Printed in Brazil - ©2005 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
A
r
ti
c
le
* e-mail: [email protected]; [email protected]
Configuration Interaction Simulation of the NEXAFS Photoabsorption Spectrum of
Naphthalene
Eduardo Hollauer*,a, Elísia dos Santos Prucolea, Maria Luiza M. Rocco*,b, Annibal D. Pereira Nettoa, Achim Schöllc and Rainer Finkd
a
Instituto de Química, Universidade Federal Fluminense, Outeiro de São João Batista, s/n Valonguinho, 24020-150 Niterói - RJ, Brazil
b
Instituto de Química, Universidade Federal do Rio de Janeiro, Cidade Universitária, 21949-900 Rio de Janeiro - RJ, Brazil
c
Experimentelle Physik II, Universität Würzburg, Am Hubland, D-97074 Würzburg, Germany
d
Physikalische Chemie II, Universität Erlangen, Egerlandstrasse 3, D-91058 Erlangen, Germany
O espectro NEXAFS do naftaleno foi analisado sob a aproximação do estado final. Cálculos ROHF/SCF foram realizados para os estados localizados do caroço, segundo o procedimento de Bagus-Schaffer para cada canal de excitação. Cálculos CI foram realizados com excitações simples e duplas do carbono 1s para os dez orbitais virtuais de menor energia. As energias e forças do oscilador foram calculadas e o espectro final foi gerado por convolução de picos gaussianos com 0,5 eV de largura a meia altura, com ponderação estatística dos canais. Os resultados teóricos mostram excelente concordância com o espectro NEXAFS de alta resolução, obtido no BESSY II. Trabalho se encontra em andamento de modo a controlar de maneira adequada o problema do colapso variacional em nível CI através da metodologia MEG-CI, desenvolvida para o tratamento de estados de simetria quebrada. Esta metodologia oferece uma forma clara e transparente de considerar o problema da relaxação, da energia de correlação e da especificidade orbital necessária para o problema da predição do espectro NEXAFS de compostos poliaromáticos.
The near-edge X-ray absorption fine structure (NEXAFS) spectrum of naphthalene was analysed theoratically using the final state approximation rule. ROHF/SCF calculations were carried out for a localized core-hole cationic state under the Bagus-Schaffer scheme for each channel of inequivalent excitations. CI calculations were performed allowing single and double excitations from carbon 1s electrons to the first ten virtual orbitals. The energies and oscillator strengths were calculated. The final spectrum was generated by convolution of Gaussian peaks with 0.5 eV as FWHM of the statistically-averaged calculated transitions. The theoretical result shows remarkable agreement with the experimental high-resolution NEXAFS data measured at BESSY II. Work is in progress to get better control of the variational wave function collapse problem at CI level through the use of symmetry breaking core-hole excited states (MEG-CI). This methodology might offer a clear and transparent way to take into account the relaxational and correlation energy, the virtual orbital specificity and the required restricted configuration interaction needed to the NEXAFS prediction problem.
Keywords:ab initio calculations, photoabsorption spectroscopy, NEXAFS, naphthalene
Introduction
Poly-p-phenylene and polyacene compounds have been chosen as prototype molecular probes for several conceptual patterns in the interpretation of the carbon K-edge X-ray absorption spectrum.1 Earlier studies observed that NEXAFS
For several molecular systems the NEXAFS spectrum is a typical fingerprint of the molecular system, a problem which poses a great deal of controversial questions such as: the nature of the observed peaks, the lack of dependency of chain length increase, the diminished oscillator strength for the HOMO-LUMO transition, the nature of the final state and whether it possesses localized or delocalized character, and the role of the relaxational processes. Currently, at least three different interpretations of the NEXAFS spectra can be found.
Ma et al.2 measured the NEXAFS spectra of ethylene
and benzene and its isotopomers, and interpreted the near-edge structure as a progression of many in- and out-of-plane CH bendings, CC and CH stretchings modes showing intense vibronic coupling to the inner-shell electronic states. In this study the authors reported clear indications of symmetry breaking processes in core-excited states.
A second alternative interpretation to the complex features of near-edge excitations has been proposed on empirical grounds and is known as the initial state proposition.1 For large molecular systems, built from
heterogeneous block units the whole spectrum might be convoluted from the spectra of the basic block units. This model suggests an additive pattern among these block units but their results do not take into account the molecular coupling among the block units. Consequently, the NEXAFS spectrum is expected to be an approximate replica of the DOUS. Therefore, from the calculational point of view, it would be enough to calculate the fundamental state and its orbital structure to estimate the NEXAFS spectra from the DOUS. Earlier calculations showed a perfect agreement among the NEXAFS spectra and the calculated DOUS for few long chains p-conjugated systems. The buckyball C60 and graphite are examples of few systems for which an exact agreement was found,3
however, for other strongly delocalized molecular systems this approach fails. Nowadays, it is recognized the limitations of this proposition that doesn’t consider the electronic relaxation associated to the core-hole effect.
A third alternative, of increasing acceptance, is known as the final state proposition.2,4,5 Here a major role is
dedicated to the relaxation processes between the core-hole and the valence orbitals. In this formalism the complex spectral features are interpreted as a consequence of the major chemical shifts caused by the relaxation of the core states in each of the C-1s sites. The final NEXAFS calculated spectrum results from the convolution of the different K-edge absorption spectra of the different sites.
Many authors proposed slightly different schemes to deal with the strong relaxation processes. Aagren et al.5,6
suggested the static exchange hamiltonian (STEX)
approach where the core hole orbital and its orbital energies are determined through a SCF calculation. The optical transition elements are evaluated through a non-orthogonal dipole-transition element calculation between the ground and core-excited states. Such a procedure often leads to symmetry broken states and exhibits an intense lowering of relaxation and correlation energies. NEXAFS spectra of benzene, naphthalene, anthracene, tetracene and pyrene were studied with the STEX formalism.6
Oji et al.7 employed a modification of the improved
virtual orbital technique (IVO) of Hunt and Goddard8 where
the final state energy is directly evaluated from atomic core-hole states. By using site-specific virtual orbitals they observed that the virtual orbitals are stabilized by several eV and accordingly the oscillator strengths are concentrated close to the low-lying virtual orbitals. Moreover, the rich K-edge NEXAFS structure was interpreted as being a consequence of the intense chemical shifts observed in the carbon sites. NEXAFS spectra of benzene, chrysene, perylene and coronene were studied by the final state methodology7
and a remarkable agreement between the experimental spectra and the calculated values was observed.
Recently Bergmann et al.9 and Gordon et al.10 reported
final state calculations for benzene and several poly-aromatic molecules, such as naphthalene, anthracene, phenanthracene, triphenylene, pyrene and 1,2-ben-zanthracene. Their studies were concerned to the dipole limit, which was found valuable and showed good agreement with inner-shell data. The assignments presented there show a perfect match with our results.
Intense work has been carried out in recent years by Hollauer and coworkers to understand the gains of stepping further and employing fully-symmetrical wavefunctions built from multistructural generalized configuration interaction calculations (MEG-CI)11 to describe inner-shell
excitations. This procedure was successfully used to study the He2+ molecular ion,12 and nitrogen and carbon dioxide
inner-shell excitations.13 In all cases it was possible to show
that reliable oscillator strengths and excitation energies can be obtained by this procedure.
Methodology
One process that is recognized of major importance to satisfactorily describe the NEXAFS spectrum is the molecular relaxation. However, most of the methods currently employed for the prediction of NEXAFS spectra deal with this property in limited ways by: a) allowing relaxation of the core-hole orbital; b) artificially imposing a core hole over a specific site; or c) calculating the virtual orbitals with the previous restrictions. Here, we present a procedure, which possibly can be carried out with many of the available quantum molecular programs to obtain NEXAFS spectrum. The main points of the adopted procedure are sketched below.
The ground state is calculated with a basis set of, at least, double-zeta quality and a full use of symmetry. The molecular ground state wavefunction is then represented as a CI calculation with double excitations from the core orbital to the first few virtual orbitals of the system.
Core hole states for the cationic species are optimized through the Bagus and Schaffer methodology, where the ionized state is calculated through a sequence of SCF calculations with successive frozen-restriction optimizations. Due to their characteristics, the final virtual orbitals of the cationic species are the best virtual orbitals for the neutral core-hole excited state. This procedure is conducted for all non-equivalent core-hole states within a molecular frame. The orbitals obtained within this procedure are employed for the localized CI calculation described below.
Several restricted CI calculations are carried out, one for each site considered. Within these CI calculations all double excitations were allowed from the C-1s core active inner-shell orbital to the first ten virtual orbitals of the system.
Oscillator strengths were calculated through the two CI calculations previously mentioned within the dipole gauge. Recent work has shown the validity of this procedure.10
The final spectrum is obtained by a statistical convolution from each of the site spectra, using gaussian functions with 0.5 eV of full width at half maximum (FWHM). The employed weights are determined by the symmetry equivalent sites present in the system.
Experimental
The high-resolution photoabsorption spectrum at the C K-edge of naphthalene condensed on Ag(111) was taken at the U49/1-PGM undulator beamline of BESSY II.14 An
energy resolving power (E/DE) better than 10.000 at the nitrogen K-edge could be demonstrated with a photon flux of about 3 x 1010 ph/s/100 mA (fixed-focus constant c
ff = 10,
exit slit 10 mm). The beam line is equipped with an
ultrahigh-vacuum spectroscopy chamber with a SCIENTA XES200 electron energy analyzer for photoemission experiments and a retarding field analyzer for partial electron yield detection. Saturation effects can be completely neglected due to the fact that the films are not too thick. Neither in total nor in partial yield detection mode one would expect saturation since the secondary electrons that are used for detection in either case should reach the detector.1 The Ag(111) crystal
was subjected to several sputtering/annealing cycles in order to achieve the desirable clearance. The cleanliness of the substrate was checked through photoemission recorded at the carbon and oxygen K-edges as well as at the silver 3d-edge. High-purity naphthalene purchased from Aldrich (Germany) was used without further purification. The naphthalene multilayer films were prepared in situ by sublimation at a substrate temperautre of 150K.
Calculations
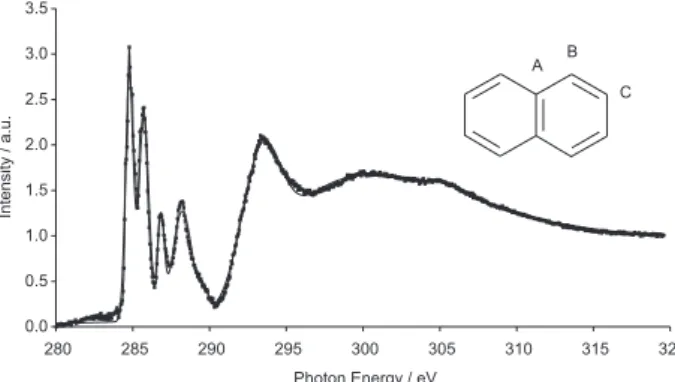
The naphthalene NEXAFS spectrum has been subjected to a deconvolution process aiming to obtain the best spectral fit (Figure 1). We employed Gaussian and Lorentzian bands to describe the major peaks and a Gaussian step function to represent the ionization channel. All calculations employed the FIT program15 previouly
developed to deconvolute spectra resulting from Gaussian, Lorentzian and Gaussian step functions. The program employs few iterations with regular steepest descent methods and then turns to Davidon-Fletcher-Powell/ Broyden-Fletcher-Goldfarb-Shanno variable metric methods.16 During the convergence process few restrictions
have been imposed, however by the end of the process a self-consistent set of bands have been obtained for which energies, band widths and intensities were fully optimized. The naphthalene spectrum shows noticable features. The Lorentzian band present at 301 eV, which is the most intense band in the 280 - 315 eV energy region, was chosen as the reference band for the determination of intensities. Therefore, all peak intensities actually report relative areas, as can be seen in Table 1. Two intense and sharp peaks found at 284.8 and 285.7 eV with 0.21 and 0.23 relative intensities, respectively, are related to core excitations to the LUMO π* orbitals. Two transitions follow at 286.8
All calculations employed double-zeta basis of Dunning-Huzinaga.17 No diffuse, neither polarization, basis
functions have been added to constrain the calculation size limits. For the ground state, D2h symmetry was employed while C1 symmetry was used for the cationic core-hole states. For the core-hole states the ROHF/SCF wavefunction was carried out through the Bagus and Schaefer methodology, where the ionized state is calculated through a sequence of SCF calculations with successive frozen-restriction optimizations. For convention, in this paper the tertiary carbon atom of naphthlene was denoted as A carbon, its neighboor secondary carbon as B carbon and the next as the C carbon (Figure 1). The GAMESS-GVB program18 was
used for the SCF procedure, with the mixing restriction among different orbital classes. This procedure was repeated along all equivalent ionization sites. The calculated ionization energies for each site are presented in Table 2.
For each channel SCF calculations for the occupied as well as for the virtual orbitals were carried out and the corresponding orbitals were collected for the CI calculations. As the final virtual orbitals, in this case, were obtained from the SCF calculation for the cationic species, we must point out that this procedure makes them the best virtual orbitals for the neutral core-hole excited state.13
Configuration interaction calculations were carried out for each channel by using its localized occupied and virtual orbitals obtained from the cationic state calculations. Single and double excitations from the ground-state determinant were performed. Excitations were allowed from the inner-shell orbital to the first ten low-lying unoccupied orbitals. All the other orbitals were kept frozen as expectators during these calculations, which led to a CI representation of the ground state and an ensemble of CI representations for the core-hole excited states. Their energies and oscillator strengths for the ground state were calculated. The results are presented in Table 3, along with its channel contributions.
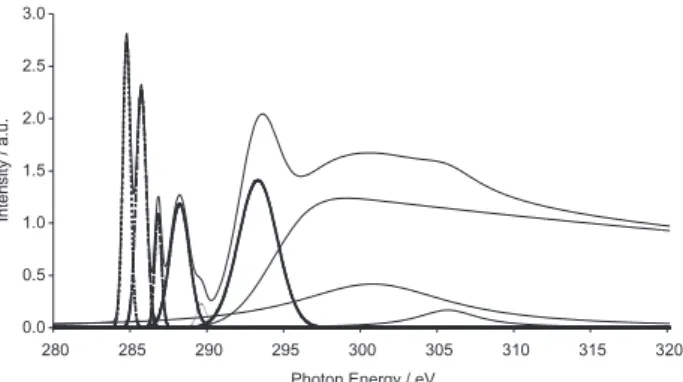
The final spectrum was generated by a convolution of Gaussian peaks with 0.5 eV as FWHM of the statistical-averaged calculated transitions (Figure 2). An energy shift of 3.9 eV was introduced in the convoluted spectrum to describe the relaxational behavior of the expectator electrons.
The results clearly show that the first sharp intense peak at 284.8 eV must be assigned to a mixture of two excitations from the B and C 1s carbon orbitals to the π*
LUMO orbital. The second sharp peak placed at 285.7 eV clearly results from the 1s excitation from the A carbon to the same π* orbital. The third convoluted peak, calculated Table 1. Deconvolution process carried out with six Gaussian
func-tions, two Lorenztian functions and a Gaussian step function. All energies are given in eV and the areas in the last column have been normalized to the largest peak area
Function type Energy (eV) A Area Relative Area
Gaussian 284.77 8.10 1.69 0.21
Gaussian 285.73 4.66 1.85 0.23
Gaussian 286.83 10.07 0.60 0.07
Gaussian 288.23 1.27 1.85 0.23
Gaussian 289.65 5.07 0.18 0.02
Gaussian 293.32 0.32 4.44 0.55
Gaussian Step 294.29 4.98
Lorentzian 300.79 5.71 8.12 1.00 Lorentzian 305.70 2.47 1.29 0.16
Table 3. Oscillator strengths and energies for the most important inner-shell excitation channel transitions. All energies have been subtracted by 3.9 eV
Trans. Energy (eV) A Channel B Channel C Channel (Total Intensity) (Channel (Channel (Channel
Intensity) Intensity) Intensity)
284.9 (0.057) 0.014
285.2 (0.060) 0.015
285.8 (0.038) 0.019
286.5 (0.005) 0.001
287.9 (0.016) 0.002 0.003
290.3 (0.116) 0.029
290.4 (0.116) 0.029
290.5 (0.048) 0.005 0.007
291.3 (0.024) 0.006
291.7 (0.048) 0.009 0.003
292.0 (0.060) 0.008 0.007
292.2 (0.008) 0.004 292.5 (0.063) 0.038
Table 2. Ionization potentials of naphthalene at SCF and CI levels
State E (SCF) Hartree IP (eV)
Ground State -383.2508020
Core Hole State IP (SCF) eV
A -372.4944 292.70
B -372.5055 292.40
C -372.5055 292.40
at 286.8 eV, shows a small intensity when compared to the experimental results. However, it results from C carbon 1s excitation. The fourth convoluted peak (288.2 eV) has a major contribution from B and A carbons. All the previously mentioned peaks show an agreement to the experimental excitation energies within the 0.30 eV error, which is a significant task considering the experimental energy range. The final assignment for the NEXAFS C-1s spectrum of naphthalene is shown in Table 4.
Conclusions
Several simulations have been carried out in order to interpret the carbon 1s NEXAFS spectrum of naphthalene. A temptative calculation, based on frozen core calculations and symmetry based orbital excitations to IVO orbitals, revealed that the simple initial state proposition, where the NEXAFS spectrum is expected to be an approximate replica of the density of unoccupied states, fail in the naphthalene case.
The simulations carried out in this work reveals also that the naphthalene spectrum might be interpreted as single particle excitations from localized core hole at the 1s carbon orbital to the first few low-lying unoccupied π*
orbitals of naphthalene, based on the so-called final state proposition. Within our procedure, a CI calculation based on state specific self-consistent calculations explicitely treat two of the major difficulties concerning inner-shell valence transition calculations; the orbital relaxation and the strong virtual orbital dependency with respect to the core-hole state. The results show a remarkable agreement to the naphthalene NEXAFS spectrum.
Work is in progress to get better control of the variational wave function collapse problem at the CI level. In the present work this problem has been overridden by a restricted configuration selection. Superposition of symmetry breaking core-hole excited states are currently subject of work in our group and might offer a clear and transparent way to take into account the relaxational correlation energy, the virtual orbital specificity and wide configuration interaction to the required solution to the variational collapse in this kind of calculation.
Acknowledgments
MLMR would like to thank DAAD, CNPq and FAPERJ for fellowship supports. EH and MLMR acknowledge CNPq for finantial support. ESP acknowledges CNPq for a PIBIC undergraduate grant. Part of this project was supported by Projeto Jovem Cientista from FAPERJ (E-26/150118/2001) and Projeto PAI/FAPERJ/CNPq (520499/01-8). The experimental work was financed by the BMBF under contract 05 KS1WWA/5. The experimental assistance of the BESSY staff is gratefully acknowledged.
References
1. Stohr, J.; NEXAFS Spectroscopy, Springer: Berlin, 1992. 2. Ma, Y; Sette, F.; Meigs, G.; Modesti, S.; Chen, C.T.; Phys. Scr.
1990, 41, 833. Table 4. Final assignment based on inner-shell localized excitation
model for the near-edge C-1s spectrum of naphthalene
Transition Energy (eV) Assignments
284.80 C(B,C) to LUMO π orbital 285.75 C(A) to LUMO π orbital 286.85 C(C) to LUMO π orbital 288.25 C(B,A) to LUMO π orbitals
289.67 C(B,C) to LUMO orbitals
293.35 C(A) excitations to CH σ* orbitals
Figure 3. The convoluted spectra obtained from SCF/CI calcula-tions. At the top the density of unoccupied states are shown. In the middle part we present the convoluted spectrum, compared with the NEXAFS spectrum in the 284 - 289 eV energy range (bottom). The first experimental band, placed at 285 eV, has contributions from B and C carbon sites, while the second band has sole contribution from the A site. The convoluted band at 287 eV results from C excitations, and shows small intensity. B and C sites contribute to band shown at 288 eV.
3. Yokoyama, T.; Seki, K.; Morisada, I.; Edamatsu, K.; Ohta, T.; Phys. Scr.1990, 41, 189.
4. Ma, Y; Sette, F.; Meigs, G.; Modesti, S.; Chen, C.T.; Phys. Ver. Lett.1998, 19, 2044.
5. Aagren, H.; Carravetta, V.; Vahtras, O.; Pettersson, L.G.M.; Chem. Phys. Lett. 1994, 222, 75.
6. Aagren, H.; Vahtras, O.; Carravetta, V.; Chem. Phys. 1995, 196, 47.
7. Oji, H.; Mitsumoto, R.; Ito, E.; Ishii, H.; Ouchi, Y.; Seki, K.; Yokoyama, T.; Ohta, T.; Kosugi, N.; J. Chem. Phys.1998,
109, 10409.
8. Hunt, W. J.; Goddard III, W.; Chem. Phys. 1969, 54, 414. 9. Bergmann, U.; Groezin, H.; Mullins, O.C.; Glatzel, P.; Fetzer,
J.; Cramer, S.P.; Chem. Phys. Lett. 2003, 369, 184.
10. Gordon, M. L.; Tulumello, D.; Cooper, G.; Hitchcock, A.P.; Glatzel, P.; Mullins, O.C.; Cramer, S.P.; Bergmann, U.; J. Phys.
Chem. A2003, 107, 8512.
11. Hollauer E.; Nascimento, M. A. C.; J. Chem. Phys. 1993, 99, 1207.
12. Hollauer E.; Nascimento, M. A. C.; J. Chem. Phys.Lett. 1991,
184, 470.
13. Bielschowsky, C. E.; Nascimento, M. A. C.; Hollauer, E.; Phys.
Rev. A1992, 45, 7942.
14. Rocco, M.L.M.; Schöll, A.; Fink, R.; Umbach, E. (unpublished work).
15. Hollauer, E.; Prucole, E.S.; FIT Program: A small Fortran program to deconvolute spectra resulting from Gaussian, Lorenztian and Gaussian step functions. The program works with steepest descent methods, and Davidon-Fletchr-Powell and Broyden-Fletcher-Goldfarb-Shanno variable metric methods. Optimization of energies, half-width and intensity contributions can be done. Restrictions on the optimization variables can be established by the users.
16. Press, W. H.; Flannery, B. P.; Teukolsky, S. A.; Vetterling, W. T.; Numerical Recipes, Cambridge University Press: Cambrigde, 1986.
17. Dunning Jr., T. H.; J. Chem.l Phys. 1970, 53, 2823. 18. Schmidt, M. W.; Balridge, K. K.; Boatz, J. A.; Elbert, S. T.;
Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, M.; Nguyen, K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Montgomery, J. A.; J. Comp. Chem.1993, 14, 1347.
Received: October 21, 2003