as IS estão frequentemente associadas a plasmídeos transmissíveis ou a bacteriófagos, funcionando estes como seus vectores. As IS desempenham assim um papel importante ao nível evolutivo, facilitando o rearranjo interno dos genomas. É de substancial interesse e importância determinar como estes acontecimentos genéticos ocorrem [34], pois poderá ajudar a compreender os mecanismos de adaptação dos microrganismos a mudanças no ambiente, especialmente no caso de adaptação a ambientes extremos, bem como se a presença de sequências de inserção intimamente relacionadas entre si, em diferentes espécies ou géneros, pode indicar que poderão ter ocupado o mesmo nicho a uma dada altura e trocaram DNA [12].
1.1. SEQUÊNCIAS DE INSERÇÃO
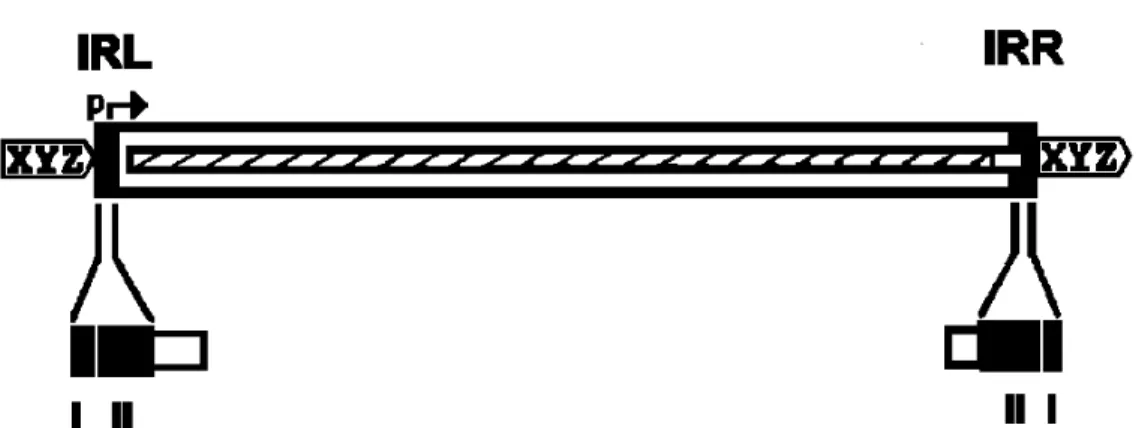
AS IS são ET de pequena dimensão (750 a 2500 pb), com uma organização muito compacta e que codificam, em geral, apenas as funções ligadas à sua mobilidade. Na sua maior parte, apresentam curtas sequências inversamente repetidas (10 a 40 pb), os inverted repeats (IR), que estão localizados nas suas extremidades. São reconhecidos pela maquinaria de transposição [2, 24] e contêm as sequências de DNA activas na recombinação. Estes IR apresentam uma fase de leitura aberta (ORF), que codifica a Tpase do elemento, enzima que vai reconhecer e recombinar as extremidades envolvidas no mecanismo de transposição [16].
Figura 1. Organização típica de uma IS. Esta encontra-se representada como uma caixa aberta,
na qual os terminais IR se encontram representados como caixas negras e rotulados de IRL (inverted repeat esquerdo) e IRR (inverted repeat direito). Uma única ORF que codifica a transposase, está indicada como uma caixa ao longo de todo o comprimento da IS, estendendo-se até ao interior da sequência IRR. XYZ no interior da caixa pontiaguda no flanco da IS representa curtas sequências DR geradas no DNA alvo como consequência da inserção. O promotor da transposase, p, que se encontra parcialmente localizado no IRL, está representado por uma seta horizontal. A estrutura (caixas negras) típica do domínio dos IR está indicada na zona inferior. O domínio I representa os pares de bases terminais na extremidade do elemento cujo reconhecimento é necessário para a clivagem da transposase. O domínio II representa os pares de base necessários para o reconhecimento específico da sequência e ligação da transposase.
Adaptado de [23].
Em alguns casos, a Tpase é codificada por duas fases de leitura, e é apenas produzida num processo não convencional, a deslocação da fase em -1. Esta segunda proteína é uma versão truncada da transposase e comporta-se como regulador da transposição [13]. É o caso por exemplo da IS1 e dos membros da família IS3. Em todo o caso, as fases de leitura abertas representam a quase totalidade do elemento. Por convenção, a IR que comporta o promotor e que vai permitir a expressão das fases abertas de leitura é denominada de IR esquerda (IRL), e a situada no final do gene é designada por IR direita (IRR) [31].
As IS não apresentam um modo único de transposição. Algumas são não replicativas (IS10), outras replicativas (IS6) ou mistas (IS1). Em todos os casos, as frequências de transposição são baixas e controladas pelo elemento ou pelo hospedeiro [31].
Uma particularidade importante das IS é que estas podem colaborar entre si na sua transposição. Esta propriedade permite mover toda a sequência de DNA compreendida entre duas cópias duma mesma IS, em orientação directa ou inversa. A estrutura deslocada é designada transposão composto, sendo os
exemplos mais conhecidos Tn5 (IS50), Tn9 (IS1) e Tn10 (IS10), a que estão associados genes de resistência a antibióticos [31].
A colaboração entre duas cópias de uma mesma IS pode provocar rearranjos múltiplos e complexos dos genomas, em função das extremidades implicadas e da localização do alvo.
Um evento de transposição intermolecular (i.e., em que o transposão e o alvo não existem no mesmo replicão) poderá conduzir a produtos de transposição directa se as IR activas forem as IR externas, ou produtos de transposição inversa se são as IR internas a serem movidas. Neste último caso, os marcadores sequestrados entre os IR internos do DNA dador serão perdidos.
Um evento de transposição intramolecular (em que o replicão é portador do transposão e do alvo) conduzirá sistematicamente à perda de DNA situado entre os IR internos. Segundo a polaridade do evento, gerar-se-ão delecções adjacentes ou inversões/deleções. Este último tipo de produto corresponde à formação de um novo transposão composto [31].
1.2. TRANSPOSASES
1.2.1. DOMÍNIOS ESTRUTURAIS
Transposases são proteínas multifuncionais com um papel estrutural (manter unido o transposão) e catalítico (cortar DNA e trocar fragmentos de DNA), emergindo uma estrutura geral de organização funcional do número limitado de Tpases [23]. Após a fixação específica às extremidades do ET, as duas extremidades serão aproximadas por interacções proteína-proteína para formar um complexo designado transpososoma. Em alguns casos, a sequência alvo é requisito para que esta união tenha lugar e nos outros casos ela é apenas necessária após a formação do complexo nucleoproteico. Nesta etapa podem igualmente intervir as proteínas ditas acessórias, codificadas pelo hospedeiro ou pelo ET, que desempenharão um papel estrutural e/ou de regulador da reacção. Após a formação desta estrutura, poderão intervir as reacções catalíticas. Esta
multifuncionalidade traduz-se numa organização do domínio das Tpases, em que a cada domínio corresponde uma função específica. No seio de uma família de ET encontra-se uma organização comum das Tpases associadas [31].
1.2.1.1. DOMÍNIO DE LIGAÇÃO AO DNA (DBD)
A Tpase liga-se especificamente aos IR onde cataliza as clivagens das cadeias de DNA e ocorrem as reacções de transferência no local alvo, pelo que o reconhecimento e ligação ao IR é um factor fundamental da sua actividade [30]. Na grande maioria dos casos, este domínio encontra-se na região N-terminal das Tpases. Este arranjo permite a ligação preferencial de polipeptídeos de Tpase a locais de ligação vizinhos. A afinidade da porção N-terminal da proteína pelos IR é superior à afinidade da Tpase completa; o que sugere que a extremidade C-terminal pode mascarar a actividade de ligação ao DNA da região N-terminal [4, 23].
O DBD N-terminal apresenta dois motivos distintos e bem definidos. O primeiro, leucine-zipper (LZ, motivo fecho-de-leucina), está envolvido na ligação ao DNA e tem a função de assegurar as interacções proteína-proteína para estabelecer a ligação aos IR. O segundo, o motivo helix-turn-helix (HTH, motivo hélice-volta--hélice) - motivo mais frequentemente utilizado [31] - é responsável pela especificidade da sequência de ligação do DNA, sendo portanto uma região essencial à actividade da Tpase [30, 13]. Os factores responsáveis pela variação da especificidade de ligação (que varia significativamente de elemento para elemento), podem estar relacionados com o reconhecimento de uma sequência específica, o tamanho da sequência, os conteúdos G+C ou A+T, o grau de superenrolamento, replicação e transcrição de DNA, direcção da transferência, proteínas mediadoras do alvo, proteínas de exclusão da região de controlo transcricional ou inserção perto do sequências semelhantes com o terminal do transposão [21]. A natureza do alvo e a sua imunidade podem também ser factores adicionais [23].
1.2.1.2. DOMÍNIO CATALÍTICO
Historicamente, o alinhamento das Tpases da família IS3 com as integrases retrovirais, pôs em evidência a existência de uma tríade conservada de aminoácidos com um espaçamento característico. Esta tríade é conhecida por motivo DD(35)E [20], em que o resíduo glutamato (E) está geralmente separado dos dois resíduos aspartato (D) por 35 aminoácidos, e está implicado na ligação de dois iões metálicos bivalentes [4]. Noutras posições conservadas foram igualmente identificadas, particularmente um resíduo K (que pode ser substituído por um R). Esta assinatura foi também encontrada noutras Tpases, nomeadamente Mu (MuA), Tn7 (TnsA), IS50 e as da família Tn3. Actualmente, a grande maioria das transposases de IS está incluída nesta grande família.
Ainda que o espaçamento entre os resíduos DDE, e a sua conservação estrita seja variável, a conservação deste motivo é notável. Uma função essencial do motivo DDE é coordenar os catiões bivalentes, indispensáveis à catálise. A título exemplificativo, tal como foi observado no caso específico do Fago Mu, o Mg2+ retido pelo motivo DDE interage então com as extremidades do elemento e fragiliza uma ligação fosfodiéster que se torna sensível a um ataque nucleófilo por uma molécula de água. A reacção liberta as extremidades 3`-OH do DNA fágico que encetam um segundo ataque nucleófilo sobre as ligações fosfodiéster do DNA alvo e libertam as extremidades 5`-P. Nesta segunda reacção, as duas extremidades 3` de Mu estão ligadas às duas extremidades 5` da sequência alvo. O segundo ataque nucleófilo não ocorre sobre o DNA alvo, mas sim sobre o primeiro corte, libertando o transposão na sua forma livre. A cada extremidade do elemento, um novo ataque nucleófilo liberta de novo uma extremidade 3`-OH que ataca o alvo [24].
A importância do motivo DDE para a catálise foi demonstrada por mutagénese dirigida em diversos casos, MuA, TnsAB, IS10, e IS911, por exemplo. A reacção de substituição nucleófila não necessita de cofactor energético (ATP) e não necessita de intervenção de um intermediário covalente entre o DNA e a transposase [24]. A primeira reacção, que conduz à excisão do ET, vai utilizar uma
molécula de água nucleofílica, formando extremidades 3`-OH para o ET . São estas extremidades 3`-OH que vão servir de nucleófilos da reacção de integração [9].
Contudo, nem todas as IS apresentam uma tríade DDE bem definida. Por exemplo, as Tpases de um grupo de elementos, a família IS91, mostram semelhanças significativas com as enzimas associadas a replicões, que utilizam o mecanismo de formação círcular para replicação, ou o facto dos locais activos para a família ISL3 ainda não terem sido definidos [23].
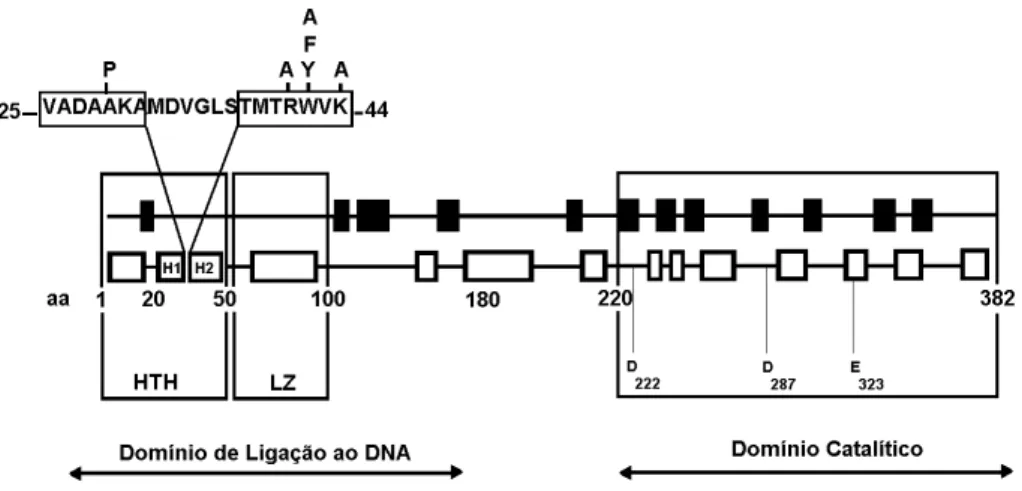
Figura 2. Organização conhecida da transposase (ORF AB) da IS911. As caixas brancas
representam α-hélices; as caixas negras representam cadeias β. O domínio de ligação ao DNA é composto pelo motivo helix-turn-helix (HTH; H1, hélice 1; H2, hélice 2) e leucine-zipper (LZ). O domínio catalítico apresenta o motivo DDE. Adaptado de [30].
1.2.1.3. DOMÍNIO DE MULTIMERIZAÇÃO
A multimerização é outra propriedade extremamente bem conservada nas Tpases [26]. Esta é importante para a união do complexo sináptico, de forma a permitir a colaboração entre monómeros necessários à catálise e permitindo igualmente regular a actividade da Tpase [24]. Contudo, não existe uma assinatura de domínio comum a todas as Tpases devido a esta função, que foi demonstrada bioquimicamente para as Tpases de Mu, IS50 e IS911 [31].
No caso específico da Tpase de IS50, uma proteólise parcial evidenciou duas regiões distintas, capazes de interagir com a proteína nativa. Uma destas regiões sobrepõe-se ao domínio catalítico, a outra está situada na região C-terminal e as mutações nesta região afectam a actividade de transposão [31].
Para a Tpase de IS911, foram identificadas três regiões distintas que permitem a associação. Além do domínio catalítico capaz de dimerizar, um motivo do tipo leucine-zipper permite igualmente a multimerização que vai permitir à Tpase fixar--se aos IR. Além disso, uma região próxima deste motivo está implicada na união da sinapse. Este tipo de motivo (LZ) é comum à maioria das Tpases da família IS3 de que faz parte IS911 [31].
1.2.2. PROTEÍNAS ASSOCIADAS
É geralmente aceite que as IS bacterianas necessitam apenas da Tpase para assegurar todas as etapas da reacção. A situação é mais complexa para os transposões ou fagos mutagénicos. É na verdade frequente que uma ou mais proteínas codificadas para a ET cooperem com a Tpase e a assistam na reacção. É por exemplo o caso do transposão Tn7. É igualmente frequente encontrar reguladores negativos da transposição, quer repressores transcricionais, quer inibidores de transposição. Este tipo de regulação negativa é frequentemente encontrado nas IS, particularmente naquelas que apresentam duas fases de leitura sobrepostas [31].
Um certo número de proteínas codificadas pelo hospedeiro pode ser igualmente utilizado pelos ET para a sua transposição. Numerosos ET requerem a presença de moléculas tipo histonas (IHF, HU e FIS por exemplo), mas também de chaperonas, que vão induzir mudanças conformacionais na Tpase, conduzindo à sua activação (IS1 por exemplo). As proteases celulares podem igualmente desempenhar uma função reguladora de transposição proteolizando a transposase (efeito repressor) ou um inibidor da transposase (efeito activador) [31].
1.3. ACTIVIDADE E EXPRESSÃO
Os promotores da Tpases são geralmente fracos e muitos estão parcialmente localizados nos IR terminais, permitindo que a Tpase se autorregule por ligação. A sua expressão e actividade estão portanto sob controlo de diversos mecanismos, como a produção de repressores transcricionais ou de inibidores de tradução. Para além destes, que como mecanismos clássicos se encontram mais documentados, existem outros mecanismos com papéis importantes na expressão das Tpases. Assim, mecanismos como sequestro de sinais de início de tradução, alteração do quadro de leitura traducional programado, terminação de tradução, transcrição invasiva a partir de um elemento localizado a montante, estabilidade da Tpase e actividade em cis, operam também na sua actividade e expressão [23].
Como a actividade transposicional é frequentemente modulada por vários factores do hospedeiro, estes são frequentemente específicos para cada elemento. Apesar do seu modo de acção não ser actualmente conhecido, várias proteínas hospedeiras estão implicadas na transposição e podem também estar envolvidas na regulação de Tpases [23].
Uma característica importante de alguns elementos transponíveis consiste na baixa afinidade de inserção de uma segunda cópia numa molécula alvo que já contém uma cópia desse elemento [15]. Esta propriedade designa-se imunidade transposicional. Apesar de não estar claramente demonstrado que esta estratégia é adoptada por IS, existem evidências que sugerem que elementos da família IS21 a podem seguir. Nesta família de IS, são necessárias duas ORFs consecutivas para ocorrer a transposição. Os produtos das duas ORFs, IstA que transporta um motivo tipo-DDE e IstB com um potencial ATP-binding site, respectivamente, têm a capacidade de distinguir e evitar DNA já existente no elemento [35].
1.4. FAMÍLIA ISL3
As relações filogenéticas das transposases e o seu mecanismo de transposição, bem como algumas características a este associadas, como por exemplo, os inverted repeats das extremidades da maioria dos elementos [34, 10], os locais alvo e a organização das ORFs, permitem que as sequências de inserção sejam organizadas em grupos, que são designados por famílias [12].
A família ISL3 é actualmente composta por 48 membros conhecidos [1] (tabela 1), cujas dimensões se encontram compreendidas entre 1186 pb (ISSg1) e 1553 pb (IS1165), com as excepções da ISCx1 para a qual apenas se encontra disponível apenas uma sequência parcial e para a IS1069 que é significativamente maior. A família ISL3 é também caracterizada por transportar IR fortemente relacionados, que variam de 15 a 39 pb e geram direct target repeats de 8 pb com geometria palindrómica [3, 8, 18, 32]. Apresenta preferência por regiões ricas em A+T e até ao momento não foi possível observar qualquer especificidade por uma determinada sequência alvo [10, 18, 19, 34]. Contém também um grande quadro de leitura, que codifica proteínas com 400 a 440 aminoácidos, apresentando bom alinhamento, particularmente na região C-terminal [23].
Os motivos [D (154º a.a.), D (227º a.a.), E (365º a.a.) e K (372º a.a.)] e [D (154º a.a.), D (228º a.a.), E (360º a.a.) e K (367º a.a.)] foram identificados nas ORFs de dois elementos pertencentes à família ISL3, as IS667 e IS668. Estes motivos, que são designados motivos DDE e são conservados na maioria das transposases [37] e de outras enzimas capazes de catalizar a clivagem de cadeias de DNA [33] apresentam uma correspondência directa com o motivo DDE tipo da família ISL3: DD(129)EK [1].
Apesar de ter já sido demonstrada a transposição dos elementos pertencentes à família ISL3, o mecanismo de transposição apresenta variações e em alguns casos permanece mesmo por clarificar [16, 23]. Os mecanismos de transposição relatados incluem transposição via formação circular (IS1409 em que se verifica a formação de uma molécula precursora com a forma de 8, na qual apenas uma das cadeias foi alvo de clivagem e de transferência para a extremidade oposta, resultando a circularização de uma cadeia simples) [17, 27], via formação
cointegrada (nos casos das IS31831, ISPs1 [10] e ISAfe1 [14]); E no caso da IS1096 que contém uma resolvase putativa [23].
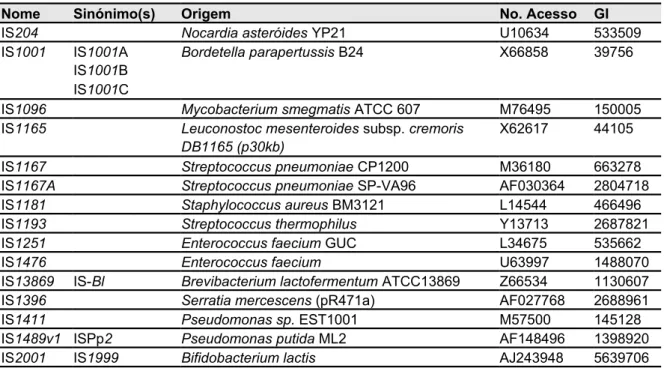
Tabela 1. Sequências de Inserção pertencentes à família ISL3. Adaptado de [1].
Nome Sinónimo(s) Origem No. acesso
IS204 Nocardia asteróides YP21 U10634
IS1001 IS1001A Bordetella parapertussis B24 X66858 IS1001B
IS1001C
IS1096 Mycobacterium smegmatis ATCC 607 M76495
IS1165 Leuconostoc mesenteroides subsp. cremoris DB1165 (p30kb) X62617
IS1167 Streptococcus pneumoniae CP1200 M36180
IS1167A Streptococcus pneumoniae SP-VA96 AF030364
IS1167L Streptococcus pneumoniae 13868 Z83335
IS1167R Streptococcus pneumoniae 13868 Z83335
IS1181 Staphylococcus aureus BM3121 L14544
IS1182 Staphylococcus aureus BM3121 L43082
IS1193 Streptococcus thermophilus Y13713
IS1193D IS1193 Streptococcus thermophilus CNRZ368 Y13713
IS1207 ISbB1 Corynebacterium glutamicum Bl15 ND
IS1251 Enterococcus faecium GUC L34675
IS1476 Enterococcus faecium U63997
IS13869 IS-Bl Brevibacterium lactofermentum ATCC13869 Z66534
IS1396 Serratia mercescens (pR471a) AF027768
IS1411 Pseudomonas sp. EST1001 M57500
IS1489v1 Pseudomonas putida ML2 AF148496
IS2001 IS1999 Bifidobacterium lactis AJ243948
IS31831 IS-Cg Corynebacterium glutamicum ATCC 31831 D17429
IS466A Streptomyces coelicolor A3(2) M145 AB032065
IS469 Streptomyces coelicolor A3(2) M145 AB032065
IS1651 Bacillus halodurans C-125 NC 002570
IS1652 Bacillus halodurans C-125 NC 002570
ISAe1 ISAE1 Ralstonia eutropha H1-4 M86608
ISAsp1 Anabaena sp. strain PCC7120 U13767
ISBli1 Brevibacterium linens AF052055
ISBli3 Brevibacterium linens
ISBma1 ISBm1 Burkholderia mallei AF285635
ISCx1 Corynebacterium xerosis M82B (pTP10) U21300 [P] ISL3 Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842 X79114 ISLdl1 Lactobacillus delbrueckii subsp. lactis AJ302652
ISM1 Methanobrevibacter smithii X02587
ISPp2 Pseudomonas putida ML2 U25434
ISPsp2 Tn401 Pseudomonas sp. strain EST1001 (pEST1226) M57500
ISPst2 ISPs1 Pseudomonas stutzeri M1 AJ012352
ISRso15 Ralstonia solanacearum NC 003295
ISSg1 Streptococcus gordonii M5 ND
IST445 Thiobacillus ferrooxidans Y18309
ISBlo6 Bifidobacterium longum NCC2705 NC 004307
Nome Sinónimo(s) Origem No. acesso
ISSth1 Streptococcus thermophilus AY376237
ISPpu12 Pseudomonas putida AY128707
ISTfe1 IST1, ISAfe1 Acidithiobacillus ferrooxidans U66426
ISP1 Lactobacillus plantarum NC 004567
ISFnu5 Fusobacterium nucleatum subsp.nucleatum NC 003454
ISLjo2 Lactobacillus johnsonii NC 005362
2. OBJECTIVOS DO PRESENTE TRABALHO
Tendo em conta a existência de bases de dados informatizadas de sequências nucleotídicas e de proteínas, bem como de programas de computador de utilização simples para analisar este tipo de dados, foi decidido fazer uma actualização do conhecimento respeitante às transposases contidas em sequências de inserção da família ISL3. A realização deste trabalho pretendeu alcançar de forma sequencial os seguintes objectivos:
a) Efectuar um levantamento das sequências de transposases da família ISL3 disponíveis em bases de dados informáticas.
b) Efectuar a análise comparada das sequências das transposases da família ISL3.
c) Analisar a existência de domínios conservados em sequências de bactérias Gram-positivas com elevado conteúdo em G+C.
3. MATERIAL E MÉTODOS
3.1. OBTENÇÃO DAS SEQUÊNCIAS DAS TRANSPOSASES
A identificação das sequências de inserção pertencentes à família ISL3 foi efectuada on line numa base de dados cujo endereço é http://www-is.biotoul.fr/. Esta base de dados disponibiliza uma lista de sequências de inserção isoladas de Eubactérias e Arquiobactérias. Encontra-se organizada em ficheiros individuais que contêm as características gerais das sequências de inserção (designação, dimensão, origem e famílias, entre outras), bem como informações sobre o seu DNA e sequências proteicas potenciais. Apesar da maioria das entradas ter sido identificada como elementos individuais, um número crescente está a ser incluído mediante a detecção da sua presença em genomas bacterianos completamente sequenciados.
O motor de busca deste serviço informático permite pesquisar sequências de inserção individuais e grupos de sequências de inserção, com base na combinação das suas características gerais, e estão disponíveis dois níveis de pesquisa. A pesquisa simples, que permite ao utilizador pesquisar elementos, utilizando um número limitado de itens básicos; e a pesquisa extensiva, que disponibiliza um conjunto adicional de possibilidades, tais como comparações de sequências de terminal inverted repeats e uma variedade de diferentes representações da pesquisa. Aqui recolheu-se o número de acesso das sequências de inserção que foi utilizado no GenBank (base de dados de sequências genéticas do National Institutes of Health, que contém uma colecção anotada de todas as sequências de DNA públicas disponíveis e a que se pode aceder on line no endereço http://www.ncbi.nlm.nih.gov/) para se obterem as sequências de nucleótidos e de aminoácidos.
As sequências de aminoácidos foram guardadas em formato FASTA num ficheiro cujo conteúdo se apresenta no anexo I e foram identificadas pelo “GenInfo
Identifier” sequence identification number (GI) obtido também no GenBank. Foram excluídas as transposases detectadas em sequências de genomas completos para evitar redundâncias, bem como sequências de transposases incompletas ou ambíguas.
3.2. ALINHAMENTO DAS SEQUÊNCIAS DE AMINOÁCIDOS DAS TRANSPOSASES
O alinhamento das sequências de aminoácidos foi efectuada usando a aplicação informática ClustalW, disponível on line no endereço da European Bioinformatics Institute. A EBI é uma organização académica com fins não lucrativos de investigação e prestação de serviços em bioinformática; de entre as suas principais actividades, constam a gestão de bases de dados que contenham informação biológica como ácidos nucleicos, sequências proteicas e estruturas macromoleculares. O endereço de acesso a estas aplicações é: em
http://www.ebi.ac.uk/clustalw/. Esta aplicação permite efectuar o alinhamento de
sequências de DNA ou de proteínas, produzindo um alinhamento múltiplo de sequências do qual se podem derivar árvores filogenéticas verdadeiras.
A sua utilização pode ser feita de duas formas: interactivamente ou por correio electrónico. Na primeira situação, i.e. interactivamente, o utilizador deve esperar que os resultados sejam disponibilizados na janela do browser; na segunda os resultados estão unicamente disponíveis após serem enviados para o endereço de correio electrónico indicado pelo utilizador.
Desta forma, após as sequências se encontrarem num formato aceite pelo programa (NBRF/PIR, EMBL/UniProt, Pearson (FASTA), GDE, ALN/ClustalW, GCG/MSF, RSF), acedeu-se ao serviço para introdução das sequências. A sua introdução foi efectuada num único ficheiro e com as sequências colocadas umas a seguir às outras, usando os comandos copiar/colar; pode também ser carregada integralmente a partir de um ficheiro. Para evitar que ocorram erros na execução da aplicação é muito importante que cada sequência tenha uma designação única
e que não existam linhas vazias, com espaços ou caracteres de controlo entre sequências ou no topo do ficheiro.
O alinhamento aparece na página de resultados bem como as respectivas pontuações. Este pode ser obtido ao seleccionar na opção de alinhamento do ficheiro, pode ser aberta uma janela ou guardado para um ficheiro. Por cada par de sequências que é alinhado, é calculada uma pontuação que é obtida a partir do número de identidades no melhor alinhamento dividido pelo número de resíduos comparados; são excluídas as posições em falta, designadas por gaps. Ambas as pontuações são inicialmente calculadas como pontuações de identidade percentuais (percent identity scores) e são convertidas para distâncias evolutivas através da divisão por 100 e subtracção de 1,0 para se obter o número de diferenças por local. A pontuação do alinhamento é calculada de duas formas: rápida ou lenta (forma mais precisa). As pontuações são calculadas a partir de alinhamentos de pares separados. Estes podem ser calculados usando dois métodos: Dynamic programming (lenta mas precisa) ou pelo método de Wilbur e Lipman (extremamente rápida mas aproximada), sendo este último o método escolhido para o cálculo das pontuações deste trabalho.
3.3. CONSTRUÇÃO DE CLADOGRAMAS
A partir do alinhamento das sequências de aminoácidos extraídas das bases de dados, foram derivados cladogramas representando as relações de proximidade entre elas. Os cladogramas foram realizados recorrendo à aplicação informática TreeView, um software de desenho, visualização e impressão de árvores filogenéticas, a que se pode aceder e descarregar on line no endereço
http://taxonomy.zoology.gla.ac.uk/rod/treeview.html. Este software é compatível e
tem a capacidade de ler ficheiros de árvores filogenéticas: do tipo PHYLIP, do tipo NEXUS que são gerados pelas aplicações PAUP e COMPONENT e ainda os ficheiros gerados pelo programa ClustalW, com o qual se procedeu ao alinhamento das sequências.
Este programa permite ler ficheiros que contenham árvores contendo até quinhentos taxa terminais, sendo o número de árvores limitado pela quantidade de memória disponível no computador. Tem a possibilidade de permitir que a árvore gerada contenha informação adicional como a legenda dos nós internos e as distâncias entre os terminais. Permite ainda que a ordem dos taxa terminais possa ser alterada e a árvore reorganizada.
3.4. ANÁLISE DE DOMÍNIOS CONSERVADOS
Utilizando as sequências de aminoácidos das transposases de bactérias Gram--positivas com elevado conteúdo em G+C, indicadas na tabela 4, efectuaram-se análises comparadas das sequências de aminoácidos em bases de dados proteicas, com o objectivo de verificar a existência ou não de um ou mais domínios conservados.
3.4.1. ALINHAMENTO DE SEQUÊNCIAS DE AMINOÁCIDOS
Para este fim, recorreu-se à aplicação informática WU-Blast2 que se encontra disponível on line no endereço http://www.ebi.ac.uk/blast2/asd.html. WU-Blast2 significa Washington University Basic Local Alignment Tool version 2.0 e permite pesquisar rapidamente sobre a semelhança de sequências proteicas com uma perda mínima de sensibilidade. Os resultados apresentados contêm informações a nível funcional e evolutivo sobre a função e estrutura da sequência inquirida. Esta aplicação informática realiza as pesquisas inquiridas na base de dados UniProt Knowledgebase (a que se pode aceder como UniProt no servidor EBI SRS on line no endereço http://www.ebi.uniprot.org/index.shtml) que é a base de dados central relativamente às funções e sequências proteicas, criada pela fusão da informação de UniProtKB/Swiss-Prot, UniProtKB/TrEMBL e PIR-Protein Sequence Database, e que actualiza bissemanalmente as sequências proteicas publicadas que se encontram disponíveis.
Para se obterem informações sobre uma sequência de aminoácidos, esta deverá ser introduzida na caixa de diálogo, utilizando os comandos copiar/colar, ou ser carregada integralmente a partir de um ficheiro; devendo ser introduzida no formato FASTA, que consiste na linha superior da sequência a iniciar-se com o símbolo “>”, seguido do nome da sequência, que per si, se inicia na linha seguinte. Antes de se proceder à utilização deste serviço, é recomendada a atribuição de um nome à sequência no campo respectivo. Os resultados podem ser obtidos interactivamente, forma de usar preferencial pois os resultados são entregues no servidor assim que se encontrem disponíveis – opção escolhida na obtenção de resultados – ou por correio electrónico, sendo os resultados enviados para o endereço de correio electrónico que é indicado no campo respectivo. O programa utilizado é WU-blastp, concebido para comparar a sequência inquirida com a colecção de sequências proteicas da base de dados.
A utilização deste serviço proporciona a visualização de uma tabela de resultados que poderá ser utilizada para realizar um alinhamento com o programa dbclustal após a eliminação de algumas sequências de origem dúbia, fragmentos de sequências e sequências repetidas. Ainda na tabela de resultados, encontra-se disponível uma hiperligação para a entrada de resultados que apresenta melhor pontuação e a percentagem de identidade de 100%. Abrindo essa hiperligação é possível aceder-se à base de dados UniProt (http://www.ebi.uniprot.org/entry/), onde se encontra caracterizada a sequência de aminoácidos inquirida e é permitido o acesso, através da abertura de uma outra hiperligação, à base de dados proteica Pfam.
3.4.2. CONSULTA DA BASE DE DADOS Pfam
Pfam é uma base de dados de famílias de proteínas e é constituída actualmente por 7973 famílias. Cada uma das famílias é processada manualmente e é representada por dois alinhamentos de sequências múltiplos, dois modelos de perfis-Hidden Markov (profile-HMMs) e um ficheiro de anotação. Todos os dados encontram-se disponíveis para serem descarregados em formato flatfile dos sítios
on line FTP com hiperligações a partir de cada um dos websites Pfam. As famílias Pfam são actualizadas periodicamente, sendo em média modificadas quatro vezes desde a sua criação. Os dados e características adicionais podem ser
acedidos a partir de quatro endereços on line:
http://www.sanger.ac.uk/Software/Pfam/, http://pfam.wustl.edu,
http://pfam.jouy.inra.fr e http://Pfam.cgb.ki.se/ .
Da sua colecção de dados constam famílias, domínios de proteínas, alinhamentos múltiplos de domínios ou regiões de proteínas, sendo actualmente constituída por dois conjuntos de famílias. As famílias Pfam-A, que são baseadas em alinhamentos múltiplos realizados manualmente e as famílias Pfam-B, baseadas num agrupamento automático derivado da base de dados ProDom (que compreende informação de domínios de famílias de proteínas, gerada automaticamente das bases de dados de sequências SWISS-PROT e TrEMBL). Acedendo-se a este serviço on line nos endereços acima indicados, é possível obter informações sobre o domínio ou região de um sequência proteica que seja conservada, bem como o seu alinhamento, a organização estrutural do domínio sob perspectiva gráfica, a organização do domínio por espécies e a árvore filogenética.
3.5. DETECÇÃO DE MOTIVOS CONSERVADOS EM GENOMAS PROCARIÓTICOS
3.5.1. COMPARAÇÃO DE SEQUÊNCIAS DE NUCLEÓTIDOS
Basic Local Alignment Search Tool (BLAST) é uma ferramenta informática disponibilizada pelo National Center for Biotechnology Information [NCBI – divisão da National Library of Medicine (NLM) do National Institutes of Health (NIH), organização que pretende divulgar a informação biomédica existente e que para tal disponibiliza recursos sobre biologia molecular, compila bases de dados públicas, conduz pesquisas em biologia computacional e desenvolve aplicações informáticas para analisar dados genómicos] que permite pesquisar regiões de
semelhança local entre sequências. Esta ferramenta compara sequências de nucleótidos ou proteínas com a colecção de sequências existente na sua base de dados e calcula a significância estatística dos resultados. BLAST pode ser usado para inferir sobre as relações funcionais e evolutivas entre sequências, bem como para identificar membros de famílias de genes. Esta ferramenta pode ser utilizada on line no endereço http://www.ncbi.nlm.nih.gov/BLAST/.
3.5.2. DETERMINAÇÃO DO NÚMERO DE CÓPIAS
A obtenção da sequência conservada de aminoácidos, obtida na base de dados proteicos Pfam, permitiu efectuar uma pesquisa sobre a existência de cópias dessa sequência, existentes no genoma de espécies representativas de Actinobacteriaea. A identificação e caracterização do número de cópias presentes efectuou-se acedendo-se ao serviço Genomic BLAST. Genomic BLAST é uma ferramenta gráfica que visa simplificar as pesquisas BLAST sobre sequências genómicas, completas ou incompletas. Esta ferramenta permite que o utilizador compare a sequência inquirida com a base de dados virtual de sequências de DNA e/ou proteínas do grupo de organismos seleccionado com genomas completos ou incompletos.
Os organismos da base de dados podem ser seleccionados quer utilizando uma árvore com base taxonómica, quer numa lista por ordem alfabética de sequências específicas de organismos. A primeira opção foi concebida para ajudar a explorar as relações evolutivas entre organismos dentro de um certo grupo taxonómico quando se executam as pesquisas BLAST. A utilização da lista alfabética permite ao utilizador executar um conjunto de selecções mais elaborado, consultando um número de bases de dados específicas do organismo, com genomas completos ou incompletos. Esta ferramenta encontra-se disponível on line no endereço da National Center for Biotechnology Information (
http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/genom_table_cgi) e actualmente disponibiliza mais de cento e setenta
genomas de Eubactérias e Arqueobactérias, e mais de quarenta genomas Eucariotas.
4. RESULTADOS E DISCUSSÃO
4.1. SEQUÊNCIAS OBTIDAS DO GenBank
Tal como foi referido no ponto 3.1., a identificação das IS já conhecidas e pertencentes à família ISL3 foi efectuada on line no endereço
http://www-is-biotoul.fr. Utilizando a informação obtida neste endereço da Web, efectuou-se a
busca (anexo I) das sequências de nucleótidos e aminoácidos correspondentes às transposases contidas nas sequências de inserção. Esta busca foi efectuada, também on line, a partir da base de dados GenBank (http://www.ncbi.nlm.nih.gov/).
Do grupo de IS referidas no endereço http://www-is-biotoul.fr, foram retiradas todas as sequências de inserção que se integravam em sequências completas de genomas bacterianos, de modo a evitar redundâncias, as que não apresentavam sequências de transposases publicadas no GenBank ou aquelas em que as sequências de transposases depositadas no GenBank se apresentavam incompletas ou revelavam alguma ambiguidade na sua correcta identificação. Esta filtração de resultados tem por base o facto desta base de dados não proceder à revisão das publicações. A tabela 2 mostra quais as sequências eliminadas e critério de eliminação aplicado.
Tabela 2. Sequências de inserção pertencentes à família ISL3 e não utilizadas no presente
estudo.
Nome Sinónimo (s) Origem Critério de Eliminação
IS1167L Streptococcus pneumoniae 13868 Ausente do GenBank IS1167R Streptococcus pneumoniae 13868 Ausente do GenBank IS1182 Staphylococcus aureus BM3121 Ausente do GenBank IS1193D IS1193 Streptococcus thermophilus CNRZ368 Ausente do GenBank IS1207 ISbB1 Corynebacterium glutamicum Bl15 Ausente do GenBank
ISBli3 Brevibacterium linens Ausente do GenBank
ISPp2 Pseudomonas putida ML2 Ausente do GenBank
Tabela 2. Sequências de inserção pertencentes à família ISL3 e não utilizadas no presente
Nome Sinónimo (s) Origem Critério de Eliminação
ISPsp2 Tn401 Pseudomonas sp. strain EST1001 (pEST1226) Ausente do GenBank ISSg1 Streptococcus gordonii M5 Ausente do GenBank IST445 Thiobacillus ferrooxidans Ausente do GenBank IS1651 Bacillus halodurans C-125 Genoma completo IS1652 Bacillus halodurans C-125 Genoma completo
ISRso15 Ralstonia solanacearum Genoma completo
ISBlo6 Bifidobacterium longum NCC2705 Genoma completo
ISP1 Lactobacillus plantarum Genoma completo
ISFnu5 Fusobacterium nucleatum subsp.nucleatum Genoma completo
ISLjo2 Lactobacillus johnsonii Genoma completo
De acordo com este critério, foram escolhidas as transposases das sequências de inserção que se apresentam na tabela 3, abaixo indicada, que resume as designações e números de acesso das transposases contidas em sequências de inserção que constituíram a base para a primeira fase do trabalho. Como forma de distinguir as diferentes cópias da IS466A presentes no genoma de Streptomyces coelicolor A3(2) M145, estas foram numeradas de um a cinco.
Tabela 3. Transposases codificadas em IS pertencentes à família ISL3 e utilizadas no presente estudo.
Nome Sinónimo(s) Origem No. Acesso GI
IS204 Nocardia asteróides YP21 U10634 533509
IS1001 IS1001A Bordetella parapertussis B24 X66858 39756 IS1001B
IS1001C
IS1096 Mycobacterium smegmatis ATCC 607 M76495 150005 IS1165 Leuconostoc mesenteroides subsp. cremoris X62617 44105
DB1165 (p30kb)
IS1167 Streptococcus pneumoniae CP1200 M36180 663278 IS1167A Streptococcus pneumoniae SP-VA96 AF030364 2804718 IS1181 Staphylococcus aureus BM3121 L14544 466496
IS1193 Streptococcus thermophilus Y13713 2687821
IS1251 Enterococcus faecium GUC L34675 535662
IS1476 Enterococcus faecium U63997 1488070
IS13869 IS-Bl Brevibacterium lactofermentum ATCC13869 Z66534 1130607 IS1396 Serratia mercescens (pR471a) AF027768 2688961
IS1411 Pseudomonas sp. EST1001 M57500 145128
IS1489v1 ISPp2 Pseudomonas putida ML2 AF148496 1398920 IS2001 IS1999 Bifidobacterium lactis AJ243948 5639706
Tabela 3. Transposases codificadas em IS pertencentes à família ISL3 e utilizadas no presente estudo. (continuação).
Nome Sinónimo(s) Origem No. Acesso GI
IS31831 IS-Cg Corynebacterium glutamicum ATCC 31831 D17429 790952 IS466A[1] Streptomyces coelicolor A3(2) M145 AB032065 5834493 IS466A[2] Streptomyces coelicolor A3(2) M145 AB032065 5834494 IS466A[3] Streptomyces coelicolor A3(2) M145 AB032065 5834495 IS466A[4] Streptomyces coelicolor A3(2) M145 AB032065 5834496
Nome Sinónimo(s) Origem No. Acesso GI
IS466A[5] Streptomyces coelicolor A3(2) M145 AB032065 5834497 IS469 Streptomyces coelicolor A3(2) M145 AB032065
ISAe1 ISAE1 Ralstonia eutropha H1-4 M86608 141952
ISAsp1 Anabaena sp. strain PCC7120 U13767 540078
ISBli1 Brevibacterium linens AF052055 2967829
ISBma1 ISBm1 Burkholderia mallei AF285635 9930476
ISCx1 Corynebacterium xerosis M82B (pTP10) U21300 [P] 709805 ISL3 Lactobacillus delbrueckii subsp. bulgaricus X79114 1008121
ATCC 11842
ISLdl1 Lactobacillus delbrueckii subsp. lactis AJ302652 11182138
ISM1 Methanobrevibacter smithii X02587 44520
ISPst2 ISPs1 Pseudomonas stutzeri M1 AJ012352 4803708 ISSth1 Streptococcus thermophilus AY376237 38502777
ISPpu12 Pseudomonas putida AY128707 22773654
ISTfe1 IST1, ISAfe1 Acidithiobacillus ferrooxidans U66426 8049953
ISPst3 Pseudomonas stutzeri AJ582631 37496912
4.2. ALINHAMENTO DAS SEQUÊNCIAS DE AMINOÁCIDOS DAS TRANSPOSASES
Após a selecção das transposases contidas nas sequências de inserção que estavam dentro do âmbito deste trabalho (ponto 3.1.) e depois da triagem efectuada de acordo com os critérios referidos no ponto anterior, procedeu-se ao alinhamento das respectivas sequências de aminoácidos [(alinhamento 1), (ponto 3.2.)]. Na realização trabalho preteriu-se a utilização de sequências de nucleótidos em detrimento de sequências de aminoácidos visto ser mais fácil proceder-se directamente ao alinhamento dos últimos, ignorando eventuais emparelhamentos woobble (mutações que ocorrem na terceira base de um codão da sequência codificante e que não originam normalmente alterações apreciáveis) e consequentemente o codão que foi utilizado para um dado aminoácido, centrando-se as substituições nas propriedades dos aminoácidos e não dos nucleótidos. Pesam ainda os factos das sequências de aminoácidos serem três vezes mais pequenas que as sequências de nucleótidos, a sequência de aminoácidos usar um alfabeto mais informativo e não apresentar frame-shift. O
anexo II apresenta o resultado global obtido utilizando a aplicação informática ClustalW.
4.3. CONSTRUÇÃO DE CLADOGRAMAS
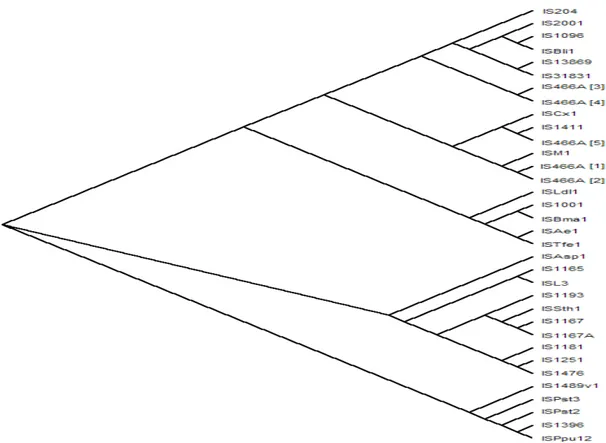
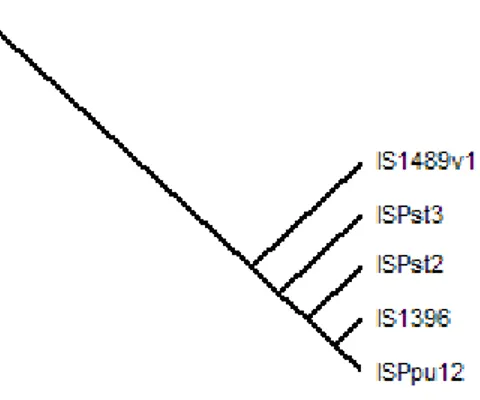
Com base no alinhamento obtido, procedeu-se à construção do cladograma correspondente (ponto 3.3.). O cladograma foi obtido usando a aplicação informática TreeView e o resultado está presente na figura 3. Esta figura evidencia as relações evolutivas existentes entre as transposases contidas em sequências de inserção pertencentes à família ISL3 alvo do estudo. A representação gráfica permite por em evidência a existência de três grandes ramos que derivam da raiz do cladograma.
Ainda da observação da figura 3, pode verificar-se que o ramo que mais cedo apresenta quer um evento cladogénico, quer um maior número de ramificações, é o superior, seguido do ramo intermédio e finalmente do inferior. Verifica-se ainda que o primeiro ramo a ter ramificações é o que mais terminais apresenta e inversamente, que o último a sofrer ramificações, é o que apresenta menos transposases terminais, portanto o ramo inferior. É também no ramo inferior que se encontra a transposase que apresenta as características genéticas mais próximas da raiz do cladograma. Trata-se da transposase da IS1489v1 (de Pseudomonas putida ML2). Esta observação baseia-se no facto da localização relativa dos pontos de ramificação (nós) ao longo do cladograma fornecer uma noção relativa do tempo de origem dos diferentes ramos e das ramificações existentes estarem associadas ao maior ou menor tempo que demorou a que esses eventos tivessem lugar, sendo eles tanto mais frequentes (e portanto com mais terminais) quanto mais cedo aconteceram esses eventos [22].
Figura 3. Cladograma que descreve as relações evolutivas entre as transposases pertencentes à
família ISL3, que se encontram depositadas na base de dados GenBank.
Em seguida descrevem-se em detalhe as características de cada um dos ramos principais do cladograma da figura 3:
a) Ramo superior:
No ramo superior do cladograma (representado em detalhe na figura 4), quatro eventos de cladogénese reorganizam as dezanove transposases em cinco grupos. Do primeiro grupo consta apenas a sequência de inserção IS204 (de Nocardia asteróides YP21). O segundo é constituído por cinco sub-ramos terminais que devido a mais quatro eventos de cladogénese resultam em dois sub-grupos, um que inclui três transposases (IS2001, IS1096 e ISBli1, Bifidobacterium lactis, Mycobacterium smegmatis ATTC 607, Brevibacterium linens, respectivamente) e um outro com as transposases das IS13869 e IS31831 (Brevibacterium lactofermentum ATCC 13869 e Corynebacterium glutamicum
ATCC 31831, respectivamente). Uma ramificação do ramo principal do terceiro grupo origina as duas transposases que o compõem, as cópias 3 e 4 da IS 466A, presentes no genoma de Streptomyces coelicolor A3 (2) M145.
Por sua vez, o quarto grupo, que é o que contém o maior número de transposases em virtude da ocorrência de cinco eventos de cladogénese, é reorganizado em dois subgrupos, constituídos respectivamente pelas sequências de inserção ISCx1, IS1411, IS466A[5] e ISM1, IS466A[1], IS466A[2] pertencentes respectivamente a Corynebacterium xerosis M82B(pTP10), Pseudomonas sp. EST1001, Streptomyces coelicolor A3 (2) M145 e Methanobrevibacter smithii, Streptomyces coelicolor A3 (2) M145. O quinto e último grupo contem um total de cinco transposases: ISLdl1, IS1001, ISBma1, ISAe1 e ISTfe1 (de Lactobacillus delbrueckii subsp. lactis, Bordetella parapertussis B24, Burkholderia mallei; Ralstonia eutropha H1-4 e Acidithiobacillus ferrooxidans respectivamente) resultado de quatro eventos de cladogénese; Sendo ISLdl1 detectada no genoma do ancestral comum de todas elas. Deste primeiro grupo de dezanove transposases, é a que é codificada em ISLdl1 a que se encontra mais próxima da raiz do cladograma, em virtude de apenas ter sido alvo de dois eventos de cladogénese.
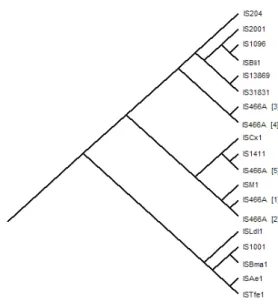
Figura 4. Ramo superior do cladograma que descreve as relações evolutivas de transposases
b) Ramo intermédio:
O ramo intermédio do cladograma (representado em detalhe na figura 5) é composto por um total de dez transposases, das quais a transposase da ISAsp1 (Anabaena sp. strain PCC7120) é o ancestral comum. É composto por um total de quatro grupos, assim organizados em virtude de nove eventos de cladogénese. As transposases IS1165 e ISL3 (de Leuconostoc mesenteroides subsp. cremoris DB1165 (p30kb) e
Lactobacillus delbrueckii subsp. bulgaricus, respectivamente) constituem o segundo
grupo e são as que apresentam maior grau de parentesco com ISAsp1. O terceiro grupo é constituído por quatro sub-ramos terminais, divididos em dois sub-grupos, um com a transposase da IS1193 (Streptococcus thermophilus) e um outro que inclui três transposases (ISSth1, IS1167 e IS1167A, de Streptococcus thermophilus,
Streptococcus pneumoniae CP1200 e Streptococcus pneumoniae SP-VA96,
respectivamente). Por sua vez, o quarto e último grupo inclui três transposases em virtude de mais dois eventos de cladogénese (IS1181, IS1251 e IS1476, de
Staphylococcus aureus BM3121, Enterococcus faecium GUC e Enterococcus faecium, respectivamente).
Figura 5. Ramo intermédio do cladograma que descreve as relações evolutivas de transposases
pertencentes à família ISL3. Adaptado da figura 3.
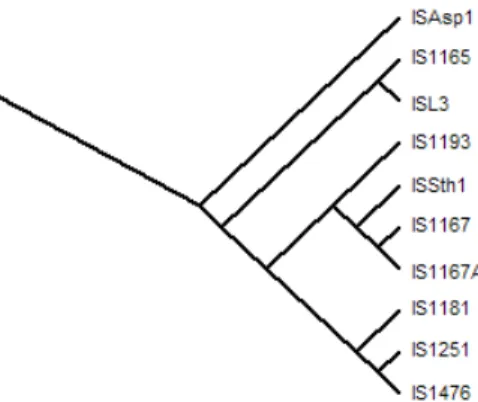
c) Ramo inferior:
O ramo inferior do cladograma, representado em detalhe na figura 6, é o ramo que apresenta menos eventos de cladogénese, apenas quatro, e consequentemente menos transposases, apenas cinco: IS1489v1 (de Pseudomonas putida ML2), ISPst3 (de Pseudomonas stutzeri), ISPst2 (de
Pseudomonas stutzeri M1), IS1396 [de Serratia mercescens (pR471a)] e ISPpu12 (de Pseudomonas putida).
À semelhança do que se verificou para o ramo intermédio, também o ramo inferior do cladograma apresenta um ancestral comum aos cinco terminais, portanto a transposase da IS1489v1.
Figura 6. Ramo inferior do cladograma que descreve as relações evolutivas de transposases
pertencentes à família ISL3. Adaptado da figura 3.
4.4. RELAÇÃO TAXONÓMICA
Com o objectivo de perceber a distribuição taxonómica dos organismos que apresentavam as sequências de aminoácidos de transposases pertencentes à família de sequências de inserção ISL3, alvo do nosso estudo, procedeu-se ao levantamento da sua classificação nos serviços on line das aplicações GenBank e Uniprot, referenciadas no capítulo material e métodos.
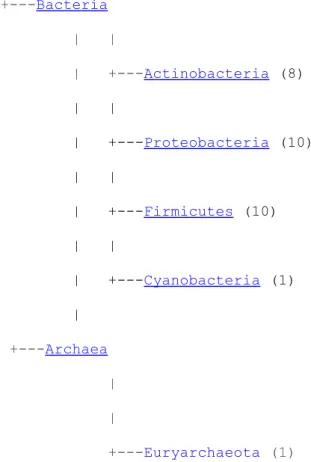
A figura 7, abaixo indicada, resume a sua distribuição taxonómica. Da sua observação, é possível verificar que estes organismos se encontram distribuídos nos domínios Eubactéria e Arqueobactéria, apresentando este último, contudo, apenas um organismo (Methanobrevibacter smithii). Todos os restantes organismos encontram-se classificados no domínio Eubactéria e distribuem-se em quatro géneros: Actinobactéria (oito organismos), Proteobacteria (dez organismos), Firmicutes (dez organismos) e Cianobactéria (um organismo). Um quadro resumo com a classificação dos organismos pode ser encontrada no anexo IV deste trabalho. Transpondo a classificação dos organismos para o
cladograma ilustrado na figura 3, é possível verificar que o ramo inferior do cladograma é constituído apenas por organismos do género Proteobactéria e o ramo intermédio por organismos do género Firmicutes, com excepção de um organismo que se encontra classificado como Cianobactéria (Anabaena sp. strain PCC7120, com ISAsp1). O ramo superior do cladograma apresenta todos os organismos classificados como Actinobactéria, para além dos restantes que se encontram classificados como (Proteobactéria, Firmicutes, Cianobactéria e Euryarchaeota, este último do domínio Arqueobactéria).
+---Bacteria | | | +---Actinobacteria (8) | | | +---Proteobacteria (10) | | | +---Firmicutes (10) | | | +---Cyanobacteria (1) | +---Archaea | | +---Euryarchaeota (1)
Figura 7. Distribuição taxonómica dos organismos contendo transposases pertencentes à família
4.5. TRANSPOSASES DE IS PRESENTES NOS GENOMAS DE ORGANISMOS PERTENCENTES AO RAMO DAS BACTÉRIAS GRAM-POSITIVAS DE ELEVADO CONTEÚDO PERCENTUAL EM G+C
De acordo com os resultados obtidos anteriormente, o cladograma representado na figura 3 contém sobretudo transposases de sequências de inserção presentes em genomas de bactérias Gram-positivas de elevado conteúdo percentual em G+C. De forma a se aprofundar um pouco mais as relações entre transposases de sequências de inserção desta família e pertencentes a um grupo restrito de géneros pertencentes ao ramo das bactérias Gram-positivas de elevado conteúdo percentual em G+C (nomeadamente os géneros Corynebacterium, Brevibacterium, Nocardia e Mycobacteryum), procedeu-se a um segundo alinhamento (alinhamento 2), e construção do respectivo cladograma, incluindo apenas as sequências de aminoácidos das transposases referidas na tabela 4. O procedimento experimental foi de acordo com o referido nos pontos 3.1., 3.2. e 3.3. respectivamente, do capítulo material e métodos.
Tabela 4. Caracterização de transposases de IS presentes em genomas de bactérias Gram--positivas de elevado conteúdo percentual em G+C, objecto do segundo alinhamento.
Nome Dimensão Origem Locus Nº. acesso
IS204 1452 pb Nocardia asteróides YP21 NAU10634 U10634 ISBli1 3501 pb Brevibacterium linens AF052055 AF052055 IS1096 2276 pb Mycobacterium smegmatis ATCC 607 MSGTNP M76495 IS1386
9 1840 pb Brevibacterium lactofermentum ATCC 13869
BLIS1369 Z66534 IS3183
1 1469 pb Corynebacterium glutamicum ATCC 31831
CORIS31831 D17429
![Tabela 1. Sequências de Inserção pertencentes à família ISL3. Adaptado de [1].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15755873.1074220/23.892.124.791.250.1116/tabela-sequências-de-inserção-pertencentes-família-isl-adaptado.webp)