Clínica Universitária de Cardiologia
Update on Arrythmogenic Right
Ventricular Cardiomyopathy
Clínica Universitária de Cardiologia
Update on Arrythmogenic Right
Ventricular Cardiomyopathy
Hugo Filipe Loureiro Cadilha
Orientado por:
Abstract
Arrythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial disease characterized clinically by ventricular arrhythmia, heart failure, sudden cardiac death (SCD), and histologically by progressive replacement of right ventricular muscle by fibrofatty tissue. ARVC was originally described as a dysplasia, as it was believed to be a developmental defect of the right ventricle. (1, 2) Currently, this clinical entity is recognized as a cardiomyopathy, as the adipose and fibrous replacement occurs gradually after birth, while being intimately related to genetic abnormalities mainly in cardiac desmosomes. (3, 4) Most recently, the term arryhtmogenic cardiomyopathy has also been used to broadly reflect variants of this condition where left ventricle involvement can be similar or even greater than right ventricular changes. (5, 6) ARVC’s estimated prevalence ranges from 1 in 5000 to 1 in 1000 individuals in some European regions. (7, 8) ARVC is a major cause of SCD in young people and athletes, and this fatal event may be the first manifestation of the disease. (9, 10) Despite ARVC’s severity, modern therapeutic approaches fail to prevent its development or to halt its progression, offering solely palliative measures. (11) This article aims to review the state of the art in arrythmogenic right ventricular cardiomyopathy, encompassing recent efforts to further our knowledge of its pathogenesis, diagnosis, treatment and prevention.
Key-words: Arrythmogenic right ventricular cardiomyopathy; Arrythmogenic
cardiomyopathy; Pathogenesis; Diagnostic; Therapy
Abbreviations and Acronyms: ARVC - arrythmogenic right ventricular cardiomyopathy; SCD – sudden cardiac death; PG – plakoglobin; VT- ventricular tachycardia; VF - ventricular fibrillation; LV - left ventricular; RV – right ventricle; RA – right atrium; RVOT – right ventricle outflow tract; ECG – electrocardiogram; Tcf/Lef - T cell/lymphoid-enhancing binding; GSK3β - Glycogen synthase kinase-3 beta; ITF - International Task Force; MRI - magnetic resonance imaging; LME - late gadolinium enhancement; EMB - endomyocardial biopsy; VUS - Variant of Uncertain Significance; S:N - signal-to-noise; AHA - American Heart Association; ACC - American College of Cardiology; HRS - Heart Rhythm Society; ESC - European Society of Cardiology; AAD - antiarrhythmic drug; ACEs – angiotensin converting-enzyme inhibitors; ARBs - angiotensin II receptor blockers; ICD - implantable cardiac defibrillator.
Resumo
A cardiomiopatia arritmogénica do ventrículo direito (ARVC) constitui uma doença hereditária do miocárdio caracterizada clinicamente por arritmia ventricular, insuficiência cardíaca, morte súbita cardíaca, assim como histologicamente por substituição progressiva do miocárdio ventricular direito por tecido fibroadiposo. A ARVC foi originalmente descrita como uma displasia devido à hipótese prevalente à altura de que esta patologia seria o resultado de um defeito durante o desenvolvimento embrionário do ventrículo direito. (1, 2) Atualmente, esta entidade clínica é reconhecida como uma cardiomiopatia, dada a substituição fibroadiposa gradual pós-natal intimamente relacionada com defeitos genéticos predominantemente associados aos desmossomas cardíacos. (3, 4) Recentemente, o termo cardiomiopatia arritmogénica tem sido usado para englobar o leque de variantes desta patologia em que o envolvimento do ventrículo esquerdo pode ser similar ou até mesmo maior do que as alterações verificadas no ventrículo direito. (5, 6) A prevalência estimada da ARVC varia entre 1 em 5000 até 1 em 1000 indivíduos em algumas regiões europeias. (7, 8) A ARVC é uma causa major de SCD em jovens e atletas, podendo esta ser a manifestação inicial da doença. (9, 10) Apesar da gravidade da ARVC, as abordagens terapêuticas modernas não impedem o seu desenvolvimento nem atrasam a progressão da doença, oferecendo unicamente medidas paliativas. (11) Este trabalho pretende rever o estado da arte relativo à ARVC, abordando esforços recentes para elucidar a sua patogénese, diagnóstico, tratamento e prevenção.
Palavras-chave: Cardiomiopatia arritmogénica do ventrículo direito; cardiomiopatia
arritmogénica do ventrículo direito; Patogénese; Diagnóstico; Terapêutica
Table of Contents
Abstract
3
Resumo
4
Pathogenesis
7
Genetics 7 Pathophysiology 10Presentation and Diagnostic
15
Epidemiology and Clinical Manifestations 15 Patient Approach and Diagnostic Criteria 16
Therapy
20
Follow-up and Clinical Management 20
Lifestyle Changes 22 Pharmacological Treatment 22 Catheter Ablation 23 ICD Therapy 24 Future approaches 24
Acknowledgements
25
References
26
Appendix
39
Pathogenesis
Genetics
Arrythmogenic right ventricular cardiomyopathy is as a genetically determined disease. This notion was first established with the identification of causative plakoglobin gene (JUP) mutations in patients with Naxos disease, an autosomal recessive disorder characterized by the triad of ARVC, palmoplantar keratoderma and woolly hair. (12)
Plakoglobin (PG) is a constituent of desmosomes and adherens junctions, which are found in both skin, hair and cardiac muscle. These structures are responsible for cell-to-cell adhesion, while also having key roles in intracellular and intercellular signal transduction. JUP mutations may compromise the functional integrity of cell-cell junctions, leading to a tissue-specific phenotype that is exacerbated by constant mechanical stress upon skin and heart tissue. (13) Plakoglobin along with plakophilin-2 are part of the armadillo protein family. These act as linker proteins between intracellular filaments and cell membrane proteins, namely through desmoplakin and desmosomal cadherins (desmoglein-2 and desmocollin-2) in the cardiac desmosome.
After causative gene mutations in PG were identified in Naxos disease patients, it was hypothesized that components of the desmosome that interact tightly with PG could be related to other forms of ARVC. (14) Subsequently, causative mutations in desmoplakin’s gene (DSP) were associated with ARVC and Carvajal syndrome, a similar autossomal recessive cardiocutaneous syndrome. (15-17)
Despite being the first to be discovered, the mutation frequency of JUP and DSP is low compared to other desmosome genes. (18, 19) ARVC is most commonly inherited in an autossomal dominant pattern, through plakophilin-2 (PKP2), desmoglein-2 (DSG2), desmocollin-2 (DSC2), as well as non-syndromic desmoplakin (DSP) gene mutations, with more than half of ARVC patients carrying causative mutations in desmosomal genes. (20) From these, PKP2 is by far the most frequently mutated, accounting for 41.6% of all pathogenic variants. (21) Furthermore, multiple mutations can be found in more than 10% of individuals with disease-causing desmosomal mutations, either in the same or different genes (compound, digenic or trigenic heterozygosity). (19, 22, 23)
Initially through linkage analysis, some causative genes have been identified that are not directly a part of the desmosome but either belong to structures of the area composita or interact closely with it. Such is the case with DES (encoding desmin), a component of the intermediate filament, TTN which encodes the myofilament titin, CDH2 (encoding N-cadherin), a component of adherens junctions, CTNNA3 (encoding α-T-catenin) which links adherens junctions and desmosomes, and FLNC (filament C) participating in Z-disk to intercalated disk interactions. (21, 24, 25)
Recently, ARVC patients harbouring pathogenic mutations in proteins related to sodium and calcium transport in myocytes (SCN5A and PLN) were identified, with incidences up to 15% in some cohorts. (26-28) Genes variants associated with the nuclear envelope (LMNA and TMEM43), and TGFB3 (transforming growth factor β-3) have also been implicated in ARVC cases. (29-31)
Penetrance can be highly variable, as exemplified by PKP2 and TMEM43 mutations, corresponding to low and very high penetrance respectively, and it is reported to be age and exercise related. (30, 32, 33)
Knowledge of the genetic background that underlies this disease has allowed for clinical correlations to be drawn, and some genotype-phenotype relationships to be enlightened. There is a clear association between desmosomal mutations and patients with a younger age of onset of ARVC, a higher incidence of T-wave inversion in leads V1-3 (see Figure 2) and a rich family history of this cardiomyopathy. (22) Moreover, the presence of more than one mutation in these genes strongly correlates to earlier occurrence of sustained ventricular tachycardia (VT) and ventricular fibrillation (VF), to lower arrhythmia-free survival, to more frequent left ventricular (LV) dysfunction, heart failure, sudden cardiac death (SCD) and to the need for cardiac transplantation. (34, 35) Specifically, DSP mutations are related to a left-dominant and possibly more severe clinical variant. This variant is characterized by marked early left ventricular dysfunction, inverted T waves in the inferolateral leads, ventricular arrhythmias with a right bundle-branch block, heart failure and are more likely to present with SCD. (34, 36) Despite ongoing efforts, phenotypic correlations of non-demosomal mutations remain feebler, likely due to smaller proband populations. Nonetheless, some families with TTN and LMNA mutations commonly displayed ARVC with conduction disease and biventricular dysfunction. (29, 37) TMEM43 double mutations may also be associated with a distinct
phenotype, featuring fetal right ventricle (RV) aneurysms and ventricular arrhythmia, although reports of single mutations vary broadly in their presentation. (38, 39) Further genotypic studies remain crucial as demonstrated by a lack of pinpointed causative molecular changes in a significant portion of ARVC patients. (21)
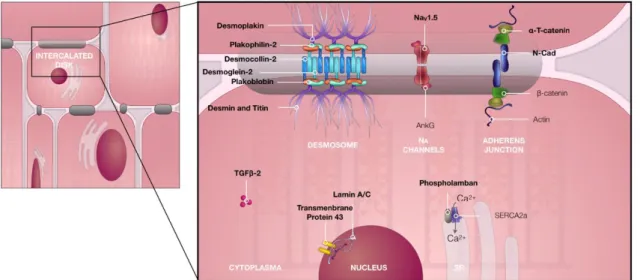
Figure 1. Schematic representation of the structures commonly involved in arrhythmogenic right ventricle
cardiomyopathy in their native configuration. Mutations in the genes encoding the proteins in bold have been implicated in ARVC. Figure based on Hoorntje et al. (40)
Pathophysiology
The pathogenic mechanisms underlying ARVC remain largely unknown. The dysontogenic, degenerative and inflammatory theories were the first to be postulated, based on autopsy examinations. Soon, the perception of ARVC as a congenital disease was abandoned, supported by pathological findings demonstrating a gradual postnatal course of this condition. (41) It was only with the establishment of a causative link between ARVC and desmosomal gene mutations that the molecular processes that drive disease progression started to be unravelled. (42)
As previously stated, desmosomes are responsible for cell-cell coupling in various tissues. Abnormal desmosomes could jeopardize mechanical cell adhesion, facilitating cardiomyocyte detachment and death, leading to the loss of cardiac muscle. (43) This mechanical hypothesis is further supported by a tight association of exercise and phenotypic manifestations. Possibly through increased pressure, afterload and wall stress leading to cell uncoupling, exercise has been shown to induce or exacerbate ARVC, with marked effects particularly on the thinner walled RV. (33) Studies showing maintenance of cell adhesion in transgenic mice with mutant desmosomal proteins suggested the existence of an alternative pathophysiologic mechanism to sheer reduced physical integrity of intercellular junctions. (44)
Desmosomes also play a key role in inter- and intracellular signalling, integrated with adherens junctions, ion channels and gap junctions in the area composita. Altered signalling pathways through abnormal desmosomes in ARVC were first demonstrated by the effect of plakoglobin in Wnt/ β-catenin signalling. PG is structurally akin to β-catenin, being also known as γ-catenin. They both interact with T cell/lymphoid-enhancing binding (Tcf/Lef) transcription factors influencing Wnt signalling, although in dissimilar manners. β-catenin enhances Wnt signalling, while an increase in nuclear PG supresses this pathway. (45) A multitude of desmosomal gene mutations, namely in DSP, PG, PKP2, DSG2, have been shown to destabilize desmosomes, increasing the nuclear translocation of PG, decreasing β-catenin activity, thereby reducing downstream Wnt activation. (46-49) A suppression of this pathway leads to an upregulation of adipogenic transcription factors and downregulation of adipogenesis inhibitors. (45)
In ARVC, these stimuli are particularly important in influencing a newly discovered cell-type that expresses desmosome genes and has been shown to be
precursors to both adipocytes and fibroblasts in cardiac tissue: fibroadipocyte progenitors cells. Wnt signalling suppression induces fibroadipocyte progenitors into adipocyte differentiation, favouring the hypothesis that this molecular mechanism might be associated with the increased amount of fatty tissue found upon histological analysis of deceased probands. (50)
Additionally, in the presence of desmosomal mutations, the Hippo/YAP and PPAR signalling pathways have been directly implicated in myocyte loss and lipogenesis, as well as through cross-talk with Wnt/β-catenin. (48, 51) Altered signalling responses induced by exercise were also hypothesized to be related to a disturbance in the aforementioned cascades, although the mechanism by which these lead to ARVC is yet to be revealed. (52)
Most recently, transcriptome analysis has revealed extensive genome-wide microRNA expression dysregulation in ARVC patients. From these, around 21 have been independently associated with diseased myocardium. Notably, upregulated miR-21-5p and miR-135b expression, as well as downregulation of miR-184 are involved in Wnt, Hippo and PPAR pathways. These act upon target genes that regulate apoptosis, cell proliferation, adipogenesis and fibrogenesis. (53, 54) miR-21-5p specifically, is one of the most significantly upregulated miRNAs in ARVC and has been associated with multiple cardiac pathologies, including hypertrophic cardiomyopathy and fibrosis. (55) An increase in its expression in response to pressure overload is related to aggravated cardiac fibrosis and heart dysfunction, providing clues to a possibly clearer molecular mechanosensation/transduction link between exercise and the cardiac phenotype, which so far remains elusive. (55)
Despite mounting evidence supporting a central role of Wnt/ β-catenin signalling, some authors underline the possibility of this disease being more heterogenous than previously thought, with neither dysfunctional PG neither supressed Wnt signalling being prerequisites to the development of ARVC. (56, 57) Ion handling abnormalities, both dependent and independent from desmosome gene mutations, also seem to play a part in disease formation, namely disrupted sodium and calcium homeostasis. Voltage-gated sodium channels co-localize and interact with cell adhesion structures. NaV1.5, a subunit of these sodium channels, is encoded by SCN5A, a gene we previously mentioned as mutated in some ARVC patients. This is consistent with reduced immunoreactive signals
of NaV1.5 and remodelling of these channels being found in human probands with this cardiomyopathy. (58) Likewise, phospholamban regulates calcium transport in the sarcoplasmatic reticulum and mutations in its gene (PLN) can be present in ARVC. Resulting calcium flux irregularities are correlated with fatal arrhythmias observed in patients with the R14del mutation in PLN. (27)
Excitingly, these advances in our understanding of the pathogenesis behind the disease might lead to novel therapeutic targets and approaches. Compounds that reverse Wnt suppression demonstrate this concept. Glycogen synthase kinase-3 beta (GSK3β) supresses Wnt signalling by degradation of β-catenin. By inhibiting GSK3β it is possible to activate the Wnt pathway, reversing desmosomal and ion channel remodelling in cellular and animal models. (49) Treatment with SB216763, an inhibitor of GSK3β, prevents and reverses cardiac phenotypic manifestations, such as cardiomyocyte injury, cardiac dysfunction and overall increases survival in zebrafish and mouse models. (49, 59) Similarly, preforming genome engineering by using targeted nucleases and combinatorial gene therapy, it was proven possible to rescue the disease phenotype in cardiomyocytes differentiated from induced pluripotent stem cells harvested from a patient with the R14del mutation. (60)
Despite the unknown exact pathogenesis of ARVC, the mechanisms previously mentioned provide essential cues to clarify the nature of the histological and anatomical findings associated with it. Histologically, the disease’s hallmark lesion is defined by cardiomyocyte loss with fibrofatty tissue replacement. The intercellular junction abnormalities stemming from affected desmosomes appear to trigger myocyte death mainly through necrosis, which is hypothesized to be the leading mechanism in disease onset and exacerbation. Apoptosis, when observed, appears to be restricted to areas of fibrosis or necrosis. (61) Autopsy reports of the vast majority of patients show areas of inflammation dominated by T-cells accompanying zones of dying myocytes. (41) In a model that mimics the disease, necrosis precedes inflammation patches, followed by injury repair with fibrous replacement suggesting a sequential pattern to these events, although this step-by-step analysis has not been possible in human probands yet. (61)
he presence of inflammatory patches led early on to the divergent hypothesis of an infective myocarditis aetiology. In fact, some studies report a greater incidence of cardiotropic viruses (e.g. coxsackie virus B3, enterovirus and adenovirus) in sporadic
cases of ARVC, although these findings were not consistent. (62-64) Myocardial damage may itself increase susceptibility to viral infection, in which case viruses might solely be bystanders in this cardiomyopathy or play instead a secondary role as a trigger or amplifier of myocyte loss. (65, 66)
Fatty infiltration was recognized as a pivotal finding in post-mortem exams early on, reflecting the underlying differentiation stimuli that act upon cardiac fibroadipocyte progenitors. However, its presence must be integrated with other clinical findings as fatty infiltration is common in normal hearts and recently its reliability as an ARVC predictor has been increasingly put to question. (67)
Overall, the significance of these histological findings in diagnosis was acknowledged by the inclusion of tissue characterization by means of right ventricle endomyocardial biopsy as a major criterion for ARVC since 1994. (68) Together, fibrous and adipose tissue act similarly to post-myocardial infarction scars, functioning as an abnormal substrate for electrical impulse conduction, facilitating macroreentry circuits and slowing conduction that translate to right bundle branch block and ventricular arrhythmia, among other electrocardiographic changes. (5) On a subcellular level and preceding these histological changes, the previously described sodium and calcium handling abnormalities may also contribute to the formation of this arrhythmogenic substrate. (69)
These histopathological features gradually progress in a wave-front pattern from the epicardium to the endocardium through a mechanism speculated to pertain to myocardial fibre architecture and wall mechanics. (61) The disease can be segmental or patchy, weakening areas of the ventricular wall and translating macroscopically into ventricular wall thinning, akinetic/dyskinectic regions and aneurisms, as depicted in Figure 2. (41, 70) Cardiac regions are differentially impacted according to their susceptibility to mechanical stress and wall thickness, with the traditionally described “triangle of dysplasia” encompassing the most frequently affected areas, namely the apex, infundibular and subtricuspid regions. (41) Recent diagnostic advances have displaced the apex from this “triangle”, as its involvement is usually restricted to advanced ARVC cases, when the RV is globally compromised. (71) In spite of the classic notion of greater right ventricle involvement, there are reports of left ventricular changes in more than 75% of ARVC cases, with disease variants of biventricular or even left dominance being
recently described. When LV involvement is present, the most affected areas seem to be the posterolateral and posteroseptal areas, consistently with their reduced thickness compared to the remainder of the LV. Conceivably since it is not a subepicardial structure, involvement of the septum is rare, although possible. (5)
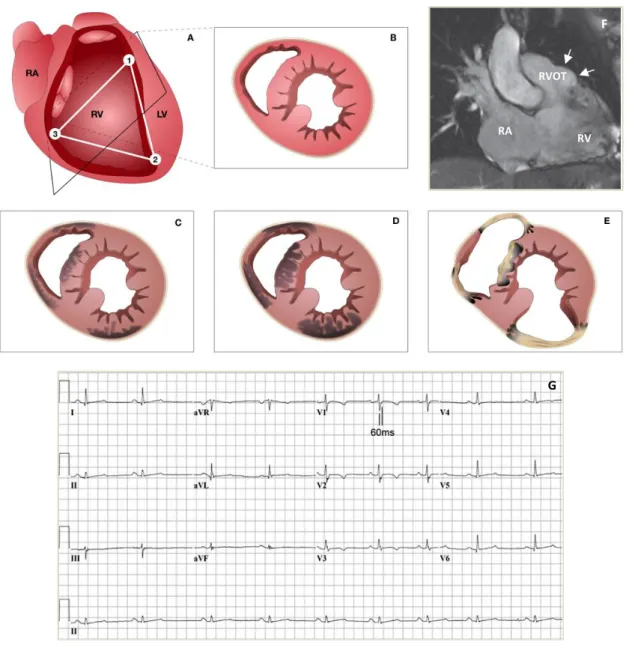
Figure 2. Schematic disease progression, electrocardiographic and imaging features in ARVC. A) The depicted
triangle represents the areas which were initially suspected to be most frequently abnormal in ARVC: the infundibular (1), apex (2) and subtricuspid (3) regions. Panels B to E show the pattern of disease progression from a phenotypically normal heart at birth (B), to subepicardial involvement (C) and transmural extension (D), followed by wall thinning and aneurysm formation (E). Panel F shows an aneurysm (arrows) of the RVOT on a cardiac MRI scan. Panel G displays an ECG with T-wave inversion in V1-V4 and prolongation of the terminal activation duration. RA=right atrium. RV=right ventricle. LV=left ventricle. RVOT=right ventricle outflow tract. MRI=magnetic resonance imaging. ECG= electrocardiogram. Figure adapted from Basso et al, Corrado et al and Marcus et al. (11, 72, 81)
F G RA RV RVOT F
Presentation and Diagnosis
Epidemiology and Clinical Manifestations
ARVC is an underdiagnosed condition, with variable phenotypic expression and an estimated prevalence ranging from 1:1000-1:5000. (7, 8) Despite being most often transmitted as an autosomal dominant trait, several studies report a higher prevalence in men and clinical manifestations being three times more frequent in males. (2, 5, 73, 74) It was hypothesized that this might be related to direct influence of sexual hormones or differences in physical activity patterns between genders. (74) Recently this notion is increasingly controversial, as an analysis of the North American ARVC Registry demonstrated similar numbers of “affected” and “borderline” male and female patients (as defined by the 2010 Task Force criteria below), suggesting that referral bias may contribute to discrepancies in gender prevalence. (75) Furthermore, there is conflicting evidence regarding gender influence on outcome and prognosis, which currently tends to favour male sex as a negative modifier of clinical course. (34, 75)
The first clinical manifestations and peak risk for life-threatening events stand between the second and fourth decade of life, although occasionally the disease has presented itself in the elderly with up to 77 years of age. (76-78) It is exceedingly rare for clinical manifestations to arise in children below the age of 10. (79) Up to 95% of patients are symptomatic at presentation, with palpitation (56%), dizziness (27%) and syncope (21%) being the most common symptoms. (79, 80) These are directly related to a history of ventricular arrhythmia (VA) which consists on the most frequent reason for clinical suspicion of ARVC. (80) Particularly, ventricular tachycardia (VT) of left bundle branch morphology and superior axis pattern, indicating right ventricular origin, should direct the clinician to a suspicion of ARVC. (81) Although heart failure is reportedly uncommon at diagnosis and follow-up, it might pertain to difficulties in differential diagnosis with conditions such as idiopathic dilated cardiomyopathy. (79, 80) SCD may be the initial manifestation of the disease occurring in previously asymptomatic young individuals and competitive athletes. (82) Despite being a relatively uncommon disease, ARVC may account for 5-20% of the cases of SCD in young individuals depending on geographical variation. (83)
The disease’s natural history has been classically described as a four-stage progression comprised of a “concealed phase” in which the patient is at risk of SCD although asymptomatic; an “overt electrical disorder” in which the patient displays symptomatic arrhythmic events; a phase characterized by right ventricular failure with severe dilation and systolic dysfunction; and a biventricular pump failure akin to dilated cardiomyopathy. It’s important to highlight that ARVC’s concealed phase is unlike other primary cardiomyopathies, resembling instead inherited ion channelopathies. (84)
Patient Approach and Diagnostic Criteria
Arrhythmogenic right ventricle cardiomyopathy is a challenging diagnosis due to its highly protean clinical presentation, even among family members with a common mutation. (85) As an early diagnosis heavily influences prognosis and the subsequent therapeutic approach, it is important to conclusively exclude or diagnose ARVC in suspected patients and their relatives. (86) This entails a detailed patient investigation as no single test can confirm the presence of the disease. As such, patients with suspected ARVC should undergo comprehensive clinical and family history, physical examination, 12-lead ECG, 24-hour Holter monitoring, signal-averaged ECG as well as morphofunctional imaging of both ventricles. (87)
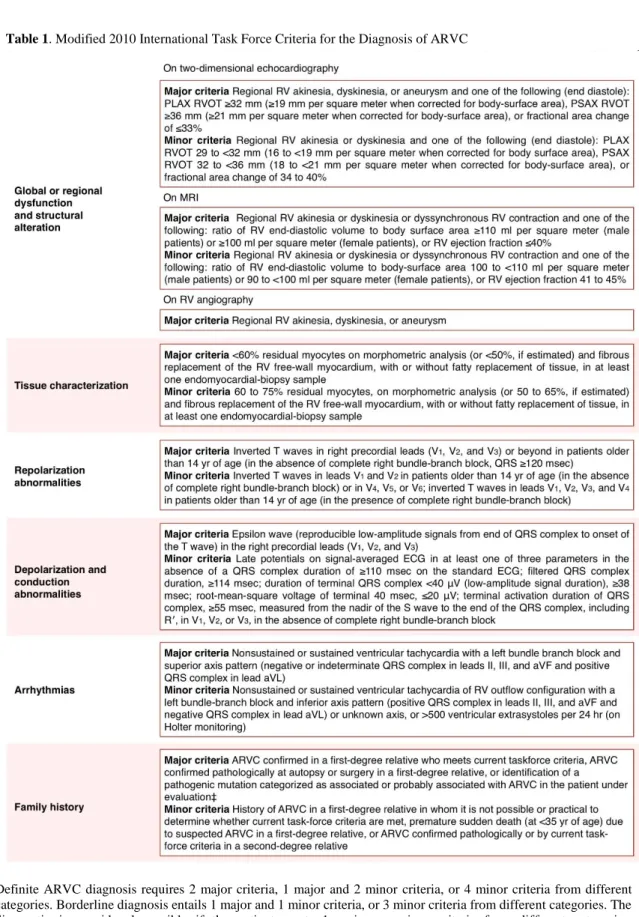
Further studies should be directed to reach a definitive diagnosis under the widely used International Task Force (ITF) criteria depicted on Table 1. These criteria were first established in 1994, being later revised in 2010 to enhance their sensitivity while maintaining diagnostic specificity by introducing quantitative criteria. (81) Accommodating the continuously expanding knowledge about phenotypic ARVC variations, these criteria also facilitate clinical diagnosis for first-degree relatives with subtle expressions of the disease. (86) As seen on Table 1, ITF criteria incorporate structural and functional abnormalities, tissue changes on endomyocardial biopsy, depolarization and repolarizations alterations, documented arrhythmic events, as well as family history and the presence of disease-causing gene mutations.
Definite ARVC diagnosis requires 2 major criteria, 1 major and 2 minor criteria, or 4 minor criteria from different categories. Borderline diagnosis entails 1 major and 1 minor criteria, or 3 minor criteria from different categories. The diagnostic is considered possible if the patient meets 1 major or minor criteria from different categories. ECG=electrocardiogram. PLAX=parasternal long-axis view. PSAX=parasternal short-axis view. RV=right ventricular. RVOT=RV outflow tract. † Hypokinesia is not included in this or subsequent definitions of RV regional wall-motion abnormalities for the proposed modified criteria. ‡ A pathogenic mutation is a DNA alteration associated with ARVC that alters or is expected to alter the encoded protein, is unobserved or rare in a large, non-ARVC control population, and either alters or is predicted to alter the structure or function of the protein or has shown linkage to the disease phenotype in a conclusive pedigree. Table adapted from Marcus et al. (81)
Echocardiography plays a central role as the first-line imaging exam, as a follow-up exam and as an initial screening exam in proband family members. (81) However, the complex RV anatomy renders two-dimensional echocardiography as inadequate to properly estimate RV volume and to visualize the outflow tract.
Cardiac magnetic resonance imaging (MRI) as an alternative imaging modality has proved to be as controversial as promising. Its high spatial and temporal resolution eliminate the need for geometrical assumptions providing improved biventricular structural and functional assessment, and coupled with late gadolinium enhancement (LME) it allows the detection of early myocardial fibrofatty scars that can precede functional changes. Overall, it has a high negative predictive value, bolstering diagnostic sensitivity. (88-90) In a recent letter, the ITF has in fact underlined that cardiac MRI is becoming the gold standard for assessing structural and functional RV abnormalities in ARVC. (91) Nevertheless, cardiac MRI has limited availability and, although highly reproducible, when a lack of expertise is involved in its interpretation reproducibility may be questionable. Its inappropriate use has been appointed as a cause for overdiagnosis. (67, 89)
RV endomyocardial biopsy (EMB) can show ARVC’s hallmark lesion of cardiomyocyte loss with fibrofatty tissue replacement, but has low associated sensitivity due to the patchy nature of the disease. As such, it should only be performed in patients whom noninvasive evaluation remains inconclusive, preferably with the guidance of high resolution imaging techniques. (68, 93, 94) Recently, it has been proposed that there might be a potential role for RV-EMB in early diagnosis, as immunohistochemical staining for GSK3β reveals characteristic ARVC patterns not present in normal hearts or other cardiomyopathies. (49)
Electrocardiographic changes in depolarization and repolarization are the earliest and most sensitive indicators for ARVC, and abnormalities such as T-wave inversion in V1-V3, as depicted in Figure 2, can sometimes be identified even in asymptomatic patients in the “concealed phase” of the disease. As such, the importance of 12-lead ECG analysis cannot be overstated. (95) Despite the fact that epsilon waves are currently considered a major criterion for diagnosis, there is considerable interobserver variability in identifying them and prolonged terminal activation duration, depicted in Figure 2, has been shown to be a more sensitive indicator of RV activation delay. (96, 97) Additionally,
it is possible to identify clues for phenotypic variants such as left-dominant arrhythmogenic cardiomyopathy by the presence of isolated T-wave inversion in V4, V5 and V6. (36)
The addition of genetic testing to the revised 2010 ITF criteria further demonstrates the importance of advances in our understanding of the underlying genetic and pathogenic mechanisms. It has proven to be one of the most significant developments in ARVC diagnosis by being responsible for the largest number of new ARVC cases, increasing criteria sensitivity without compromising specificity. (97) Genotyping allows the identification of a causative mutation in approximately 50% of probands, facilitating the screening of family members that might still be asymptomatic. It should not be used to confirm or exclude the diagnosis in borderline cases, as not all causative mutations are currently known and since nonpathological variants of these genes can be present (Table 2), therefore requiring genetic counselling for appropriate interpretation. (98)
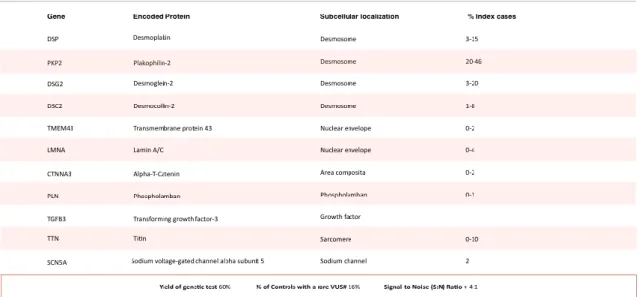
Table 2. Genes with mutations implicated in Arrythmogenic Cardiomyopathy
Yield of genetic test is an estimate derived from unrelated cases with unequivocal disease phenotype, representing the estimate from a comprehensive disease gene panel. This value represents the estimate for caucasians, due to a lack of data pertaining to other populations. % of Controls with a Rare Variant of Uncertain Significance (VUS) represents a frequency of rare amino acid substitutions found in caucasians in the major disease-associated genes that, had it been found in a case, would have been reported as a “possible disease-associated mutation.” This number does not include the frequency of rare genetic variants present in the minor disease-associated genes. The signal-to-noise (S:N) ratio is derived by dividing the yield by the background rate of VUS in controls. This provides a sense of the positive predictive value of a “positive” genetic test result. Data aggregated from Hoorntje et al and Ackerman et al. (40, 98)
Therapy
Follow-up and Clinical Management
After establishing a diagnosis of ARVC, patients should be reassessed every 1-2 years with non-invasive testing, namely 12-lead ECG, echocardiography, 24-h Holter monitoring and exercise testing to evaluate SCD-risk and optimize the therapeutic approach. (99) Familial genetic screening after the identification of a causative-mutation in a proband provides valuable insights in deciding the rate of further evaluations to the patient’s relatives. Non-carriers do not warrant additional testing or follow-up and can be regarded as healthy. (100) Asymptomatic carriers should undergo clinical testing every 2-3 years, with some studies suggesting the ages of 11/12 years old as an appropriate time to begin the aforementioned assessment. (76)
More than 50% of ARVC patients are asymptomatic before experiencing an initial life-threatening arrhythmic event, with approximately 35.5% of these having a fatal outcome. (76) This paradigm highlights the importance of identifying patients with SCD risk, as well as centring clinical management in preventing these events. Additionally, clinical management should also include strategies to prevent disease progression, increase cardiac functional capacity and improve quality of life by limiting clinical manifestations. (99)
While there is still a lack of a defined evidence-based risk stratification scheme, patients are considered at increased risk for SCD if they are young, had previous episodes of syncope, cardiac arrest or of VT with hemodynamic compromise, have two or more disease causing mutations, LV involvement, ARVC5 (a fully penetrant sex-influenced disorder caused by a missense mutation in the TMEM43 gene) and/or Naxos disease. (30, 101-103)
Generally, therapeutic options encompass lifestyle changes, pharmacological treatment, catheter ablation and ICD.
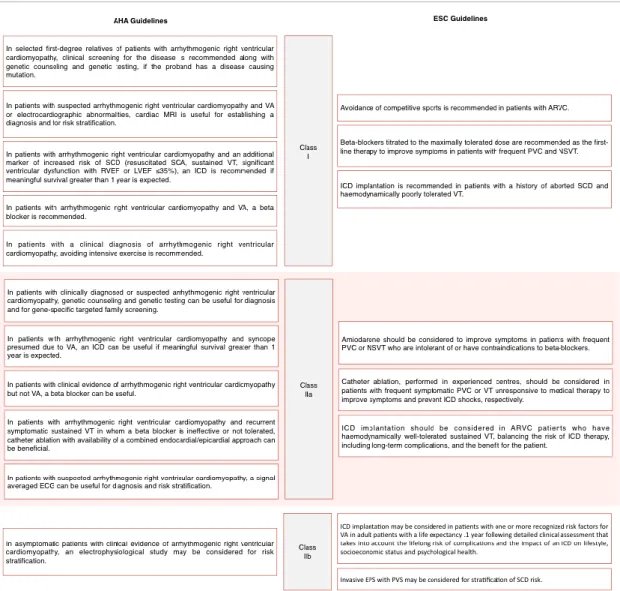
Table 3. Risk stratification and management of patients with Arrythmogenic Right Ventricular Cardiomyopathy
Side by side comparison of the recommendations for risk stratification and management of ARVC patients as seen in the 2017 American Heart Association/American College of Cardiology/Heart Rhythm Society (AHA/ACC/HRS) guidelines and 2015 guidelines by the European Society of Cardiology (ESC). Class I is defined by the existence of evidence (or general agreement) that a given procedure is beneficial, useful or effective. Class II indicates that current evidence is conflicting, regarding the usefulness/efficacy of the given treatment or procedure, with Class IIa suggesting that the weight of evidence points in favor of usefulness/efficacy, while Class IIb signifies that usefulness/efficacy as less well established. Table adapted from Priori et al and Al-Khatib et al. (23, 104)
Lifestyle Changes
As previously described, physical activity is heavily related to disease progression and phenotypic manifestations, being associated with a five-fold increase in the risk of SCD. (33, 105) It’s hard to pinpoint the intensity and frequency of physical exercise that amount to a detrimental effect in ARVC patients. ESC guidelines focus on avoidance of
competitive sports and AHA/ACC/HRS guidelines recommend avoiding intensive exercise as depicted on Table 3, while the ITF treatment consensus also suggests not engaging in endurance sports, but a widespread definition on the extent of the necessary exercise restriction is still lacking. (23, 99, 104) Furthering this, some studies underline that any activity that causes symptoms of palpitation, presyncope or syncope should be avoided. (106) Despite this apparently clear association, there are no controlled trials demonstrating the benefits of physical exercise eviction. Notably, the ITF consensus extends this recommendation to phenotype-negative family members and healthy desmosome gene mutation carriers. (99)
Pharmacological Treatment
Pharmacological treatment in ARVC patients is currently anchored in the use β-blockers, antiarrhythmic drugs and combinations classically used for hearth failure in advanced stages of the disease.
β-blockers have proven efficacy in heart failure management and potential ability to hinder disease progression by lowering ventricular workload, but controversial efficacy in VA prevention. (107, 108) Generally, the use of β-blocker therapy as first-line for all ARVC patients with VA gathers broad expert consensus, and their use should be considered irrespective of arrhythmic events. However, prophylactic treatment is not recommended in healthy gene carriers. No studies have compared the efficacy of individual β-blockers. As such, these should preferably be non-vasodilating and titrated to the maximally tolerated dose for age and weight. (23, 99, 104)
Antiarrhythmic drug (AAD) therapy has been shown not to confer adequate SCD protection in ARVC patients, while Mazzanti et al. furthered this notion reporting that neither sotalol nor amiodarone reduced the rate of life-threatening arrhythmic events (defined as SCD, aborted cardiac arrest, syncopal VT or electrical storm). (76, 107) This recent notion might be reflected in the latest AHA/ACC/HRS guidelines by omission of specific recommendations regarding AADs. Additionally, it is broadly agreed upon that healthy gene carriers or asymptomatic ARVC patients without documented VA should not take AADs regularly. (99) Nevertheless, AADs should be considered to improve symptoms in patients with frequent premature ventricular beats and/or nonsustained VT,
as well as adjunct therapy to ICD and catheter ablation, with ESC guidelines highlighting amiodarone’s superiority relative to other AADs. (107, 108)
The remaining pharmacological treatment options are usually reserved to ARVC patients with established RV or biventricular dysfunction, including angiotensin-converting-enzyme inhibitors (ACEs), angiotensin II receptor blockers (ARBs) and diuretics. ACEs and ARBs can be considered in earlier stages of the disease based on extrapolation from heart failure therapy in other diseases. Long-term oral antithrombotic therapy is solely indicated as secondary prevention in patients with previous thromboembolic complications. (99, 109)
Catheter Ablation
ARVC’s pathophysiology of fibrofatty patchy myocardial replacement that progresses in a wave-front pattern from the epicardium to the endocardium determines the rationale behind the use catheter ablation. These patchy areas serve as an abnormal substrate enabling reentry circuits, while an epicardial origin motivates attempts to conduct ablation through the pericardial space in addition to the classical endocardial approach. This combined endocardial/epicardial approach is the only situation where catheter ablation is contemplated in 2017 AHA/ACC/HRS guidelines regarding ARVC, specifically for patients with recurrent symptomatic sustained VT in whom β-blocker use was not possible. (104) Conversely, the 2015 ITF consensus reserves this combined approach for centres with experience in epicardial ablation, otherwise suggesting that initially the endocardial route should be favoured, including when VTs elicit appropriate discharges in patients with an implantable cardiac defibrillator (ICD). However, catheter ablation is associated with a high recurrence rate of VT and it does not prevent SCD, therefore configuring an inferior alternative to ICD. (99)
ICD Therapy
Implantable defibrillator therapy is a fundamental pillar of ARVC treatment. Multiple reports demonstrate that ICD therapy (both antitachycardia pacing and shocks) is effective for both primary and secondary prevention of SCD. (102, 110, 111) Up to 50% of patients experience life-saving ICD interventions during follow-up in reported
studies, with a significant amount of these discharges occurring less than a year after implantation. (101, 102, 110-112)
ICD implementation is indicated for patients with markers for increased risk for SCD, including history of aborted SCD and hemodynamically unstable VT, with recent American guidelines extending this recommendation to a history of sustained VT or RV/LV ejection fraction ≤35%, as long as survival is expected to be greater than 1 year. (23, 104) This group is considered to have an estimated rate of malignant arrhythmic events of >10% per year. (113) Otherwise, a case-by-case approach should be taken, considering clinical status, age and weighing the risk of complications and inappropriate interventions against the risk factors for SCD, with unexplained syncope, moderate RV/LV dysfunction and nonsustained ventricular tachycardia heavily favouring ICD implementation. (99)
Such an invasive procedure may have complications including pocket hematoma, lead-related problems, pericardial effusion and infection. There are two concerns related to ICD use that are unique to patients with ARVC and may account for the higher rate of complications, namely thin areas of the RV myocardium that can be perforated during placement of the RV leads and the fibrofatty changes in the RV that may interfere with adequate lead positioning and sensing of arrhythmias. (114)
Future approaches
The increased understanding of the pathogenic mechanisms underlying ARVC configures a unique opportunity to change the current treatment paradigm, shifting focus from palliative measures and SCD prevention to influencing disease progression by preventing or reversing cardiomyopathy itself. Although restricted to animal models at the moment, the inhibitor of GSK3β previously mentioned as well as the use of targeted nucleases and combinatorial gene therapy strive to achieve a curative approach. In a similar tone, myocardial regenerative medicine has gathered significant momentum in recent years, promising to restore myocardial function with stem cell therapy as multiple clinical trials are currently under way. (115) Realistically, in the future we might see attempts to adapt broadly used therapies such as furosemide or nitrates in order to reduce ventricular preload and slow exercise-induced ARVC progression. (116)
Acknowledgements
I would like extend my deepest gratitude and appreciation to my thesis advisor Prof.ª Doutora Ana Almeida. In Prof.ª Almeida I found a mentor constantly eager to bolster my efforts, to provide counsel and guidance in steering me towards the right direction
while imparting her knowledge and expertise into this review.
I am forever thankful to my family, for their incessant support and encouragement in every journey that I dared venturing into.
References
1. Frank R, Fontaine G, Vedel J, Mialet G, Sol C, Guiraudon G, et al. (1978)
Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia. Arch Mal Coeur Vaiss 71(9):963-72.
2. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et
al. (1982) Right ventricular dysplasia: a report of 24 adult cases. Circulation. 65(2):384-98.
3. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al.
(2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 113(14):1807-16.
4. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. (2008)
Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 29(2):270-6.
5. Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al.
(1997) Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 30(6):1512-20.
6. Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, et al. (2005)
Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 112(5):636-42.
7. Corrado D, Thiene G. (2006) Arrhythmogenic right ventricular
cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 113(13):1634-7.
8. Peters S, Trummel M, Meyners W. (2004) Prevalence of right ventricular
9. Finocchiaro G, Papadakis M, Robertus JL, Dhutia H, Steriotis AK, Tome M, et al. (2016) Etiology of Sudden Death in Sports: Insights From a United Kingdom Regional Registry. J Am Coll Cardiol. 67(18):2108-15.
10. Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, et al. (2005)
Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 112(25):3823-32.
11. Corrado D, Link MS, Calkins H. (2017) Arrhythmogenic Right Ventricular
Cardiomyopathy. N Engl J Med. 376(15):1489-90.
12. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar
A, et al. (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 355(9221):2119-24.
13. Rampazzo A. (2006) Genetic bases of arrhythmogenic right ventricular
Cardiomyopathy. Heart Int. 2(1):17.
14. Ohno S. (2016) The genetic background of arrhythmogenic right ventricular
cardiomyopathy. J Arrhythm. 32(5):398-403.
15. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et
al. (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 9(18):2761-6.
16. Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. (2002)
Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 71(5):1200-6.
17. Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. (2003) A
recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 42(2):319-27.
18. Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, et al. (2010)
Desmosomal gene analysis in arrhythmogenic right ventricular
dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 12(6):861-8.
19. den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, et al. (2009) Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2(5):428-35.
20. Marcus FI, Edson S, Towbin JA. (2013) Genetics of arrhythmogenic right
ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 61(19):1945-8.
21. Lazzarini E, Jongbloed JD, Pilichou K, Thiene G, Basso C, Bikker H, et al. (2015)
The ARVD/C genetic variants database: 2014 update. Hum Mutat. 36(4):403-10.
22. Xu Z, Zhu W, Wang C, Huang L, Zhou Q, Hu J, et al. (2017) Genotype-phenotype
relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: A systematic review and meta-analysis. Sci Rep. 7:41387.
23. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J,
et al. (2015) 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 36(41):2793-867.
24. Turkowski KL, Tester DJ, Bos JM, Haugaa KH, Ackerman MJ. (2017) Whole
exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 12(2):226-35.
25. Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent
V, et al. (2016) Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 68(22):2440-51.
26. Erkapic D, Neumann T, Schmitt J, Sperzel J, Berkowitsch A, Kuniss M, et al.
(2008) Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace. 10(7):884-7.
27. van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen
DJ, Wiesfeld AC, et al. (2012) Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy:
evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 14(11):1199-207.
28. van Rijsingen IA, van der Zwaag PA, Groeneweg JA, Nannenberg EA, Jongbloed
JD, Zwinderman AH, et al. (2014) Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ Cardiovasc Genet. 7(4):455-65.
29. Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, et al.
(2012) Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 33(9):1128-36.
30. Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn
JD, et al. (2008) Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 82(4):809-21.
31. Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. (2005)
Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 65(2):366-73.
32. Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K, et al. (2006)
Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 48(7):1416-24.
33. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, et al. (2013)
Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 62(14):1290-7.
34. Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, et
al. (2015) Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 36(14):847-55.
35. Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, et al. (2013)
Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 6(6):533-42.
36. Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al.
(2008) Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 52(25):2175-87.
37. Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, et al. (2011) Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 124(8):876-85.
38. Honda T, Kanai Y, Ohno S, Ando H, Honda M, Niwano S, et al. (2016) Fetal
arrhythmogenic right ventricular cardiomyopathy with double mutations in TMEM43. Pediatr Int. 58(5):409-11.
39. Baskin B, Skinner JR, Sanatani S, Terespolsky D, Krahn AD, Ray PN, et al.
(2013) TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet. 132(11):1245-52.
40. Hoorntje ET, Te Rijdt WP, James CA, Pilichou K, Basso C, Judge DP, et al.
(2017) Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc Res. 113(12):1521-31.
41. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. (1996)
Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 94(5):983-91.
42. Protonotarios N, Tsatsopoulou A. (2004) Naxos disease and Carvajal syndrome:
cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 13(4):185-94.
43. Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK,
et al. (2006) Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 27(15):1847-54.
44. Hariharan V, Asimaki A, Michaelson JE, Plovie E, MacRae CA, Saffitz JE, et al.
(2014) Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovasc Res. 104(2):280-9.
45. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury
DS, et al. (2006) Suppression of canonical Wnt/beta-catenin signaling by nuclear
plakoglobin recapitulates phenotype of arrhythmogenic right ventricular
cardiomyopathy. J Clin Invest. 116(7):2012-21.
46. Lombardi R, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT,
progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 109(12):1342-53.
47. Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, et al. (2013) Studying
arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 494(7435):105-10.
48. Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. (2014)
The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 114(3):454-68.
49. Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D,
et al. (2016) Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 1(5).
50. Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, Willerson JT,
et al. (2016) Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ Res. 119(1):41-54.
51. Takada I, Kouzmenko AP, Kato S. (2009) Wnt and PPARgamma signaling in
osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 5(8):442-7.
52. Martherus R, Jain R, Takagi K, Mendsaikhan U, Turdi S, Osinska H, et al. (2016)
Accelerated cardiac remodeling in desmoplakin transgenic mice in response to endurance exercise is associated with perturbed Wnt/beta-catenin signaling. Am J Physiol Heart Circ Physiol. 310(2):H174-87.
53. Zhang H, Liu S, Dong T, Yang J, Xie Y, Wu Y, et al. (2016) Profiling of
differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Sci Rep. 6:28101.
54. Gurha P, Chen X, Lombardi R, Willerson JT, Marian AJ. (2016) Knockdown of
Plakophilin 2 Downregulates miR-184 Through CpG Hypermethylation and Suppression of the E2F1 Pathway and Leads to Enhanced Adipogenesis In Vitro. Circ Res. 119(6):731-50.
55. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. (2008)
MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 456(7224):980-4.
56. Kant S, Krusche CA, Gaertner A, Milting H, Leube RE. (2016) Loss of plakoglobin immunoreactivity in intercalated discs in arrhythmogenic right ventricular cardiomyopathy: protein mislocalization versus epitope masking. Cardiovasc Res. 109(2):260-71.
57. Gaertner A, Schwientek P, Ellinghaus P, Summer H, Golz S, Kassner A, et al.
Myocardial transcriptome analysis of human arrhythmogenic right ventricular cardiomyopathy. Physiol Genomics. 2012;44(1):99-109.
58. Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, et al.
(2013) Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 10(3):412-9.
59. Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, et al. (2014)
Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 6(240):240ra74.
60. Karakikes I, Stillitano F, Nonnenmacher M, Tzimas C, Sanoudou D,
Termglinchan V, et al. (2015) Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun. 6:6955.
61. Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, et al. (2009)
Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med. 206(8):1787-802.
62. Grumbach IM, Heim A, Vonhof S, Stille-Siegener M, Mall G, Gonska BD, et al.
(1998) Coxsackievirus genome in myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiology. (4):241-5.
63. Bowles NE, Ni J, Marcus F, Towbin JA. (2002) The detection of cardiotropic
viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 39(5):892-5.
64. Calabrese F, Angelini A, Thiene G, Basso C, Nava A, Valente M. (2000) No
detection of enteroviral genome in the myocardium of patients with arrhythmogenic right ventricular cardiomyopathy. J Clin Pathol. 53(5):382-7.
65. Xiong D, Lee GH, Badorff C, Dorner A, Lee S, Wolf P, et al. (2002) Dystrophin deficiency markedly increases enterovirus-induced cardiomyopathy: a genetic predisposition to viral heart disease. Nat Med. 8(8):872-7.
66. Calabrese F, Basso C, Carturan E, Valente M, Thiene G. (2006) Arrhythmogenic
right ventricular cardiomyopathy/dysplasia: is there a role for viruses? Cardiovasc Pathol. 15(1):11-7.
67. Bluemke DA, Krupinski EA, Ovitt T, Gear K, Unger E, Axel L, et al. (2003) MR
Imaging of arrhythmogenic right ventricular cardiomyopathy: morphologic findings and interobserver reliability. Cardiology. 99(3):153-62.
68. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine
G, et al. (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 71(3):215-8.
69. Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, et al.
(2012) Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 95(4):460-8.
70. Indik JH, Wichter T, Gear K, Dallas WJ, Marcus FI. (2008) Quantitative
assessment of angiographic right ventricular wall motion in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). J Cardiovasc Electrophysiol. 19(1):39-45.
71. Te Riele AS, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA, et al.
(2013) Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 24(12):1311-20.
72. Basso C, Bauce B, Corrado D, Thiene G. (2011) Pathophysiology of
arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 9(4):223-33.
73. Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M,
Bhuiyan ZA, et al. (2011) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 123(23):2690-700.
74. Bauce B, Frigo G, Marcus FI, Basso C, Rampazzo A, Maddalena F, et al. (2008) Comparison of clinical features of arrhythmogenic right ventricular cardiomyopathy in men versus women. Am J Cardiol. 102(9):1252-7.
75. Choudhary N, Tompkins C, Polonsky B, McNitt S, Calkins H, Mark Estes NA,
3rd, et al. (2016) Clinical Presentation and Outcomes by Sex in Arrhythmogenic Right Ventricular Cardiomyopathy: Findings from the North American ARVC Registry. J Cardiovasc Electrophysiol. 27(5):555-62.
76. Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, et al. (2016)
Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Course and Predictors of Arrhythmic Risk. J Am Coll Cardiol. 68(23):2540-50.
77. More D, O'Brien K, Shaw J. (2002) Arrhythmogenic right ventricular dysplasia in
the elderly. Pacing Clin Electrophysiol. 25(8):1266-9.
78. Sinha AM, Ritscher G, Noelker G. (2008) Arrhythmogenic right ventricular
cardiomyopathy in the elderly: an uncommon finding using magnetic resonance imaging. Europace. 10(1):114-5.
79. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, et al.
(2015) Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 8(3):437-46.
80. Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, et al. (2009)
Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 6(7):984-92.
81. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al.
(2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 121(13):1533-41.
82. Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. (1990) Sudden death in young
competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 89(5):588-96.
83. Ruwald AC, Marcus F, Estes NA, 3rd, Link M, McNitt S, Polonsky B, et al. (2015)
Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North
American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 36(27):1735-43.
84. Thiene G NA, Angelini A, Daliento L, Scognamiglio R, Corrado D.
Anatomoclinical aspects of arrhythmogenic right ventricular cardiomyopathy. In: Baroldi G, Camerini F, Goodwin JF,eds Advances in cardiomyopathies Milano: Springer Verlag. 1990; 397:408.
85. McGregor SM, Husain AN. (2015) A Brief Review and Update of the
Clinicopathologic Diagnosis of Arrhythmogenic Cardiomyopathy. Arch Pathol Lab Med. 139(9):1181-6.
86. Femia G, Hsu C, Singarayar S, Sy RW, Kilborn M, Parker G, et al. (2014) Impact
of new task force criteria in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 171(2):179-83.
87. Orgeron GM, Calkins H. (2016) Advances in the Diagnosis and Management of
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Curr Cardiol Rep. 18(6):53.
88. Vermes E, Strohm O, Otmani A, Childs H, Duff H, Friedrich MG. (2011) Impact
of the revision of arrhythmogenic right ventricular cardiomyopathy/dysplasia task force criteria on its prevalence by CMR criteria. JACC Cardiovasc Imaging. 4(3):282-7.
89. Perazzolo Marra M, Rizzo S, Bauce B, De Lazzari M, Pilichou K, Corrado D, et
al. (2015) Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz. 40(4):600-6.
90. Sanchez-Rubio J, Carreras F, Pujadas S, Leta R, Guillaumet E, Grande C, et al.
(2005) Clinical value of cardiovascular magnetic resonance imaging in the diagnostic work-up of patients with suspected arrhythmogenic right ventricular dysplasia. Rev Esp Cardiol. 58(9):1022-8.
91. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, et al.
(2016) Response to Letter Regarding Article, "Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement". Circulation. 133(11):e437-8.
92. di Gioia CR, Giordano C, Cerbelli B, Pisano A, Perli E, De Dominicis E, et al.
(2016) Nonischemic left ventricular scar and cardiac sudden death in the young. Hum Pathol. 58:78-89.
93. Paul M, Stypmann J, Gerss J, Wirdeier S, Zumhagen S, Breithardt G, et al. (2011) Safety of endomyocardial biopsy in patients with arrhythmogenic right ventricular cardiomyopathy: a study analyzing 161 diagnostic procedures. JACC Cardiovasc Interv. 4(10):1142-8.
94. Avella A, d'Amati G, Pappalardo A, Re F, Silenzi PF, Laurenzi F, et al. (2008)
Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 19(11):1127-34.
95. Cox MG, van der Smagt JJ, Noorman M, Wiesfeld AC, Volders PG, van Langen
IM, et al. (2010) Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 3(2):126-33.
96. Platonov PG, Calkins H, Hauer RN, Corrado D, Svendsen JH, Wichter T, et al.
(2016) High interobserver variability in the assessment of epsilon waves: Implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 13(1):208-16.
97. Protonotarios N, Anastasakis A, Antoniades L, Chlouverakis G, Syrris P, Basso
C, et al. (2011) Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur Heart J. 32(9):1097-104.
98. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. (2011)
HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 8(8):1308-39.
99. Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, et
al. (2015) Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation. 132(5):441-53.
100. Sen-Chowdhry S, Syrris P, McKenna WJ. (2007) Role of genetic analysis in the
management of patients with arrhythmogenic right ventricular
101. Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, et al. (2010) Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 122(12):1144-52.
102. Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, et al. (2003)
Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 108(25):3084-91.
103. Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P, et al. (2011)
Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 123(23):2701-9.
104. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB,
et al. (2017) 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation.
105. Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. (2003) Does sports
activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 42(11):1959-63.
106. Kies P, Bootsma M, Bax J, Schalij MJ, van der Wall EE. (2006) Arrhythmogenic
right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 3(2):225-34.
107. Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, et al.
(2009) Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 54(7):609-15.
108. Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. (1992) Efficacy
of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 86(1):29-37.