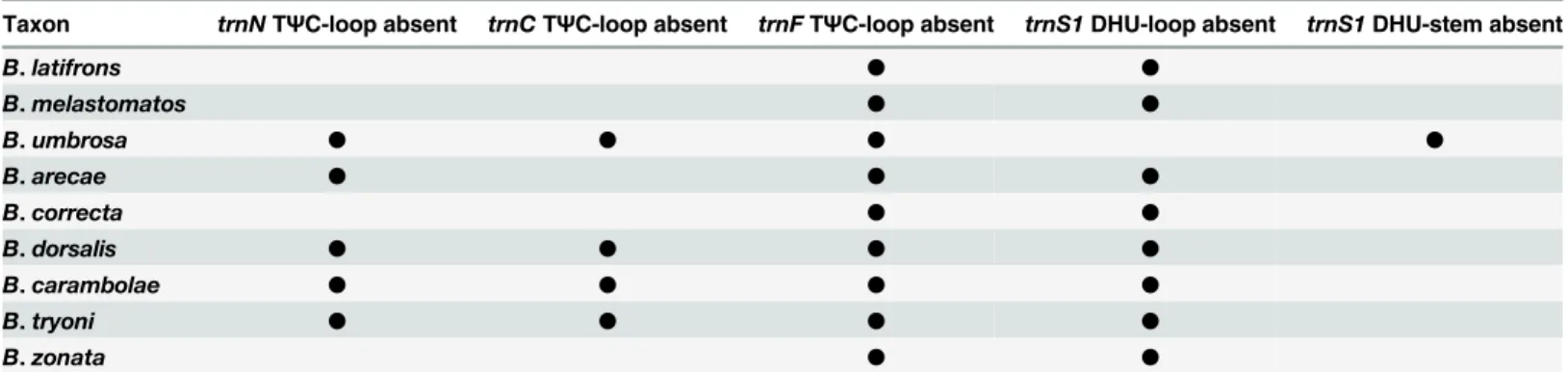
Complete Mitochondrial Genome of Three Bactrocera Fruit Flies of Subgenus Bactrocera (Diptera: Tephritidae) and Their Phylogenetic Implications.
Texto
Imagem

Documentos relacionados
Cassandra teve sua escrita marcada por tal característica, ao declarar em diversas ocasiões que seus livros não eram “picantes e nem obscenos, nem mais realistas do que outros
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
Ele complementa: “Através deste falso contraste entre arte e realidade, as mais abomináveis instituições e práticas de nossa sociedade legitimam-se e reforçam-se
openPAMPA A func¸˜ao openPAMPA vai inicializar, dentro de uma estrutura do tipo struct pampa context, todas as sockets, threads, e vari´aveis, necess´arias ao funcionamento do
This was done in order to analyze the effect of the larva development of the olive fly inside different olive culti- vars (Cobrançosa, Madural, and Verdeal Transmontana) on the
Foram meses de muito trabalho, principalmente, para mostrar aos profissionais da saúde de Foz do Iguaçu que o serviço fica muito melhor quando estamos presentes, que somos
A maximum likelihood phylogenetic analysis of the main groups of Apocrita was performed using amino acid sequence of 13 protein-coding genes, showing monophyly for the
Vainzof M, Passos-Bueno MR, Pavanello RC, Marie SK, Oliveira AS and Zatz M (1999) Sarcoglycanopathies are responsible for 68% of severe autosomal recessive limb-girdle
