UNIVERSIDADE DE LISBOA
Faculdade de Medicina de Lisboa
USE OF GENOMIC DNA-REPORTER TOOLS TO
DISSECT PATHOLOGICAL MECHANISMS
CAUSED BY GAA EXPANSIONS IN
FRIEDREICH’S ATAXIA
Ana Maria Ferreira da Silva
Orientadores: Professor Doutor Richard Wade-Martins
Professor Doutor Tiago Fleming Outeiro
Programa de Doutoramento em Neurociências
Tese especialmente elaborada para a obtenção do grau de Doutor
em Ciências Biomédicas, especialidade de Neurociências
UNIVERSIDADE DE LISBOA
Faculdade de Medicina de Lisboa
USE OF GENOMIC DNA-REPORTER TOOLS TO DISSECT
PATHOLOGICAL MECHANISMS CAUSED BY GAA
EXPANSIONS IN FRIEDREICH’S ATAXIA
Ana Maria Ferreira da Silva
Orientadores: Professor Doutor Richard Wade-Martins Professor Doutor Tiago Fleming Outeiro Programa de Doutoramento em Neurociências
Tese especialmente elaborada para a obtenção do grau de Doutor em Ciências Biomédicas, especialidade de Neurociências
Júri:
Presidente: Doutor José Luís Bliebernicht Ducla Soares, Professor Catedrático da Faculdade de Medicina da Universidade de Lisboa
Vogais: Professor Richard Wade-Martins, Professor of Molecular Neuroscience da Universidade de Oxford, United Kingdom; (Orientador)
Doutora Patrícia Espinheira Sá Maciel, Professora Associada da Escola de Ciências da Saúde da Universidade do Minho;
Doutor Luís Fernando Morgado Pereira de Almeida, Professor Auxiliar da Faculdade de Farmácia da Universidade de Coimbra;
Doutor Manuel Diamantino Pires Bicho, Professor Catedrático da Faculdade de Medicina da Universidade de Lisboa;
Doutor Mamede Alves de Carvalho, Professor Catedrático da Faculdade de Medicina da Universidade de Lisboa;
Doutor Tiago Fleming de Oliveira Outeiro, Professor Associado Convidado da Faculdade de Medicina da Universidade de Lisboa (Co-orientador).
Trabalho financiado pela Fundação para a Ciência e a Tecnologia através da bolsa de doutoramento SFRH/BD/61048/2009.
As opiniões expressas nesta publicação são da exclusiva responsabilidade do seu autor.
A impressão desta tese foi aprovada pelo Conselho Científico da Faculdade de Medicina de Lisboa em reunião de 15 de Dezembro de 2015.
vi
Acknowledgements
The work described in this Thesis would not have been possible without the support of my supervisors, colleagues, friends and family.
First and foremost, I would like to thank my supervisor Richard Wade-Martins for his time, encouragement, sharp insight and expectance of excellence. I am grateful that he entrusted me with the coolest project I could have ever hoped for, but also granted me enormous freedom to wander intellectually and pursue my own ideas. Without his support I would not have been able to see it through.
I would like to thank my co-supervisor Tiago Fleming Outeiro for his enthusiasm, constant encouragement and academic support throughout my PhD.
I am grateful for the outstanding support, knowledge and guidance of the talented Michele Lufino. I deeply enjoyed our intense and vibrant discussions which greatly boosted my intellectual growth.
My humble and sincere thanks to the exceptional minds of Jill Brown and Veronica Buckle for their scientific expertise and mentorship on the exciting field of nuclear organisation.
I am indebted to the past and present members of the Wade-Martins Laboratory for creating a supportive environment which allowed me to thrive. From a great team come greater friends: special thanks to Ruxandra Mutihac, Milena Cioroch, Heike Wobst, Tonya Taylor, Lawrence Tam and Lara Lourenço Venda.
I would like to acknowledge Eva Wegel and Ian Dobbie, from the Micron Facility, for providing microscopy training. I also thank Yaron Shav-Tal for the gift of NES-YFP-MS2-NLS construct, and Jonathan Chubb, Richard Parton and Timothy Weil for helpful discussions regarding the MS2 system.
I am grateful to Fundação para a Ciência e a Tecnologia for providing financial support during my PhD.
Lastly and most importantly, I thank my family for encouraging me in all of my pursuits and inspiring me to follow my dreams. In the words of Sir Isaac Newton, “If I have seen further, it is by standing on the shoulder of giants”.
vii
Publications and conference presentations
Publications:
Silva AM, Brown JM, Buckle VJ, Wade-Martins R and Lufino MM (2015) Expanded
GAA Repeats Impair FXN Gene Expression and Reposition the FXN Locus to the Nuclear Lamina in Single Cells. Hum. Mol. Genet. 24(12):3457-71.
Lufino MMP, Silva AM, Nemeth AH, Alegre-Albarrategui J, Russell AJ, Wade-Martins R (2013) A GAA Repeat Expansion Reporter Model of Friedreich's Ataxia Recapitulates the Genomic Context and Allows Rapid Screening of Therapeutic Compounds. Hum. Mol.
Genet. 22(25): 5173-87.
Conferences:
Silva AM, Brown JM, Buckle VJ, Wade-Martins R and Lufino MM (2015) Expanded
GAA Repeats Impair Frataxin Gene Expression and Promote Repositioning to the Nuclear Periphery at Single-Cell Level. Oral presentation at the 2015 International Ataxia
Research Conference, Windsor, UK.
Silva AM, Groves J, Chubb JR, Outeiro TF, Wade-Martins R, Lufino M (2012) Live-cell
visualization of the effect of a GAA repeat expansion on transcription of the FXN gene.
Poster presentation at the Ataxia Research Conference, Stanstead, UK.
Silva AM, Lufino M, Groves J, Wade-Martins R (2012) Epigenetic silencing induced by a
GAA repeat expansion in a cell model of Friedreich’s ataxia. Poster presentation at
Epigenomics, Keystone, CO, USA.
Silva AM, Lufino M, Russell A, Wade-Martins R (2011) Analysis of epigenetic
mechanisms induced by GAA triplet expansion in a human cell model of Friedreich’s ataxia. Poster presentation at the FARA Research Conference, Strasbourg, France.
viii
Abstract
In Friedreich’s ataxia (FRDA), abnormal GAA repeat expansions in intron 1 of the frataxin gene (FXN) cause epigenetic changes and reduce FXN mRNA levels in averaged cell samples though a poorly understood mechanism. Dissecting the silencing mechanism in FRDA in situ is crucial to improve our understanding of the disease.
Here, I use novel FRDA human cell models suitable for screening compounds able to upregulate FXN expression and to analyse the link between FXN nuclear localisation and expression in single cells. FXN-Luc, Luc, FXN-MS2-Luc and
FXN-GAA-MS2-Luc stable human clones carry a site-specific integration of a single copy of the whole FXN locus with either 6 (Luc and MS2-Luc) or ~310 (GAA-Luc and FXN-GAA-MS2-Luc) GAA repeats in intron 1. To fluorescently label the transgenic FXN
mRNA, I inserted MS2 binding sites into exon 2 of FXN-MS2-Luc and
FXN-GAA-MS2-Luc transgenes by homologous recombination. The ~310 GAA repeats recapitulate the
characteristic FXN gene repression and epigenetic changes seen in FRDA.
I report a single-cell analysis of FXN repression in which I identify the nuclear lamina (NL) as a novel and key player in FXN transcriptional impairment and silencing. Using a multidisciplinary approach, including analysis in both fixed and living single cells, I show that expanded GAA repeats increase FXN positioning at the NL, leading to decreased numbers of FXN mRNA molecules and slower transcription kinetics in the
FXN-GAA-MS2 cell model. Restoring histone acetylation reverses NL positioning. I observe the
same abnormal repositioning to the NL in carrier and FRDA patient cells and show that this tightly correlates with a marked decrease in the number of actively expressing FXN alleles. Furthermore, I show that those few active expanded FXN alleles located at the NL express at a significantly lower level than the alleles located in the interior of the nucleus. Finally, I demonstrate that expanded GAA repeats predominantly disrupt FXN transcription initiation.
Collectively, these results suggest repressive epigenetic modifications at the expanded GAA-FXN locus may lead to NL relocation, where further repression may occur. The mechanisms described may extend to other genetic diseases mediated by repeat expansions within regions of non-coding DNA.
Keywords: Friedreich’s ataxia, GAA repeat expansion, single-cell resolution,
ix
Resumo
Na Ataxia de Friedreich (FRDA), uma expansão de repetições trinucleotídicas GAA, presentes no intrão 1 do gene que codifica a proteína frataxina (FXN), causa mudanças epigenéticas e reduz os níveis médios de RNA do gene FXN em amostras celulares através de um mecanismo pouco conhecido. Com o intuito de melhor perceber a patogénese inerente à doença e, em última análise, desenvolver terapias eficientes para a FRDA, é importante criar modelos celulares que traduzam as características repressivas da doença ao mesmo tempo que permitem quantificar eficientemente os níveis de expressão do gene
FXN.
As linhas celulares reporter GAA-Luc, Luc, GAA-MS2-Luc e
FXN-MS2-Luc, descritas nesta Tese, foram especificamente criadas de modo a permitir a
comparação directa entre o efeito das repetições GAA normais e expandidas na expressão do gene FXN. Para o efeito usei: (i) todo o locus FXN com o seu promotor, intrões e exões originais e todos os elementos necessários para a expressão fisiológica do transgene, com inserção do gene luciferase no final do exão 5a; e (ii) uma única cópia de cada BAC integrado num sítio FRT especificamente localizado no cromossoma 1 de células HEK FRT, de modo a excluir efeitos contraditórios na expressão do gene FXN devido a integração de ambos os vectores em sítios diferentes.
Análise das modificações das histonas no promoter do gene FXN e regiões que ladeiam as repetições GAA a montante e a jusante revelou um decréscimo da acetilação de H3K9 e H4K8 e um aumento da metilação de H3K9me2 e H3K9me3 nas três regiões nas células FXN-GAA-Luc. Adicionalmente, as regiões a montante e a jusante das repetições GAA das células FXN-GAA-Luc apresentam um aumento da metilação do DNA em CpG específicos. As células FXN-GAA-Luc foram usadas num rastreio de compostos terapêuticos e permitiram identificar uma molécula capaz de aumentar a expressão do gene
FXN-GAA-Luc para níveis similares aos das células FXN-Luc. A análise usando a técnica
de imunoprecipitação da cromatina em células derivadas de pacientes depois de tratadas com esta molécula revelou o restauro para níveis normais de acetilação de H3K9 e H4K8 nas regiões que ladeiam as repetições GAA. Estes resultados sugerem que as repetições GAA induzem a repressão do gene FXN nas células FXN-GAA-Luc através da alteração da estrutura da cromatina no transgene, fazendo com que estas células sejam consideradas excelentes para o rastreio de moléculas capazes de aumentar a expressão do gene FXN.
x
No entanto, o modelo FXN-GAA-Luc apenas apresenta o estado provável do gene
FXN visto que os resultados provêm de experiências onde se efectuam medições médias
resultantes de amostras contento milhões de células. Consequentemente, o modelo
FXN-GAA-MS2-Luc foi criado para dissecar o mecanismo repressivo de FRDA in situ,
permitindo a visualisação e análise da localização e repressão de FXN em células fixas e vivas usando o sistema MS2.
Neste trabalho, desenvolvi um modelo celular humano para analisar a associação entre a localização e a expressão do gene FXN ao nível da célula. Os clones celulares estáveis FXN-MS2-Luc e FXN-GAA-MS2-Luc foram gerados por integração dos transgenes num local específico e contêm todo o locus FXN de 80 kb, o gene repórter luciferase no exão 5a e seis repetições ou uma expansão de ~310 tripletos GAA no intrão 1, respectivamente. Para efectuar uma marcação fluorescente do mRNA FXN transgénico e quantificar o efeito da expansão de tripletos GAA na transcrição do gene, inseri 24 repetições de locais de ligação da proteína MS2 (MBS) no exão 2 por recombinação homóloga. A expansão de ~310 GAA na linha celular FXN-GAA-MS2-Luc traduz as características repressivas do gene FXN em FRDA.
A localização do transgene FXN nas linhas FXN-MS2-Luc e FXN-GAA-MS2-Luc foi determinado por Immuno-FISH. FXN contactou com a lâmina nuclear (NL) em ~44% das células GAA-MS2-Luc quando comparado com apenas ~10% das células
FXN-MS2-Luc. Após tratamento das células com inibidores das desacetilases de histonas, apenas
o transgene FXN-GAA-MS2-Luc se reposicionou longe da NL. No entanto, ocorreu um aumento da expressão do mRNA FXN transgénico nas duas linhas celulares, sugerindo que existe uma associação complexa entre repressão do gene FXN e a sua localização intranuclear. Para aprofundar o conhecimento sobre a repressão do gene FXN, analisei o output de transcrição de alelos FXN transgénicos individuais nas células FXN-MS2-Luc e
FXN-GAA-MS2-Luc por RNA FISH e recuperação de fluorescência após a fotodegradação
(FRAP). As células FXN-GAA-MS2-Luc contêm ~5 ± 2 mRNAs por célula e as células
FXN-MS2-Luc cells contêm ~9 ± 4 mRNA por célula, indicando que ~310 tripletos GAA
reduzem o número de moléculas de mRNA em 44% ao nível celular. As curvas FRAP mostram que o tempo necessário para a recuperação completa da fluorescência após a fotodegradação é diferente nas duas linhas celulares. O transgene FXN-MS2-Luc apresentou um tempo total de recuperação de 120 segundos, enquanto o transgene
FXN-GAA-MS2-Luc apresentou uma cinética mais lenta com recuperação total de 260 segundos.
xi
células FXN-GAA-MS2-Luc e FXN-GAA-MS2-Luc, os resultados de RNA FISH e FRAP indicam que a expansão de GAA diminui a quantidade de moléculas de mRNA de
FXN-GAA-MS2-Luc através do impedimento da iniciação e/ou elongação da transcrição por
parte da polimerase de RNA II. De modo a elucidar a relação entre a localização e repressão do gene FXN, analisei a intensidade fluorescente de transgene activos em células
FXN-MS2-Luc e FXN-GAA-MS2-Luc vivas. A intensidade fluorescente de locais de
transcrição foi significativamente menor quando os transgenes estavam a expressar na periferia nuclear comparando com o nucleoplasma em células FXN-MS2-Luc e
FXN-GAA-MS2-Luc. Estes dados indicam que os dois transgenes expressam quando localizados na
periferia nuclear, embora o façam em quantidades mais pequenas. Quando comparados com transgenes FXN-MS2-Luc activos, a intensidade dos locais de transcrição dos transgenes FXN-MS2-GAA-Luc foi significativamente mais baixa apenas no interior do núcleo. No seu conjunto, estes resultados sugerem que a expansão de GAA aumenta a localização do transgene FXN-GAA-MS2-Luc na NL, onde os níveis de expressão são reduzidos quando comparados com o interior nuclear ou com os níveis de expressão do transgene FXN-MS2-Luc.
Em seguida, analisei a localização do gene no seu ambiente genómico natural em células derivadas de pessoas saudáveis, de portadores heterozigóticos de um alelo FXN mutante e de pacientes. Em células derivadas de portadores heterozigóticos, o alelo FXN contendo a expansão de GAA localiza-se preferencialmente mais próximo da periferia nuclear do que o alelo normal e contacta mais vezes com a NL. Quando se compara a localização do gene FXN em células saudáveis com células derivadas de pacientes, os resultados indicam que a expansão de GAA aumenta a probabilidade de um alelo se encontrar associado à NL e consequentemente a probabilidade de estar silenciado. Adicionalmente, quando os alelos FXN expandidos se encontravam a expressar, os níveis de expressão eram significativamente reduzidos quando se encontram no nucleoplasma, mas especialmente quando localizados na periferia nuclear. Estes resultados indicam uma relação directa entre o posicionamento do gene FXN na NL e a repressão da transcrição mediada pela expansão de GAA. A realização de uma quantificação ao nível cellular mostrou ainda que a expansão das repetições de GAA induz um défice na expressão maioritariamente ao nível da iniciação da transcrição do gene FXN, mas também induz um pequeno bloqueio na elongação da polimerase de RNA II.
No seu conjunto, estes resultados sugerem que as modificações epigenéticas repressivas no locus FXN expandido podem induzir a relocalização do gene para a NL,
xii
onde uma repressão adicional pode ocorrer. O efeito combinado da presença da expansão de GAA e relocalização do gene para a NL resultam numa redução catastrófica dos níveis de transcrição, levando à redução dos níveis da proteína frataxina e, em última análise, à manisfestação de FRDA. O trabalho descrito nesta Tese apresenta novos conhecimentos sobre as causas moleculares subjacentes à FRDA e poderá ser aplicável a outras doenças genéticas causadas por expansões de nucleótidos em regiões não codificantes do DNA.
Palavras-chave: Ataxia de Friedreich, expansão de repetições GAA, resolução ao nível da
Contents
ACKNOWLEDGEMENTS ... VI PUBLICATIONS AND CONFERENCE PRESENTATIONS ... VII ABSTRACT ... VIII RESUMO ... IX LIST OF FIGURES ... XVII LIST OF TABLES ... XIX LIST OF MOVIES ... XX LIST OF ABBREVIATIONS ... XXI
GENERAL INTRODUCTION ... 26
1.1 UNSTABLE EXPANDED REPEAT DISEASES ... 27
1.2 FRIEDREICH’S ATAXIA ... 30
1.2.1 ETIOLOGY ... 30
1.2.2 EPIDEMIOLOGY ... 31
1.2.3 CLINICAL FEATURES OF FRDA ... 32
1.3 THE FXN LOCUS ... 35
1.3.1 FXN IN HEALTHY INDIVIDUALS ... 35
1.3.2 MOLECULAR BASIS OF FRDA ... 37
1.3.2.1 GAA expansion-mediated transcriptional dysregulation ... 37
1.3.2.1.1 Abnormal DNA structures ... 38
1.3.2.1.2 Heterochromatinisation of the FXN gene ... 40
1.3.2.1.2.1 Histone modifications ... 41
1.3.2.1.2.2 DNA methylation ... 45
1.3.2.1.2.3 RNA interference (RNAi) ... 47
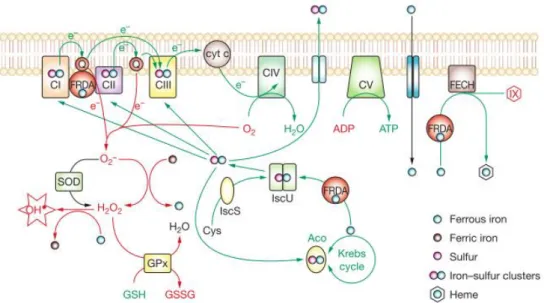
1.4 FRATAXIN PROTEIN ... 47
1.4.1 NORMAL FRATAXIN FUNCTION... 47
1.4.2 FRATAXIN DEFFICIENCY ... 51 1.5 THERAPEUTIC STRATEGIES ... 51 1.5.1 ANTIOXIDANTS ... 51 1.5.2 IRON QUELATORS ... 52 1.5.3 GENE THERAPY ... 52 1.5.4 EPIGENETIC THERAPIES ... 53
1.6 AIMS OF THIS THESIS ... 54
MATERIALS AND METHODS ... 56
2.1 CULTURE OF HUMAN CELLS ... 57
2.1.1 CELL CULTURE ... 57
2.1.2 ROUTINE SUBCULTURE ... 57
2.1.3 CRYOPRESERVATION OF CELLS ... 58
xiv
2.1.5 DRUG TREATMENT ... 58
2.2 BACTERIAL CELL CULTURE ... 58
2.2.1 PREPARATION OF ELECTROCOMPETENT DH10BE. COLI ... 59
2.2.2 ELECTROPORATION OF ELECTROCOMPETENT CELLS ... 59
2.2.3 TRANSFORMATION EFFICIENCY OF ELECTROCOMPETENT CELLS ... 59
2.3 DNA MANIPULATION ... 60
2.3.1 MINIPREP ... 60
2.3.2 MAXIPREP ... 60
2.3.3 GENOMIC DNA EXTRACTION FROM HUMAN CELLS ... 61
2.3.4 POLYMERASE CHAIN REACTION (PCR) ... 62
2.3.4.1 Standard PCR ... 62
2.3.4.2 Amplification of GAA repeats ... 62
2.3.4.3 qPCR ... 63
2.3.4.4 Small-pool PCR ... 63
2.3.4.5 Colony PCR ... 64
2.3.5 RESTRICTION DIGESTION OF PLASMID DNA ... 64
2.3.6 AGAROSE GEL ELECTROPHORESIS ... 64
2.3.7 PULSED-FIELD GEL ELECTROPHORESIS ... 65
2.3.8 PURIFICATION OF DNA FRAGMENTS FROM AGAROSE GELS ... 65
2.3.9 DNA LIGATION ... 65
2.3.10 DNA SEQUENCING ... 65
2.3.11 HOMOLOGOUS DNA RECOMBINATION IN E. COLI ... 66
2.3.12 CRE-LOXP RECOMBINATION FOR RETROFITTING ... 68
2.4 DELIVERY OF DNA INTO HUMAN CELLS ... 68
2.4.1 LIPOFECTION ... 68
2.4.2 GENERATION OF STABLE CLONES BY SITE-SPECIFIC INTEGRATION ... 69
2.4.2.1 X-gal staining ... 69 2.5 EPIGENETIC ANALYSIS ... 70 2.5.1 BISULPHITE SEQUENCING ... 70 2.5.2 CHROMATIN IMMUNOPRECIPITATION... 70 2.6 ANALYSIS OF FXN EXPRESSION ... 71 2.6.1 RT-PCR ... 71 2.6.2 QRT-PCR ... 71
2.7 ANALYSIS OF FRATAXIN PROTEIN LEVELS ... 72
2.7.1 LUCIFERASE ASSAY ... 72
2.8 FLUORESCENCE IN SITU HYBRIDISATION (FISH) ... 72
2.8.1 PROBES FOR DNAFISH ... 72
2.8.2 2DFISH ... 73
2.8.3 3DFISH ... 74
xv
2.8.5 RNAFISH ... 75
2.9 FLUORESCENCE RECOVERY AFTER PHOTOBLEACHING (FRAP) ... 77
2.10 IMAGING ACQUISITION AND ANALYSIS ... 77
EPIGENETIC SILENCING INDUCED BY A GAA REPEAT EXPANSION IN A HUMAN CELL MODEL OF FRDA ... 80
3.1 INTRODUCTION ... 82
3.1.1 FRDA CELL MODELS ... 82
3.1.2 FRDA MOUSE MODELS ... 82
3.1.3 EPIGENETIC CHANGES AT THE EXPANDED FXN ALLELE ... 83
3.1.4 AIMS OF THIS CHAPTER... 87
3.2 RESULTS ... 87
3.2.1 CHARACTERISATION OF THE FXN-LUC AND FXN-GAA-LUC CELL LINES ... 87
3.2.2 EPIGENETIC CHARACTERISATION OF FXN-LUC AND FXN-GAA-LUC CELL LINES ... 90
3.2.3 C5 RESTORES HISTONE ACETYLATION LEVELS AROUND THE GAA REPEAT EXPANSION ... 95
3.3 DISCUSSION ... 96
VISUALISATION OF THE EFFECT OF A GAA REPEAT EXPANSION ON FXN GENE NUCLEAR LOCALISATION AND EXPRESSION AT SINGLE-CELL RESOLUTION ... 100
4.1 INTRODUCTION ... 102
4.1.1 IN SITU HYBRIDISATION (ISH) ... 102
4.1.2 MRNA IMAGING IN LIVING CELLS ... 103
4.1.2.1 The MS2 system: improvements and applications ... 104
4.1.3 AIMS OF THIS CHAPTER... 107
4.2 RESULTS ... 108
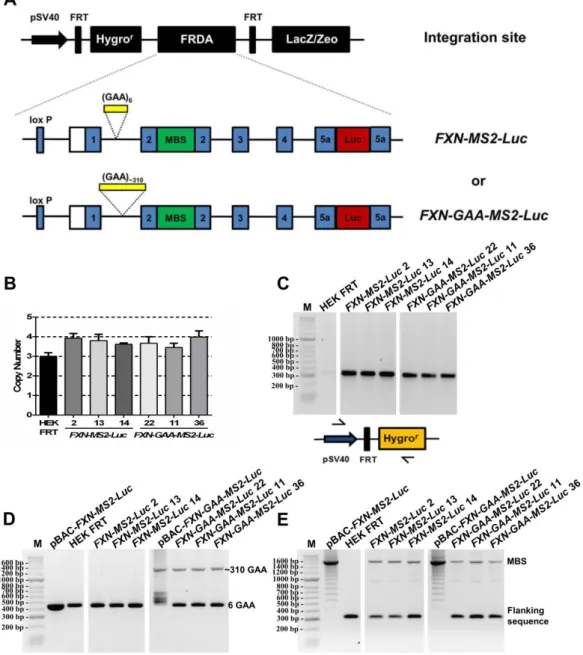
4.2.1 CONSTRUCTION OF PBAC-FXN-MS2-LUC AND PBAC-FXN-GAA-MS2-LUC VECTORS BY HOMOLOGOUS RECOMBINATION ... 108
4.2.2 TRANSIENT EXPRESSION OF THE FXN-MS2-LUC CONSTRUCTS ... 117
4.2.3 IMAGING OPTIMISATION ... 119
4.2.4 GENERATION OF STABLE FXN-MS2-LUC CELL LINES... 122
4.2.5 GAA REPEAT EXPANSION INCREASES FXN-GAA-MS2-LUC POSITIONING AT THE NL ... 124
4.2.6 GAA-EXPANDED REPEATS DECREASE THE NUMBER OF FXN MRNA MOLECULES AND SLOW TRANSCRIPTION KINETICS IN SINGLE FXN-GAA-MS2 CELLS ... 126
4.3 DISCUSSION ... 129
FXN GENE NUCLEAR LOCALISATION AND EXPRESSION IN HEALTHY, CARRIER AND FRDA PATIENT-DERIVED CELLS ... 131
5.1 INTRODUCTION ... 133
5.1.1 NUCLEAR ORGANISATION ... 133
5.1.1.1 Nuclear compartments involved in gene expression ... 134
5.1.1.2 Nuclear compartment involved in gene repression ... 136
5.1.2 GENE REGULATION AT THE NP ... 137
xvi
5.1.3 AIMS OF THIS CHAPTER... 140
5.2 RESULTS ... 140
5.2.1 EXPANDED FXN ALLELES LOCALISE PREFERENTIALLY CLOSE TO THE NP IN CARRIER CELLS ... 140
5.2.2 FXN GENE POSITIONING RELATIVE TO NUCLEAR SUBDOMAINS IN CARRIER CELLS ... 141
5.2.3 NL IS A KEY PLAYER IN FXN TRANSCRIPTIONAL IMPAIRMENT AND SILENCING ... 144
5.2.4 EXPANDED GAA REPEATS IMPAIR PREFERENTIALLY FXN TRANSCRIPTION INITIATION IN SINGLE FRDA CELLS ... 147
5.3 DISCUSSION ... 149
GENERAL DISCUSSION AND FUTURE DIRECTIONS ... 152
REFERENCES ... 159
ANNEXES ... 195
ANNEX 1 ... 196
xvii
List of figures
Figure 1.1 Main sites of organ dysfunction in FRDA. ... 33
Figure 1.2 Correlation between age at onset and the number of GAA repeats in the smaller FXN allele ... 34
Figure 1.3 The 5' end of the FXN gene ... 36
Figure 1.4 Distribution pattern of histone modifications involved in transcription regulation ... 42
Figure 1.5 Maturation of the frataxin protein ... 49
Figure 1.6 Frataxin function and oxidative stress in FRDA ... 50
Figure 3.1 Characterisation of the FXN-GAA-Luc and FXN-Luc cell lines ... 89
Figure 3.2 Workflow of Chromatin Immunoprecipitation (ChIP) ... 91
Figure 3.3 Histone modifications on the FXN locus ... 93
Figure 3.4 DNA methylation analysis of the FXN locus ... 94
Figure 3.5 Effect of C5 on a FRDA patient-derived cell line ... 96
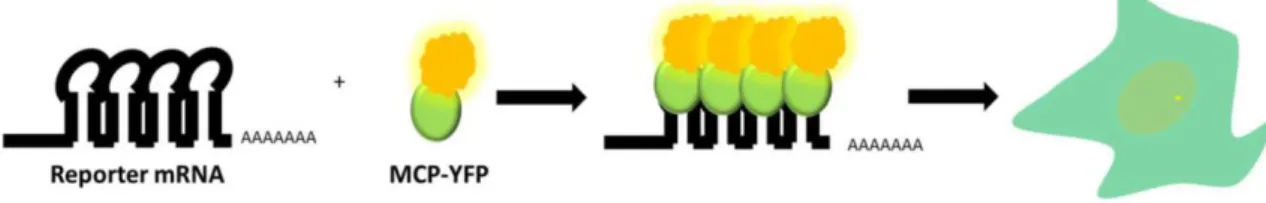
Figure 4.1 The MS2 system allows real-time imaging of RNA synthesis at a single-cell level ... 104
Figure 4.2 Traditional and novel uses of MS2-like systems to investigate mRNA biology ... 107
Figure 4.3 Experimental outline for vector construction of pBAC-FXN-GAA-MS2-Luc and pBAC-FXN-MS2-Luc using a selection/counter-selection homologous recombination approach in E.coli. ... 109
Figure 4.4 Insertion of rpsL-neo into exon 2 of the FXN-GAA-Luc gene by homologous recombination. ... 111
Figure 4.5 Construction of Exon2-MBS for homologous recombination ... 112
Figure 4.6 Replacement of the rpsL-neo cassette by 24 MBS in the second round of homologous recombination ... 113
Figure 4.7 Insertion of rpsL-neo into intron 1 of the FXN-GAA-MS2-Luc gene to generate FXN-MS2-Luc by homologous recombination ... 114
Figure 4.8 Replacement of the rpsL-neo cassette by 6 GAA repeats in the second round of homologous recombination ... 115
Figure 4.9 Cre-loxP mediated retrofitting of pBAC-FXN-MS2-Luc and pBAC-FXN-GAA-MS2-Luc vectors to pH-FRT-Hy ... 116
Figure 4.10 Confirmation of successful construction of the pBAC-FXN-MS2-Luc and pBAC-FXN-GAA-MS2-Luc vectors from pBAC-FXN-Luc and pBAC-FXN-GAA-Luc ... 117
Figure 4.11 Transient expression of the FXN-MS2-Luc vectors in HEK293 cells ... 118
Figure 4.12 Construction of pHGCX-Ex2-MS2 as a positive control for MS2 RNA FISH. ... 120
Figure 4.13 MS2 imaging optimisation in fixed HEK 293 cells ... 121
Figure 4.14 Live-cell imaging optimisation in HEK FRT cells ... 122
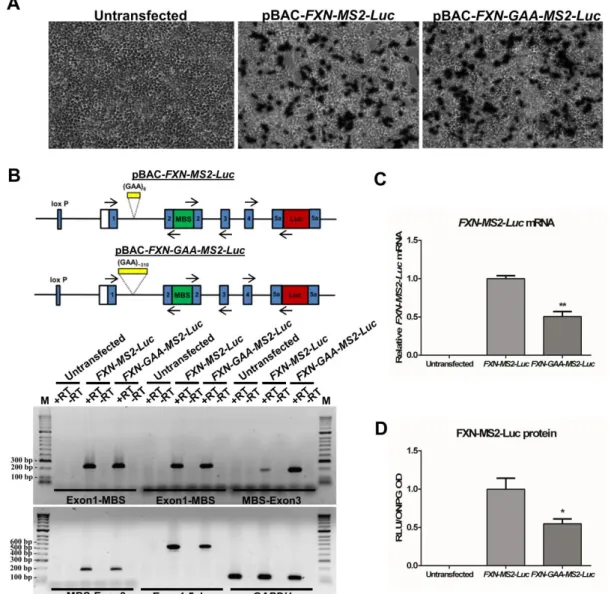
Figure 4.15 FXN-MS2-Luc and FXN-GAA-MS2-Luc cell models to study FXN localisation and expression 123 Figure 4.16 Expression of FXN-MS2-Luc mRNA and protein in FXN-MS2-Luc cell lines ... 124
Figure 4.17 The expanded GAA repeat FXN transgene associates with the NL more frequently in an FXN-GAA-MS2-Luc cell model... 125
Figure 4.18 The GAA repeat expansion reduces GAA-MS2-Luc transcriptional output in fixed FXN-GAA-Luc cells... 127
xviii
Figure 4.20 The GAA repeat expansion reduces FXN-GAA-MS2-Luc transcriptional output and impedes
transcription in living FXN-GAA-Luc cells ... 129
Figure 5.1 Nuclear organisation ... 134
Figure 5.2 Illustration of genome-NL interactions in mammalian cells ... 139
Figure 5.3 FXN mRNA expression in helathy, carrier and FRDA patient-derived cells ... 141
Figure 5.4 Expanded FXN alleles localise closer to the NP in carrier cells ... 142
Figure 5.5 Expanded FXN allele preferentially sits at the NL in carrier cells ... 143
Figure 5.6 GAA repeat expansion increases FXN localisation at the NL, reduces the number of active FXN alleles and downregulates transcription from active FXN in FRDA cells ... 145
Figure 5.7 Nascent FXN RNA FISH spot intensities in healthy and FRDA patient cells ... 146
Figure 5.8 GAA repeat expansion disrupts transcription initiation and elongation in FRDA cells. ... 148
Figure 5.9 Chromosome location of the FXN locus ... 150
Figure 6.1 Model of GAA repeat-mediated FXN repression and the repressive role of NL in FXN transcriptional impairment ... 155
xix
List of Tables
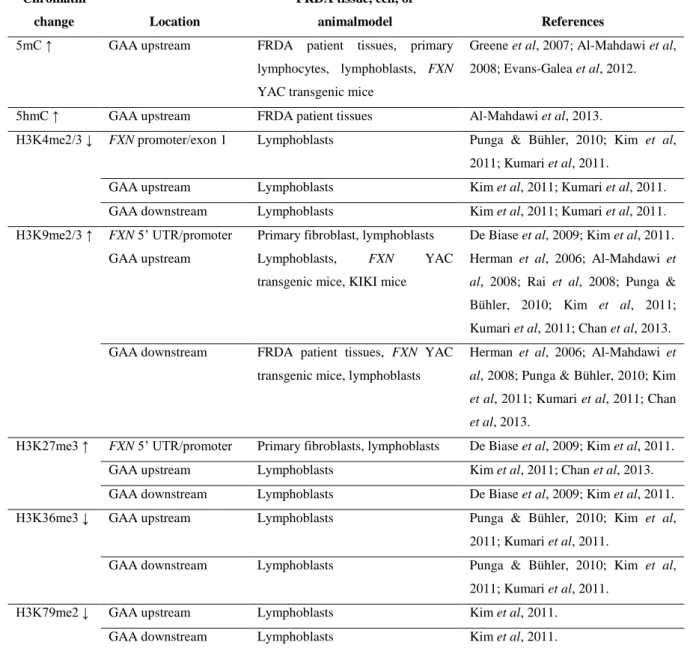
Table 1.1 List of unstable repeat expansion neurodegenerative diseases ... 28 Table 1.2 Prevalence of FRDA in Caucasians ... 32 Table 2.1 Custom Stellaris FISH probes TSS, UpGAA and DownGAA used in RNA FISH experiments. ... 76 Table 3.1 Epigenetic changes in FRDA patient cells, tissue, cellular and mouse models ... 84
xx
List of movies (available on CD)
Movie 1: Live-cell imaging of pBAC-FXN-MS2-Luc transcription in HEK FRT cells. Movie 2: Live-cell imaging of pBAC-FXN-GAA-MS2-Luc transcription in HEK FRT cells. Movie 3: Real time visualization of FXN-MS2-Luc transcription.
Movie 4: Real time visualization of FXN-GAA-MS2-Luc transcription. Movie 5: FRAP of a FXN-MS2-Luc transcription site.
xxi
List of abbreviations
5hmC 5-hydroximethylcytosine
5mC 5-methylcytosine
AAV Adeno-associated virus
ActD Actinomycin D
AID Activation-induced cytidine deaminase
ANOVA Analysis of variance
ATP Adenosine 5′-triphosphate
ATXN8 Ataxin 8 gene
ATXN8OS Ataxin 8 opposite strand gene
BAC Bacterial artificial chromosome
BAF Barrier to autointegration factor
BCA Bicinchoninic acid solution
Bp Base pairs
BSA Bovine serum albumin
BTF BCL-2 associated transcription factor
CCD Charge-coupled device
CFU Colony forming units
ChIP Chromatin immunoprecipitation
CMV Human cytomegalovirus
CT Chromosome territory
CTCF CCCTC-binding factor
DAM DNA adenine methyltransferase
DAPI 4′,6-diamidino-2- phenylindole
dATP Deoxyadenosine triphosphate
dCTP Deoxycytidine triphosphate
dGTP Deoxyguanosine triphosphate
DM Myotonic dystrophy
DMEM Dulbecco’s modified Eagle’s medium
DMSO Dimethyl sulphoxide
DNMT DNA methyltransferase
xxii DRG Dorsal root ganglia
DTT Dithiothreitol
EDTA Ethylenediaminetetraacetic acid
EGR3 Early growth response factor 3
EGTA Ethylene glycol tetraacetic acid
EMCCD Electron multiplying charge-coupled device
FAST1 Frataxin antisense transcript 1
FBS Fetal Bovine Serum
FISH Fluorescence in situ hybridisation
FPN Ferroportin
FRAP Fluorescence recovery after photobleaching
FRAXA Fragile X syndrome
FRAXE Fragile X mental retardation associated with the FRAXE site
FRDA Friedreich’s ataxia
FRT Flp recombinase target
FRTs Ferritins
FTD/ALS Frontotemporal dementia and amyotrophic lateral sclerosis
FXTAS Fragile X tremor and ataxia syndrome
FXN Frataxin gene
GCL Germ-cell-less
GFAP Glial fibrillary acidic protein gene
HAT Histone acetyltransferase
HD Huntington’s disease
HDAC Histone deacetylase
HDM Histone demethylase
HEK Human embryonic kidney 293 cells
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HMT Histone methyltransferase
HP1 Heterochromatin protein 1
HSV-1 Herpes simplex virus type 1
i-FXN Intermediate frataxin
INM Inner nuclear membrane
xxiii
iPSC Induced pluripotent stem cell
ISCs Iron-sulphur clusters
ISH In situ hybridisation
K Lysine
KDa Kilodalton
KIKI Knock-in mice
KIKO Knock-in – knockout mice
LAD Lamina-associated domains
LB Luria broth
LBR Lamin B receptor
LEM Lap2β, Emerin and MAN1
LINEs Long interspersed nuclear elements
LN Long normal
MBS MS2-binding site
MCP MS2 coat protein
MCS Multiple cloning site
MECP2 Methyl-CpG-binding protein 2
m-FXN Mature frataxin
MIR Mammalian-wide interspersed repeats
MMR Mismatch repair proteins
MPP Mitochondrial processing peptidase
MSH MutS homologs
NA Numerical aperture
NCoR Nuclear corepressor complex
NES Nuclear export signal
NL Nuclear lamina
NLS Nuclear localisation signal
NOR Nucleolus organiser region
NP Nuclear periphery
NTC No template control
ONM Outer nuclear membrane
ORF Open reading frame
xxiv PBS Phosphate-buffered saline
PcG Polycomb group
PEV Position effect variegation
PFA Paraformaldehyde
PIP5K1B phosphatidylinositol 4-phosphate 5-kinase β type I gene
PML Promyelocytic leukemia
PCP Phage PP7 coat protein
PCR Polymerase Chain Reaction
PRC Polycomb repressive complexes
R Purine
RAN Repeat-associated non-ATG
RISC RNA-induced silencing complex
RNAi RNA interference
RNAP RNA polymerase
ROI Region of interest
SAP Shrimp alkaline phosphatase
SBMA Spinal and bulbar muscular atrophy
SCA Spinocerebellar ataxia
SDS Sodium dodecyl sulfate
SEM Standard error of the mean
SINEs Short interspersed nuclear elements
SN Short normal
snRNPs small nuclear ribonucleoprotein particles
SRF Serum responsive factor
SSC Saline sodium citrate
TALE Transcription activator-like effector protein
TBE Tris-borate EDTA
TDG Thymine DNA glycosylase
TE Tris-EDTA
TET Ten-eleven translocation methylcytosine dioxygenase
TFAP2 Transcription factor AP2
TFR1 Transferrin receptor 1
xxv UTR Untranslated region
WT Wild-type
Y ·Pyrimidine
YAC Yeast artificial chromosome
Yfh1 Yeast frataxin homolog
CHAPTER 1
27 1.1 Unstable expanded repeat diseases
Abnormally expanded DNA repeats are associated with several neurodegenerative diseases (Table 1.1). The majority of these diseases are caused by expanded trinucleotide repeats, but tetra-, penta-, and hexanucleotide repeat expansions have also been identified. Expanded repeats can be found either in coding or non-coding regions of a given locus. In the first group, exonic repeats code for amino acid homopolimers, as occurs in diseases mediated by polyglutamine and polyalanine runs in proteins, for example Huntington’s disease (HD) and oculopharyngeal muscular dystrophy, respectively. In the second group, repeats can be located in: (i) the 5ʹ untranslated regions (5ʹ UTRs), as in fragile X syndrome (FRAXA), fragile X mental retardation associated with the FRAXE site (FRAXE), fragile X tremor and ataxia syndrome (FXTAS), and spinocerebellar ataxia (SCA) 12; (ii) the 3ʹUTRs, as in myotonic dystrophy (DM) 1, SCA8 and HD-like 2; (iii) introns, as in DM2, Friedreich’s ataxia (FRDA), SCA10, SCA31; SCA36 and C9ORF72-related frontotemporal dementia and amyotrophic lateral sclerosis (FTD/ALS) (reviewed in Gatchel & Zoghbi, 2005; Mirkin, 2007; La Spada & Taylor, 2010; Polak et al, 2013).
Although each particular disease is caused by a mutation in a different locus, several features are shared. Alleles are present in three states: normal, premutated and fully mutated. While healthy individuals carry less than 30 repeats, patients harbor longer and unstable tracts of repeats on mutant alleles, with a strong tendency to expand rather than contract. Repeats show intergenerational and somatic instability. As longer repeat tracts are transmitted through generations, disease symptoms become more severe and appear at an earlier age, a phenomenon known as anticipation (reviewed in Dion & Wilson, 2009; Castel et al, 2010). Repeat instability seems to be triggered by formation of unusual DNA structures, such as hairpins, slipped-strand structures, triplexes and quadruplexes, during DNA replication, repair, recombination and transcription, where there is a transient separation of complementary DNA strands, or exposure of a single DNA strand. The DNA repair machinery is then thought to bind to these aberrant structures, stabilizing instead of repairing them, which results in repeat expansions or contractions (Mirkin, 2007; Polak et
al, 2013). Changes in the epigenetic landscape of the mutated locus, such as aberrant DNA
methylation, histone modifications, and chromatin remodelling also play a role in the molecular pathogenesis of several unstable expanded repeat diseases (reviewed in Evans-Galea et al, 2013).
28
Table 1.1 List of unstable repeat expansion neurodegenerative diseases (Gatchel & Zoghbi, 2005; La
Spada & Taylor, 2010; Walsh et al, 2014).
Repeat length Disease Main clinical features Mutation Location Normal Expanded
Loss of protein function
FRDA Ataxia, sensory loss, weakness, diabetes mellitus, cardiomyopathy
(GAA)n FXN (Intron 1) 6–32 70–1700
FRAXA MR, facial dysmorphism, autism
(CGG)n FMR1 (5′ UTR) 6–52 >200
FRAXE MR, hyperactivity (GCC)n FMR2 (5′ UTR) 4–39 200–900
Toxic gain of RNA function
SCA10 Ataxia and seizures (ATTCT)n ATXN10 (Intron 9) 10–29 400–4500
SCA31 Ataxia, dysarthria, nystagmus
(TGGAA)n TK2/BEAN (Intron) 0 ≥100
SCA36 Ataxia, eye movement abnormalities, tongue fasciculations, upper and
lower motor neuron disease (GGCCTG)n NOP56 (Intron 1) 3–14 650–2500 DM1 Muscle weakness, myotonia, cardiac conduction deficit, MR (CTG)n DMPK (3′ UTR) 5–37 50–1000 DM2 Muscle weakness, myotonia, cardiac conduction deficit (CCTG)n ZNF9 (Intron 1) 10–26 75–11,000
FXTAS Ataxia, intention tremor, parkinsonism, cognitive
deficit
(CGG)n FMR1 (5′ UTR) 6–52 55–200
Toxic gain of protein function
DRPLA Ataxia, epilepsy, choreoathetosis, dementia
(CAG)n ATN1 (Exon 5) 7–34 49–88
HD Severe movement abnormalities, chorea, dystonia, cognitive decline, psychiatric features (CAG)n HTT (Exon 1) 6–28 36–180
SBMA Motor weakness, swallowing difficulty,
gynecomastia, hypogonadism
(CAG)n AR (Exon 1) 9–36 38–62
SCA1 Ataxia, dysarthria, spasticity, cognitive
impairment
(CAG)n ATXN1 (Exon 8) 6–39 41–83
SCA2 Ataxia, slow eye movement, hyporeflexia,
polyneuropathy, motor neuropathy, infantile
variant
(CAG)n ATXN2 (Exon 1) 15–24 32–200
SCA3 (MJD)
Ataxia, dystonia, lower motor neuron disease
29
SCA6 Ataxia, dysarthria, nystagmus, tremor
(CAG)n CACNA1A (Exon 47) 4–20 20–33
SCA7 Ataxia, retinal degeneration, cardiac involvement in infantile
variant
(CAG)n ATXN7 (Exon 3) 4–35 37–306
SCA17 Ataxia, behavioural changes or psychosis, intellectual deterioration,
seizures
(CAG)n TBP (Exon 3) 25–44 47–63
Toxic gain of function at both the protein and RNA levels
SCA8 Ataxia, dysarthria, nystagmus
(CTG)n*(CAG)n ATXN8OS (3′ UTR)
and ATXN8 (Exon 1)
15–50 71–1300
Haploinsufficiency and toxic gain of function at both the protein and RNA levels
FTD/ALS Cognitive impairment, psychosis/Upper and lower
motor neuron disease, paralysis
(GGGGCC)n C9ORF72
(5’UTR/Intron 1)
2–19 30–2000
Unknown pathogenic mechanism
SCA12 Ataxia, tremor, dementia (CAG)n PPP2R2B (5′ UTR) 7–28 55–78
HDL2 Similar to HD (CTG)n*(CAG)n JPH3 (exon 2A) 7–28 66–78
Abbreviations: AR, androgen receptor; ATXN, ataxin; BEAN, brain-expressed, associated with Nedd4;
CACNA1A, calcium channel, voltage-dependent, P/Q type, α1A subunit; DM1, myotonic dystrophy type 1;
DM2, myotonic dystrophy type 2; DMPK, dystrophia myotonica protein kinase; DRPLA, dentatorubral-pallidoluysian atrophy; DM, dystrophia myotonica; FMR1, fragile X mental retardation 1; FMR2, fragile X mental retardation 2; FRAXA, fragile X syndrome; FRAXE, fragile X mental retardation associated with the
FRAXE site; FRDA, Friedreich ataxia; FTD/ALS, frontotemporal dementia and amyotrophic lateral sclerosis; FXN, frataxin; FXTAS, fragile X tremor/ataxia syndrome; JPH3, junctophilin 3; HD, Huntington’s disease;
HDL2, Huntington’s disease-like 2; HTT, huntingtin; MJD, Machado–Joseph disease; MR, mental retardation; NOP56, nucleolar protein 56; PPP2R2B, protein phosphatase 2 (formerly 2A) regulatory subunit B; SBMA, spinal and bulbar muscular atrophy; SCA, spinocerebellar ataxia; TBP, TATA box binding protein; TK2, thymidine kinase 2; ZNF9, zinc-finger protein 9.
There are three key pathological mechanisms by which expansions result in disease: (i) loss of function typically by disrupting transcription of the mutated gene (FRDA, FRAXA, FRAXE); (ii) RNA toxic gain of function through aberrant interactions of the respective RNAs with proteins controlling RNA metabolism (SCA10, SCA31, SCA36, DM1, DM2, FXTAS); and (iii) protein toxic gain of function via abnormal protein interactions and accumulation of aggregates (polyglutamine diseases, for example HD) (reviewed in Nelson et al, 2013). Interestingly, some diseases have more than one repeat-mediated mechanism at play, for example SCA8 and C9ORF72-related FTD/ALS. In SCA8, two overlapping genes expressed in opposite directions are affected by the expansion, with a (CAG)n expansion in the ataxin 8 gene (ATXN8) and a (CTG)n expansion
in the non-coding 3’ UTR of the ataxin 8 opposite strand gene (ATXN8OS). As a result,
30
CUG expansion mRNAs which accumulate as RNA foci and sequester RNA-binding proteins (Moseley et al, 2006; Daughters et al, 2009). In C9ORF72-related FTD/ALS, the (GGGGCC)n hexanucleotide repeat expansion leads to disease by: (i) formation of DNA
and RNA G-quadruplexes and RNA/DNA hybrids (R-loops) which lead to abortive transcription; (ii) formation of RNA foci that bind to ribonucleoproteins, preventing RNA-processing factors from functioning normally in the nucleus; and (iii) repeat-associated non-ATG (RAN) translation, which results in the production of dipeptide repeat proteins that also form inclusions in affected cells (DeJesus-Hernandez et al, 2011; Renton et al, 2011; Mori et al, 2013; Haeusler et al, 2014).
In the following sections, I review the major findings in FRDA and also highlight the similarities with other repeat expansion diseases with special emphasis on non-coding repeats at the DNA and RNA level.
1.2 Friedreich’s ataxia
Friedreich’s ataxia (FRDA; OMIM 229300) is a progressive neurodegenerative disease and the most common form of recessive ataxia (Campuzano et al, 1996). The disease is named after Nikolaus Friedreich (1825–1882), a German pathologist and neurologist, who first described its clinical features (Friedreich, 1863a, 1863b, 1863c) and hereditary nature (Friedreich, 1876, 1877). An abnormal GAA trinucleotide–repeat expansion in intron 1 of the frataxin gene (FXN) causes FRDA. Approximately 96% of FRDA patients are homozygous for a GAA repeat expansion and the remaining patients (4%) are compound heterozygous with one expanded allele and a classical mutation (nonsense, missense, deletions, insertions) in the second allele (Campuzano et al, 1996; Bidichandani et al, 1997; Cossée et al, 1999). FXN alleles in healthy individuals contain <36 GAA repeats, whereas in FRDA patients GAA expansions ranging from ~70 to 1700 GAA repeats lead to FXN mRNA deficiency and subsequent reduced levels of frataxin, a nuclear-encoded mitochondrial protein essential for life (Campuzano et al, 1996; Cossée et
al, 2000).
1.2.1 Etiology
Normal FXN alleles are divided in 2 groups: short normal (SN, <10 GAA repeats) and long normal (LN, from 12 to 60 GAA repeats). LN FXN alleles account for ~17% of normal alleles. Haplotype analysis of five markers (FAD1-ITR4-F5225-ITR3-CS2) close to the GAA repeats revealed that a single founder event that occurred on the major AT2CC
31
haplotype is at the origin of the LN alleles and the expanded alleles. The two second most frequent haplotypes AT3CC and AT2CT are most likely derived from the major haplotype by marker mutation rather than by independent events on different haplotypes (Cossée et
al, 1997). Expanded FXN alleles are thought to have arisen through a two-step process. A
singular event was responsible for the transition of SN to LN GAA repeats as these alleles only share the same haplotype rarely. This is thought to have occurred through DNA polymerase “stuttering” during DNA replication, originating LN alleles of (GAA)18 by
duplication of SN alleles of (GAA)8. After a number of small increases in size because of
slippage events, larger LN alleles served as a pool for further repeat expansions once the instability threshold (~34 GAA repeats) was reached (Montermini et al, 1997a; Labuda et
al, 2000). Therefore, repeat numbers between 35 and 66 are referred as premutations. In
line with this, all LN and expanded alleles share a common haplotype (Cossée et al, 1997). A single initial event is also thought to have occurred in DM1, HD, DRPLA and MJD where haplotypes associated preferentially or uniquely with expanded alleles are also preferentially associated to LN alleles (Imbert et al, 1993; Rubinsztein et al, 1995; Takiyama et al, 1995; Yamagata et al, 1996; Yanagisawa et al, 1996). Furthermore, as observed in FRDA, the prevalence of DM1, HD and DRPLA in a given population is correlated with the frequency of the LN alleles (Novelli et al, 1994).
The FRDA founding mutation is dated between 9,000 and 24,000 years ago and went through a population bottleneck (Colombo & Carobene, 2000) which coincides with the glacial period in Europe. This may provide an explanation for the highest prevalence of FRDA in the surviving northern Spain ice age refuge (Polo et al, 1991). Further support comes from the observed distribution of FRDA in the remaining European regions where the prevalence decreases from west to east and correlates with the frequency gradient of the chromosomal R1b marker, a genetic marker for the population that originated from the Cantabrian refuge (Tambets et al, 2004). It has been hypothesized that the FRDA distribution in Europe is derived from Palaeolithic migrations out of the Franco-Cantabrian ice age refuge (Vankan, 2013).
1.2.2 Epidemiology
FRDA is detected in individuals from Europe, the Middle East, North Africa and India (Labuda et al, 2000). The prevalence of the disease is closely correlated with the frequency of LN FXN alleles in the population, and is highest in Western Europe, with more than 1 case per 30,000 individuals (1/30,000) reported in north-west Spain and
32
Ireland (Schulz et al, 2009; Vankan, 2013). LN alleles are virtually absent in individuals from East Asia and in American Indians (Labuda et al, 2000).
The frequency of heterozygous mutation carriers in Europe shows a decreasing gradient from south to north-east, ranging from 1:78 in France to 1:89 in Germany and 1:500 in Finland (Cossée et al, 1997; Epplen et al, 1997; Labuda et al, 2000; Juvonen et al, 2002; Schulz et al, 2009; Vankan, 2013). The prevalence of FRDA in Caucasians is shown in Table 1.2.
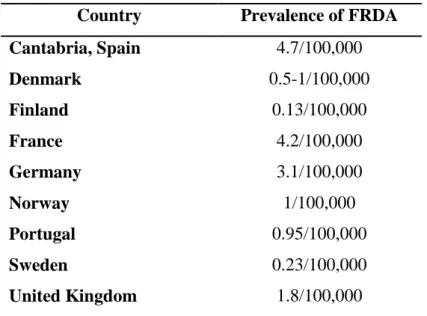
Table 1.2 Prevalence of FRDA in Caucasians (source: Schulz et al, 2009; Vankan, 2013).
Country Prevalence of FRDA
Cantabria, Spain 4.7/100,000 Denmark 0.5-1/100,000 Finland 0.13/100,000 France 4.2/100,000 Germany 3.1/100,000 Norway 1/100,000 Portugal 0.95/100,000 Sweden 0.23/100,000 United Kingdom 1.8/100,000
1.2.3 Clinical features of FRDA
The healthy FXN gene is expressed in all cells but at variable levels. In adult humans,
FXN mRNA is most abundant in the heart and spinal cord, mild in the cerebellum,
pancreas, liver, brown fat and skeletal muscle, and low in other tissues, such as the cortex (Campuzano et al, 1996; Koutnikova et al, 1997). In FRDA, residual levels of frataxin vary according to the length of the expansion and the cell type. Affected cells, such as sensory neurons and cardiomyocytes, are known to have a high mitochondrial content and high metabolic rate, making them more sensitive to frataxin deficiency.
The first neurologic symptoms usually appear around puberty, but early-onset and late-onset variants also exist. Patients experience progressive gait and limb ataxia, dysarthria and lower limb areflexia, resulting from the loss of large sensory neurons in the dorsal root ganglia (DRG), atrophy of the large sensory fibres in peripheral nerves, degeneration of the posterior columns, spinocerebellar and corticospinal motor tracts in the
33
spinal cord and dentate nucleus in the cerebellum (Fig. 1.1). Hearing disability and loss of vision are also common. Non-neurological symptoms include hypertrophic cardiomyopathy, diabetes mellitus, kyphoscoliosis and pes cavus. Life expectancy is reduced to an average of 40-50 years (Filla et al, 1990; Dürr et al, 1996; Pandolfo, 2009; Schulz et al, 2009).
In most cases, compound heterozygous patients are clinically indistinguishable from patients that are homozygous for the GAA repeat expansions, but a few missense mutations (e.g., G130V, D122Y, R165P, L106S) may cause an atypical or milder clinical phenotype due to an adverse effect on protein expression, function and/or stability (Cossée
et al, 1999; Gellera et al, 2007).
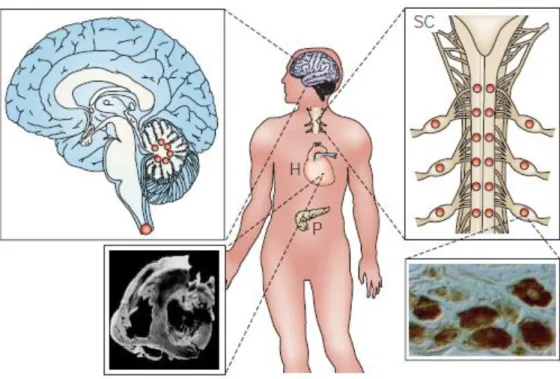
Figure 1.1 Main sites of organ dysfunction in FRDA. Large dots indicate more severe neuronal loss.
Pathological features are shown for the brain, spinal cord (SC), heart (H) and pancreas (P). Loss of large primary neurons in dorsal root ganglia is a prominent and early hallmark. As the disease progresses, the posterior columns of the spinal cord degenerate. This associated with atrophy of the spinocerebellar tracts, the corticospinal motor tracts of the spinal cord and the large sensory fibers in peripheral nerves. Non-neurological symptoms include hypertrophic cardiomyopathy and diabetes (adapted from Taroni & DiDonato, 2004).
In homozygous patients, the GAA repeat length of the shorter of the 2 FXN expanded alleles correlates with residual frataxin protein levels, earlier onset (Fig. 1.2) and confinement to a wheelchair, and increased phenotypic severity of the disease (Dürr et al, 1996; Filla et al, 1996; Montermini et al, 1997b). The GAA expansion length only
34
accounts for ~50% of the variability in age of onset (Filla et al, 1996). Phenotypic variability among FRDA patients and within affected families can be also due to intergenerational instability of the GAA expansion, somatic mosaicism, interruptions in the repeat sequence, changes in expansion size over life and other modifying genes or environmental factors (Dürr et al, 1996; Montermini et al, 1997b; Pandolfo, 2009).
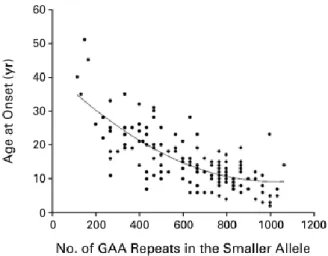
Figure 1.2 Correlation between age at onset and the number of GAA repeats in the smaller FXN allele.
Larger GAA expansions in the smaller FXN allele correlates with earlier age at onset (Dürr et al, 1996).
For most of the unstable expanded repeat diseases, the repeat sequence shows intergenerational and age-dependent somatic instability. In FRDA, DM1, FRAXA and SCA8, expansions and contractions are equally likely after maternal transmission, whereas contractions are favoured after paternal transmission (Malter et al, 1997; Monrós et al, 1997; Pianese et al, 1997). In FRDA, expanded alleles are highly unstable in somatic cells
in vivo. While expanded FXN allele with <250 GAA repeats tend to expand, FXN alleles
with >500 GAA repeats show contractions in peripheral leukocytes from FRDA patients. Due to somatic instability, carriers carrying LN FXN alleles (44–66 triplets) can develop a FRDA phenotype (Sharma et al, 2002, 2004). Somatic instability in FRDA is mostly postnatal, progresses throughout life and is tissue-specific (De Biase et al, 2007a, 2007b). Expanded GAA repeats undergo progressive expansion in tissues primarily affected in FRDA: the DRGs, cerebellum and heart of FRDA patients (De Biase et al, 2007a), and in the DRGs, cerebellum and brain of mouse models (Al-Mahdawi et al, 2004; Clark et al, 2007; Anjomani Virmouni et al, 2015). Therefore, the progressive accumulation of large expansions coupled with a lower frequency of large contractions in specific cell types, such as DRGs, suggests that somatic instability may contribute to the selective vulnerability of
35
these cells in FRDA. Although the molecular mechanism of somatic instability in FRDA is still poorly understood, a few studies have suggested that mismatch repair (MMR) proteins may be involved (section 1.3.2.1.1).
In FRAXA and DM1, large expansions arise in non-dividing oocytes during arrest in meiotic prophase I, and in terminally differentiated somatic cells in females; in males, the full mutation alleles tend to contract in the dividing male spermatogonia, during weeks 13– 17 of fetal development, and small gains and losses in the pre-mutation range can be observed in both spermatogonia and in somatic cells (McMurray, 2010).
1.3 The FXN locus
1.3.1 FXN in healthy individuals
The human FXN locus is located on the positive strand of chromosome 9q21.11 and contains seven exons (1–4, 5a, 5b, and non-coding 6). Alternative splicing results in multiple transcript variants with exons 1–5a generating the 1.3 kb major transcript that encodes the 210-amino acid frataxin protein (Campuzano et al, 1996). Two major transcription start sites (TSS) were identified: TSS1 is located 221 bp upstream of the ATG translation start site (Campuzano et al, 1996), whereas TSS2 is located 62 bp upstream of the ATG and is considered the major TSS in Epstein Barr virus-transformed lymphoblastoid cell lines. (Kumari et al, 2011).
The FXN promoter (Fig. 1.3) is enriched in repetitive DNA elements. These include L2 (LINE) and Alu (SINE) elements as well as MIRb and mariner DNA transposon downstream of TSS1 and in intron 1 (Greene et al, 2005). The presence of these different retroelements suggest that the FXN promoter sequence has been remodelled repeatedly during the evolution of mammals and fairly recently in the primate lineage (Greene et al, 2005). Although their contribution to FXN regulation is unclear, deleting these elements significantly impairs FXN expression (Greene et al, 2005). Alu and MIR elements are transcribed from internal RNA polymerase (RNAP) III promoters and may exert a positive
cis-acting enhancer effect on RNAPII-transcribed genes (Oliviero & Monaci, 1988;
Tomilin, 1999). Moreover, Alu elements are also thought to affect gene expression by altering nucleosome positioning (Englander & Howard, 1995) and providing direct binding sites for hormone receptors (Norris et al, 1995) and transcription factors (Deininger, 2011). The region between 121-221 bp upstream of the start of the frataxin open reading frame (ORF) shows homology between primates and rodents and contains important regulatory
36
elements for FXN expression (Greene et al, 2005). Interestingly, the FXN promoter does not present typical motifs of most mammalian promoters, such as a TATA box, and is not controlled by either the initiator or the downstream promoter-like elements found 24 bp downstream of TSS1 (Greene et al, 2005; Kumari et al, 2011).
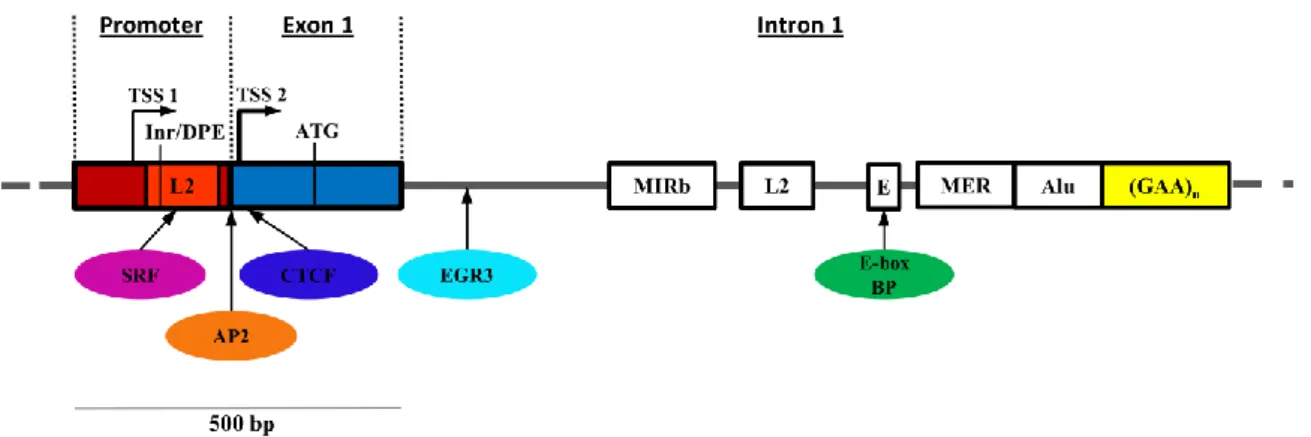
Figure 1.3 The 5' end of the FXN gene. Schematic representation of the the 5’end FXN gene showing its
regulatory elements and protein binding sites. The repetitive DNA elements identified at the FXN locus include L2 (LINE) and Alu (SINE) elements as well as MIRb and mariner DNA transposon (MER). FXN is transcribed from TSS1, which is located 221 bp upstream of the ATG, and TSS2, located 62 bp upstream of the ATG. TSS2 is the major TSS in lymphoblastoid cell lines. The region between TSS1 and exon 1 is thought to be a TATA-less downstream promoter, which contains the initiator/downstream promoter-like elements (Inr/DPE). Binding sequences were identified for transcription factors SRF, TFAP2 and EGR3, and for the insulator protein CTCF. An E-box is present in the region upstream of the GAA repeats and can potentially be a target for MyoD or c-myc (adapted from Kumari & Usdin, 2012).
A few studies have identified transcription factors that influence FXN expression (reviewed in Yandim et al, 2013). Li and colleagues showed that SRF (serum responsive factor) and TFAP2 (transcription factor AP2) directly bind to sequences within and immediately downstream of the FXN promoter region conserved between rodents and primates due to the presence of the L2 retrotransposon-like sequence (Greene et al, 2005; Li et al, 2010). TFAP2 upregulates FXN mRNA expression in several cell lines (Li et al, 2010) and is important in the balance between cell proliferation and differentiation during embryogenesis (Eckert et al, 2005). SRF increases FXN expression only in specific cell-lines and activate genes involved in synaptic activity and plasticity (Knöll & Nordheim, 2009). Moreover, the chromatin insulator CCCTC-binding factor (CTCF) binds between 154–173 bp downstream of TSS1 (De Biase et al, 2009). CTCF is implicated in diverse roles in gene regulation, including context-dependent promoter activation/repression,
37
enhancer blocking and/or barrier insulation, hormone-responsive silencing, genomic imprinting, and long-range chromatin interactions (reviewed in Phillips & Corces, 2009).
The intronic region downstream of exon 1 is also important for FXN regulation. Deletion of the binding site for the early growth response factor EGR3 (Li et al, 2010) or of an E-box element (Greene et al, 2007) induced a decrease in FXN expression using reporter assays. The E-box element can potentially be bound by proteins of the large family of basic Helix-Loop-Helix that includes the muscle-specific factor MyoD and c-myc (Greene et al, 2007).
A polymorphic (GAA)n repeat sequence located approximately 1.3 kb downstream of
TSS2 is embedded in the centre of an Alu element in intron 1 and is preceded by a poly(A) tract characteristic of Alu elements. FXN alleles in healthy individuals contain <36 GAA repeats, whereas in FRDA patients GAA expansions ranging from70 to 1700 GAA repeats lead to FXN mRNA deficiency (Campuzano et al, 1996; Cossée et al, 1997; Clark et al, 2004; Monticelli et al, 2004).
1.3.2 Molecular basis of FRDA
1.3.2.1 GAA expansion-mediated transcriptional dysregulation
In FRDA, expanded GAA repeats in intron 1 of the FXN gene reduce steady-state
FXN mRNA levels to 4% to 29% of normal (Campuzano et al, 1997) through a still poorly
understood mechanism. This severe decrease in FXN mature mRNA content could potentially be due to impairment in transcription, pre-mRNA processing or decay.
RNA splicing differences between normal and expanded FXN alleles have only been shown when using a FXN minigene construct containing a human cytomegalovirus (CMV) promoter, FXN exon 1, part of intron 1 and all of exon 2, transfected into mammalian cells (Baralle et al, 2008); or when inserting expanded GAA repeats into an intron of the URA3 gene in yeasts (Shishkin et al, 2009). However, these results failed to replicate when studying the endogenous FXN transcript in FRDA patient cells (Bidichandani et al, 1998; Punga & Bühler, 2010). Moreover, there is no evidence showing differences in the decay rate of mature FXN transcripts from normal and expanded FXN alleles (Punga & Bühler, 2010).
Therefore, it seems plausible to assume that the FXN mRNA decrease seen in FRDA results from FXN transcriptional impairment. However, how the mutation interferes with the transcription machinery at the FXN gene remains unclear. Two non-exclusive
38
hypotheses have been proposed (reviewed in Kumari & Usdin, 2012; Yandim et al, 2013). Firstly, expanded GAA repeats may form non-B DNA structures, such as triplexes or sticky DNA (Bidichandani et al, 1998; Sakamoto et al, 1999), and RNA/DNA hybrids (R-loops; Grabczyk et al, 2007; Groh et al, 2014) which impair RNAPII elongation. Secondly, GAA repeat expansions can induce the formation of heterochromatin (Saveliev et al, 2003; Herman et al, 2006), leading to increased DNA methylation at specific CpG sites (Greene
et al, 2007; Al-Mahdawi et al, 2008; Evans-Galea et al, 2012), reduced histone acetylation
(H3/H4ac) and increased levels of methylated histones H3K9me2 and H3K9me3 (Herman
et al, 2006; Al-Mahdawi et al, 2008). It has been suggested that these epigenetic changes
surrounding the GAA expansion impair RNAPII elongation (Punga & Bühler, 2010), but also spread upstream towards the FXN promoter, inducing a non-permissive chromatin configuration for transcription initiation, altering nucleosome positioning and preventing insulator protein CTCF binding (De Biase et al, 2009; Kumari et al, 2011; Chutake et al, 2014; Groh et al, 2014).
1.3.2.1.1 Abnormal DNA structures
Repetitive DNA sequences are prone to adopting unusual DNA secondary structures due to sequence symmetry, base composition, DNA supercoiling and cellular ambient conditions (Mirkin, 2007). These structures contribute to the instability of disease-associated repeats by impacting on DNA metabolism, for example stalling replication, altering repair or causing unusual recombination, inducing the formation of hairpins, triplexes and quadruplexes. During transcription, repeats can form RNA/DNA hybrids (Castel et al, 2010).
Of all the repeat expansions associated with unstable expanded diseases, only GAA repeats do not form hairpins. Single-stranded CNG repeats form hairpin structures that consist of both Watson–Crick base pairs and mismatched base pairs, with a decreasing stability in the order CGG > CTG > CAG = CCG. In addition to hairpins, single-stranded CGG, CCG and GGGGCC repeats can fold into G-quadruplexes (Fry & Loeb, 1994; Marquis Gacy et al, 1995; Haeusler et al, 2014).
Expanded GAA repeats form non-B DNA conformations, such as triplexes, that can affect FXN transcription in a length and orientation-dependent manner (Bidichandani et al, 1998; Ohshima et al, 1998). Triplexes are three-stranded DNA structures formed at polypurine·polypyrimidine (R·Y) tracts, where the third strand forms Hoogsteen base pairs between the R or Y bases with the purines already paired with pyrimidines in the duplex
39
DNA. R·R·Y triplexes, unlike Y·R·Y, form more rapidly and at neutral pH. In FRDA, GAA repeat expansions form a R·R·Y triplex containing two GAA repeat strands along with a single strand of TTC repeats (Mariappan et al, 1999; Grabczyk & Usdin, 2000; Potaman et al, 2004). These structures are stabilised by negative supercoiling and can self-associate, forming complexes known as sticky DNA which can only be resolved at high temperatures (80°C) and with the addition of EDTA to remove divalent metal ions (Sakamoto et al, 1999; Wells, 2008). Sticky DNA can impede virtually all biological processes in vitro, including transcription, replication, repair and recombination (Wells, 2008).
Another model suggests that expanded GAA repeats can form R-loops, a shared feature with CGG, CCG, CAG, CTG and GGGGCC repeats (Reddy et al, 2011; Colak et
al, 2014; Groh et al, 2014; Reddy et al, 2014). These RNA/DNA hybrids are formed
during transcription when the nascent RNA hybridizes to the DNA template behind the elongating RNAPII. Indeed, R-loops have been shown to form at expanded GAA repeats following in vitro transcription (Reddy et al, 2011), in E. coli (Grabczyk et al, 2007), and on endogenous FXN alleles from FRDA patients cells (Groh et al, 2014).
All these unusual structures could impair FXN transcription by creating a physical blockage to the RNAPII elongation through the GAA repeat sequence and act as an initial trigger to promote FXN silencing (Sakamoto et al, 2001; Groh et al, 2014).
Repeat-mediated aberrant DNA structures may be subsequently processed by proteins of the DNA repair machinery, potentially inducing repeat instability (reviewed in Castel et al, 2010; Iyer et al, 2014). In healthy conditions, these proteins stabilize the genome by correcting DNA replication errors, attenuating chromosomal rearrangements, and mediating the cellular response to certain types of DNA damage. Eukaryotic MutS homologs (MSHs) detect specific lesions by distinct heterodimeric complexes: human MSH2 forms a heterodimer in solution with MSH6 (MutSα) or MSH3 (MutSβ). MutSα recognizes and initiates removal of base–base mispairs, a subset of insertion–deletion loops and certain types of lesions caused by DNA damaging agents, whereas MutSβ almost exclusively recognizes loops. Mismatch recognition by MutSα or MutSβ is followed by recruitment and activation of a latent endonuclease function in MutLα (heterodimer of MLH1 and PMS2) in the presence of ATP and DNA-loaded replication sliding-clamp proliferating cell nuclear antigen (PCNA), resulting in single-strand breaks flanking both sides of the mismatch in the newly synthesized DNA strand. Mismatch removal then occurs by processive 5’–to–3’ hydrolytic activity of MutSα-activated Exo1, which is