2018
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
The role of MEG3 lncRNA in imprinting and pluripotency
Inês Filipa Banha Godinho
Mestrado em Biologia Molecular e Genética
Dissertação orientada por:
Doutor Simão José Teixeira da Rocha
Professora Doutora Gabriela Rodrigues
i
Agradecimentos
O primeiro agradecimento é, como não podia deixar de ser, para o meu orientador durante este intenso ano de aprendizagem, o Doutor Simão Teixeira da Rocha. Por toda a disponibilidade e paciência para as minhas dúvidas e indecisões, por nunca me ter cobrado nenhum erro e me ter ensinado a relativiza-los tantas vezes, por ter acreditado em mim outras tantas e me ter confiado a oportunidade de desenvolver este projeto tão enriquecedor.
À minha orientadora interna, Dr. Gabriela Rodrigues, deixo também um agradecimento especial por toda a disponibilidade, simpatia e interesse que manifestou em acompanhar o meu trabalho.
Da mesma forma, agradeço à Prof. Maria do Carmo Fonseca, por me ter recebido e permitido desenvolver a minha dissertação no seu laboratório, junto do seu grupo, ao qual manifesto também aqui o meu obrigada, por toda a simpatia, ensinamentos e ajuda em todos os momentos. Destacando os técnicos Sérgio e Carolina que de forma mais direta me ajudaram enumeras vezes.
Dentro do grupo, um muitíssimo obrigado à Ana Raposo, não pela paciência, mas por tudo o que me ensinou mesmo sem ela, por toda a atenção e tempo que me dispensou, por todos os conselhos e partilhas, e sobretudo, por ter estado comigo e me ter transmitido segurança nos pontos críticos de todos os protocolos e desta etapa. Ao Duarte por todos os ensinamentos e apoio inicial. À Maria, por todas as gargalhadas e alegria, por toda a preocupação e todas as opiniões, pela companhia nos dias bons e menos bons, resumindo, por passar de colega a amiga. Aos bioinformáticos, Rui, Kenny e Pedro, pelo bom ambiente, pela boa disposição e pelo entusiasmo com que encaram e discutem os seus trabalhos e se tornam tão inspiradores. E também ao Pedro Prudêncio, à Teresinha, à Marta e à Rita pela amabilidade e pela prontidão que sempre demonstraram para esclarecerem as minhas dúvidas e por todas as sugestões que me foram dando ao longo deste tempo e me fizeram melhorar.
Um agradecimento muito especial também para todas as minhas amigas, por todo o apoio e compreensão pela falta de tempo. Por toda a paciência e confiança que me transmitiram ao longo desta fase e, sobretudo, por ficarem sempre incondicionalmente e independentemente do quão difícil sejam os tempos. Dori e Rita, pelo apoio de todos os dias e por todos os dramas que me aturam. Joana, por ser sempre a festa e a alegria. Beatriz e Carina, por tornarem o longe perto e estarem sempre presentes. Maria e Carolina, por me acompanharem há tantos anos e ainda continuarem a festejar comigo as minhas conquistas como se fossem as vossas. Cindy e Cristiana, por apesar de terem chegado há pouco tempo se terem tornado tão importantes para mim e juntamente com a Sofia, a Marta e a Catarina, terem feito com que esta etapa fosse muito mais fácil. Ana Marta, por tudo, sempre. Obrigada!
Por último, mas não menos importante, um enorme obrigada à minha família. À minha tia Maria, por ser a esperança, a crença e o orgulho incondicional em mim. Ao meu padrinho e à Ana, por toda a alegria, descomplicação e amor que acrescentam à minha vida, através do Manel Maria e da Clarinha. E, sobretudo, aos meus Pais, por me alimentarem desde sempre todos os sonhos, por não me deixarem desistir nunca das minhas utopias, por acreditarem em mim tantas e tantas vezes mais do que eu, pelo orgulho e confiança inabaláveis, por serem os meus exemplos de todos os dias e referências em todos os momentos.
ii
Resumo
Desde que se descobriu que as células estaminais embrionárias, descritas como autorrenováveis e pluripotentes, podiam ser obtidas a partir de embriões pré-implantados e expandidas indefinidamente
in vitro, muito se especulou sobre as suas possíveis utilizações na medicina regenerativa e na criação de
modelos celulares de doenças. Contudo, apesar destas esperanças iniciais, as considerações éticas relativas ao uso de embriões humanos e o risco de imuno-rejeição após transplante inviabilizaram a sua posterior aplicação. A solução para ultrapassar estas limitações parecia ser a criação de células pluripotentes oriundas de células somáticas do próprio paciente, através de um processo de reprogramação celular. Neste sentido, em 2006, foram descritas as primeiras células estaminais pluripotentes induzidas, conhecidas na sigla inglesa por iPSCs, que, por serem semelhantes às células estaminais embrionárias na morfologia e nas capacidades de pluripotência, vieram renovar as expectativas nas suas promissoras aplicações.
O processo de reprogramação, no qual é baseado a formação de iPSCs, implica a superação da barreira epigenética das células diferenciadas. Por este motivo, e apesar dos vários avanços na tecnologia da reprogramação, este continua a ser um processo pouco eficiente e propenso a erros epigenéticos, tendo um impacto relevante em fenómenos epigenéticos como é o caso do “genomic imprinting”.
O “genomic imprinting” é um processo que ocorre em alguns domínios genómicos e que resulta na expressão génica de apenas um dos alelos parentais. O alelo expresso é determinado com base na diferença de marcas epigenéticas, nomeadamente ao nível da metilação do DNA, entre ambos os alelos. Erros no “imprinting” ocorrem frequentemente durante a reprogramação das iPSCs e têm um impacto negativo na sua capacidade de pluripotência. Contudo, é essencial que o estado de “imprinting” da célula dadora seja preservado ao longo da reprogramação para que a sua utilização futura seja segura. Isto é especialmente importante quando se geram iPSCs derivadas de pacientes com doenças associadas ao “genomic imprinting”, onde é fundamental que o estado de “imprinting” seja mantido, para que estas células possam ser utilizadas na criação de modelos celulares das doenças. Um dos domínios sujeito a “imprinting” descrito como sendo um dos mais suscetíveis a este tipo de erros é o DLK1-DIO3.
O domínio DLK1-DIO3 encontra-se localizado no cromossoma 14 humano e no cromossoma 12 murino. Este domínio contém três genes codificantes, DLK1, RTL1 e DIO3, expressos a partir do alelo paterno e vários genes não codificantes expressos a partir do alelo materno, incluindo os RNA não codificantes longos (lncRNA) MEG3, RTL1-antisense (RTL1-AS) e MEG8, assim como RNAs não codificantes pequenos, snoRNAs e microRNAs. O principal elemento envolvido na regulação do “imprinting” é a IG-DMR, que consiste numa região intergénica diferencialmente metilada (DMR) entre os dois alelos, encontrando-se metilada no alelo paterno e desmetilada no alelo materno. Para além da IG-DMR, outras DMRs estão descritas neste domínio, como por exemplo a MEG3-DMR, localizada na zona do promotor do lncRNA MEG3. A desregulação do “imprinting” neste locus causa duas síndromes em humanos, conhecidas como síndrome de Temple (associada à perda de expressão de genes codificantes paternos e sobre-expressão dos RNA não codificantes maternos), e síndrome de Kagami-Ogata (associado à perda de expressão dos genes não codificantes maternos e sobre-expressão dos genes paternos) caracterizados por múltiplos problemas de desenvolvimento físico e intelectual. A desregulação do “imprinting” é também muito frequente em iPSCs, onde o alelo materno tem tendência em adquirir hipermetilação, o que conduz à perda de expressão do lncRNA MEG3 e dos outros RNAs não codificantes. Consequentemente, estas iPSCs têm menores capacidades de pluripotência e diferenciam menos eficientemente em determinados tipos celulares. A contribuição de cada gene desregulado nestas iPSCs, incluindo o silenciamento do lncRNA MEG3, para este fenótipo é desconhecido.
iii Tal como ocorre noutros domínios “imprinted”, acredita-se que também neste seja um dos seus lncRNAs o responsável pela regulação do “imprinting”. O candidato principal é o MEG3 e vários estudos anteriores, onde se realizou “Knockouts” para este gene, apontam para esta possibilidade. Contudo, estas abordagens não permitem garantir que seja efetivamente a molécula de lncRNA do
MEG3 a responsável, pois os “Knockouts” causam a eliminação da sequência do DNA do gene MEG3
(incluindo também sequências adjacentes), e, portanto, a atribuição do impacto somente ao lncRNA transcrito por esta região não é correta. Desta forma, o principal objetivo desta dissertação é averiguar o papel deste lncRNA na regulação do ”imprinting” deste domínio e na pluripotência através da modulação dos níveis de expressão deste RNA.
Como ponto de partida para o estudo da regulação do “imprinting”, utilizámos iPSCs isogénicas que apresentavam diferentes padrões de metilação no IG-DMR: uma linha iPSC com IG-DMR normal (alelo paterno metilado e alelo materno desmetilado), e outra linha iPSC com IG-DMR hipermetilado (ambos os alelos parentais metilados). Assim, começámos por avaliar o impacto da hipermetilação do IG-DMR no estado de metilação do MEG3-DMR, tal como na expressão do próprio MEG3 e de outros RNAs não codificantes como o lncRNA MEG8. Observámos que a hipermetilação do IG-DMR resultou na hipermetilação do MEG3-DMR, e que nestes casos não havia, como esperado, expressão do lncRNA
MEG3, nem do lncRNA MEG8. De seguida, quisemos perceber como estas alterações afetavam o
“imprinting” do DLK1, ou seja, se este se tornaria bialélico na linha iPSC com IG-DMR hipermetilado e sem expressão do MEG3. Como esperado, verificou-se que esta linha iPSC apresentava expressão bialélica do DLK1. Contudo, ao contrário da nossa previsão, observámos que a linha iPSC com padrão de metilação normal, exibia também expressão bialélica do DLK1. Enquanto esta investigação decorria, um estudo recentemente publicado sobre células estaminais embrionárias de ratinho permitiu-nos entender este resultado inesperado. Nesse estudo foi descrito que células no estádio estaminal expressavam bialelicamente um nível muito baixo de Dlk1, e que apenas durante a diferenciação neuronal é que os níveis de expressão do Dlk1 aumentavam no alelo paterno, sendo que no alelo materno os níveis se mantinham sempre muito baixos através de um mecanismo de regulação que possivelmente envolve o lncRNA Meg3.
Neste sentido, o passo seguinte foi induzir a diferenciação neuronal nas iPSCs para avaliar se ocorria o aumento da expressão do DLK1 e, se na ausência do MEG3, a expressão se tornava bialélica. Observámos que efetivamente, havia um pico de expressão muito significativo no dia 17 da diferenciação e que na linha celular hipermetilada e sem expressão do MEG3, a expressão era claramente bialélica, enquanto que na linha celular com padrão e expressão de MEG3 normais, os resultados indicaram, preferencialmente, uma tendência para a expressão do alelo paterno. Pensamos que esta tendência apenas não foi traduzida numa expressão claramente monoalélica, devido ao facto de dentro do conjunto de células analisadas a maioria ter uma expressão monoalélica do DLK1, enquanto que a minoria restante pudesse ter expressão bialélica devida a uma incorreção do estado de metilação do IG-DMR.
A nossa análise na linha iPSC com IG-DMR hipermetilado e sem expressão do MEG3 é coerente com a possibilidade do lncRNA MEG3 ter um papel fulcral na regulação do “imprinting” e, como consequência, na pluripotência das iPSCs. Para entendermos especificamente o papel desempenhado pelo lncRNA MEG3 no “imprinting”, decidimos silenciar a expressão deste lncRNA. Para tal, utilizámos “Antisense Locked Nucleic Acids (LNA) GapmeRs”, que demonstrámos serem uma ferramenta bastante eficiente no silenciamento da expressão do MEG3 em iPSCs indiferenciadas. Verificámos que a par do silenciamento do lncRNA MEG3, também o lncRNA MEG8 foi silenciado, o que contribuí para a ideia de que ambos fazem parte da mesma unidade de transcrição policistrónica.
Em seguida, decidimos realizar a experiência do silenciamento do MEG3 no dia 17 da diferenciação neuronal na linha iPSC com preponderância de expressão do DLK1 a partir do alelo paterno. O propósito desta experiência foi entender se o silenciamento do lncRNA MEG3 revertia a expressão
iv maioritariamente paterna em expressão bialélica, onde ambos os alelos parentais se expressassem de forma equitativa. Infelizmente, esta experiência não funcionou por motivos técnicos, muito provavelmente, devido à natureza e densidade celulares durante este estádio de diferenciação.
Por essa razão, decidimos então proceder ao silenciamento do MEG3 em fibroblastos, as únicas células estudadas sobre as quais sabíamos que o DLK1 era exclusivamente expresso pelo cromossoma paterno. Nesta experiência, o silenciamento do MEG3 foi muito eficaz (<5% da expressão normal) e levou a uma reativação modesta do alelo materno do DLK1. Este resultado sugere a participação de RNAs não codificantes como o MEG3 ou mesmo o MEG8, também silenciado nesta experiência, na regulação do “imprinting” deste gene.
Em suma, com este trabalho foi possível testar pela primeira vez o impacto real do lncRNA MEG3 na regulação do “imprinting” do domínio DLK1-DIO3. Os resultados aqui apresentados, sugerem que o MEG3 e/ou outros RNAs não codificantes expressos no alelo materno, como, por exemplo, o MEG8, podem ter um papel importante na regulação do “imprinting” em todo o domínio. No futuro, será interessante investigar através da abordagem de silenciamento com LNAs GapmeRs contra o MEG3 ou até mesmo contra o MEG8, o impacto noutras linhas celulares com níveis mais abundantes de expressão monoalélica do DLK1. Também será importante estudar os mecanismos de atuação destes e de outros lncRNAs, de modo a garantir a manutenção correta do “imprinting” neste locus.
Palavras-chave: Epigenética; Metilação do DNA; “Genomic Imprinting”; Pluripotência; lncRNA
v
Abstract
In 2006, Takahashi and Yamanaka demonstrated that it was possible to convert a differentiated cell into an induced pluripotent stem cell (iPSC) with self-renewing and pluripotent capabilities and enormous biomedical potential. However somatic reprogramming into iPSCs is still inefficient and prone to epigenetic errors which impact on phenomena such as genomic imprinting.
Genomic imprinting is a process that occurs in some genomic domains and results in a monoallelic expression of genes depending on their parental origin. This is determined by a differential epigenetic marking, by DNA methylation of the two parental alleles. DLK1-DIO3 domain represents an example of a genomic region regulated by genomic imprinting. This domain contains several protein-coding genes, such as DLK1, expressed from the paternal allele and several non-protein-coding genes from the maternal homolog, including the long non-coding RNAs (lncRNA) MEG3 and MEG8. The main regulatory element involved in imprinting is an intergenic methylated region (IG-DMR), that is methylated in the paternal allele and unmethylated in the maternal one. In addition, other DMRs are described in this domain, such as MEG3-DMR, believed to be hierarchically dependent on IG-DMR. Deregulation of imprinting at this locus causes two human syndromes known as Temple and Kagami-Ogata syndromes. Deregulation of imprinting is also very common in iPSCs, where the maternal allele tends to acquire hypermethylation which leads to loss of non-coding RNAs expression, including
MEG3. Consequently, these iPSCs have lower pluripotency potential. The contribution of each
deregulated gene, including silencing of the MEG3 lncRNA to this phenotype in iPSCs is unknown. It is believed that one of the lncRNAs is responsible for the imprinting regulation in this domain.
MEG3 is the main candidate based on the results from several MEG3 KO studies. However, these
approaches did not ensure that the MEG3 lncRNA molecule is indeed the responsible. Thus, the main objective of this dissertation is to investigate the role of this lncRNA in the imprinting regulation of this domain and pluripotency by modulating its expression levels.
We initially used isogenic iPSCs with different methylation patterns at IG-DMR: an iPSC line with normal IG-DMR and another iPSC line with hypermethylated IG-DMR to understand the mechanisms of imprinting regulation of this locus in iPSCs. Hypermethylation of IG-DMR resulted in the hypermethylation of MEG3-DMR, and consequently to the lack of lncRNA MEG3 expression. As expected, we also detected that the hypermethylated line had biallelic DLK1expression. Interestingly, the iPSC line with normal IG-DMR also exhibit low, but biallelic DLK1 expression, a finding corroborated by others in mouse embryonic stem cells.
We then induce neuronal differentiation in the iPSCs and observed a very significant peak of
DLK1 expression at day 17 of differentiation with a bias towards the paternal allele in normal iPSCs,
but clearly biallelic in iPSC with hypermethylated IG-DMR and no MEG3 expression. We felt this time point provide a good system to enquire about the role of MEG3 on DLK1 imprinting during differentiation of iPSCs. To specifically understand the role played by MEG3 lncRNA, we downregulate its expression using Antisense Locked Nucleic Acids (LNA) GapmeRs. This approach proved to efficiently downregulate MEG3 in undifferentiated iPSCs. Interestingly, LNAs against MEG3 also downregulate MEG8 lncRNA contributing to the idea that both are part of the same unit of polycistronic transcription. Unfortunately, MEG3 LNAs did not work at day 17 of neuronal differentiation for unforeseen reasons. We then decide to downregulate MEG3 in fibroblasts, the only cells presenting exclusive paternal DLK1 expression. In this experiment, effective MEG3 downregulation led to a modest re-activation of the maternal DLK1 allele. This result suggests the participation of non-coding RNAs such as MEG3 or even MEG8, also silenced in this experiment, in the regulation of DLK1 imprinting.
In summary, with this work it was possible to test for the first time the real impact of the MEG3 lncRNA on the regulation of the imprinting of the DLK1-DIO3 domain. In the future, it will be
vi interesting to investigate the impact of MEG3 or even MEG8 on other cell lines with more abundant levels of monoallelic expression of DLK1 through silencing with LNAs GapmeRs. It will also be necessary to understand from the mechanistic point of view how these lncRNAs act to ensure the correct maintenance of imprinting at this locus.
vii
Table of Contents
Agradecimentos i Resumo ii Abstract v List of Figures ix List of Tables ix List of Abbreviations x 1. Introduction 1Stem Cells – from Embryonic to Induced Pluripotent Stem Cells 1
Epigenetics 2
Genomic imprinting 2
DLK1-DIO3 domain 4
Paternally Expressed Genes, PEGs 4
Maternally Expressed Genes, MEGs 5
Imprinting regulation of DLK1-DIO3 domain 6
Role of MEG3 in imprinting regulation and gene regulation in general 7
Imprinting disorders associated with DLK1-DIO3 domain 7
Temple Syndrome (TS14) 7
Kagami-Ogata Syndrome (KOS14) 8
Imprinting defects of DLK1-DIO3 domain in iPSCs and impact on pluripotency 9
Aims of the study 10
2. Materials and Methods 11
Biological Samples and tissue culture 11
Fibroblasts 11
hiPSCs 11
Antisense LNA GapmeRs Transfections 12
Molecular Biology Techniques 13
Allelic Specific Imprinting Assessment 13
DNA extraction 13
DNA Methylation Analysis 13
Nested PCR 14
Combined Bisulfite Restriction Analysis for MEG3-DMR 15
RNA extraction 15
RT-qPCR and RT-PCR 16
DLK1 allelic-specific expression analysis 16
RNA Fluorescent in situ hybridization (RNA FISH) 17
3.Results and Discussion 18
MEG3-DMR methylation status in hiPSCs 18
MEG3 Expression in iPSCs with normal and abnormal DLK1-DIO3 methylation patterns 19 Imprinting of DLK1 in iPSCs with normal and abnormal DLK1-DIO3 methylation patterns 20
Differentiation of iPSC into cortical neurons 21
viii
4. Conclusion and future perspectives 25
5.
Bibliography 27ix
List of Figures
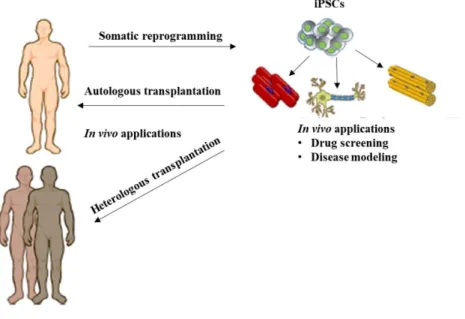
Figure 1.1 - Generation of hiPSCs and their possible applications 1
Figure 1.2 - Schematic representation of the DLK1-DIO3 Domain 4
Figure 1.3 - Schematic representation of defects at the DLK1-DIO3 locus in TS14 8 Figure 1.4 - Schematic representation of defects at the DLK1-DIO3 locus in KO14 9
Figure 2.1 – Bisulfite conversion process 13
Figure 2.2 - MEG3-DMR amplified sequence 15
Figure 2.3 - Schematic representation of the DLK1 17
Figure 3.1 – MEG3-DMR methylation status 18
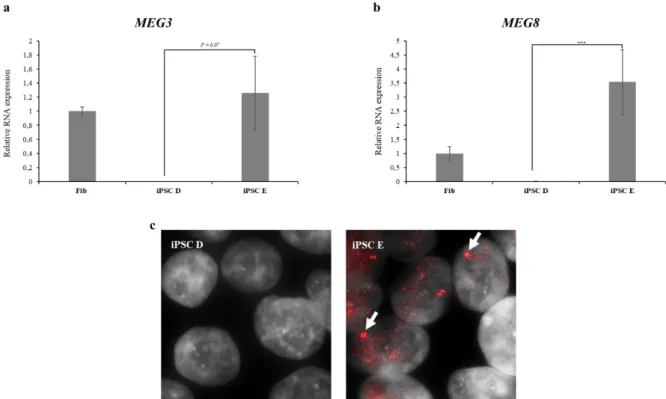
Figure 3.2 - MEG3 Expression 19
Figure 3.3– Allelic-specific expression to address DLK1 imprinting status 20 Figure 3.4 – DLK1 and MEG3 expression analysis during neuronal differentiation of iPSC D
and iPSC E lines 22
Figure 3.5 - Impact of MEG3 lncRNA on the regulation of DLK1-DIO3 imprinting at D17 of
neuronal differentiation of iPSCs lines 23
Figure 3.6 - Impact of MEG3 RNA on the regulation of DLK1-DIO3 imprinting.in fibroblasts 24
List of Tables
Table 2.1 - Antisense LNAs GapmeRs for MEG3 12
Table 2.2 - Primers and conditions used for MEG3-DMR Nested PCR 14
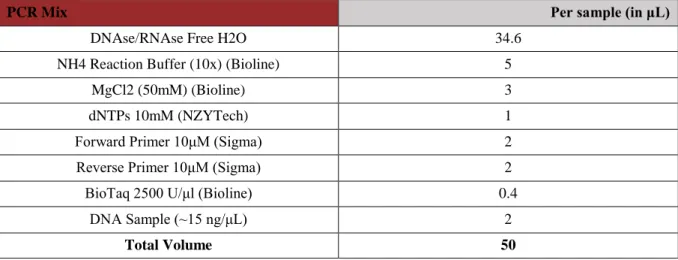
Table 2.3 - PCR Mix with Biotaq 14
Table 2.4 - Primers and conditions used for RT-qPCR 16
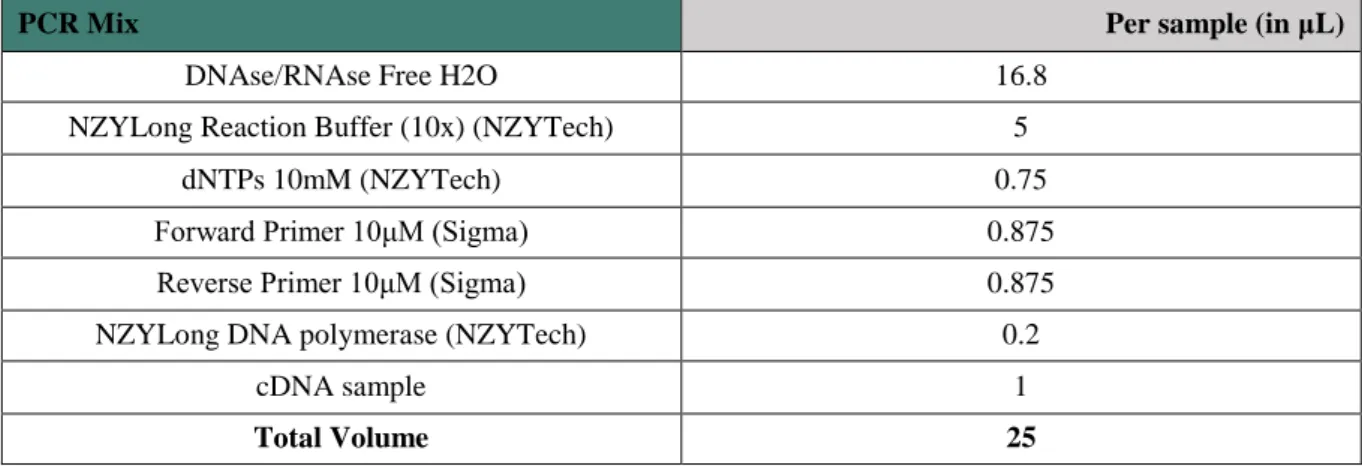
Table 2.5– PCR Mix with NZYLong DNA polymerase 16
x
List of Abbreviations
AS – Angelman Syndrome CRISPRi – CRISPR interference
COBRA – Combined Bisulfite Restriction Analysis DAPI - 4’,6-diamidino-2-phenylindole
DMR – Differentially Methylated Region DMSO – Dimethyl Sulfoxide
DNMT – De Novo Methyltransferases EDTA – Ethylenediaminetetraacetic Acid ESC – Embryonic Stem Cell
FA – Formamide
FBS – Fetal Bovine Serum
FISH – Fluorescent In Vitro Hybridization hESC – Human Embryonic Stem Cell
hiPSC – Human Induced Pluripotent Stem Cell ICR – Imprinting Control Region
IF – Immunofluorescence
IG-DMR – Intergenic Differentially DNA-Methylated Region iPSC – induced Pluripotent Stem Cell
KO – Knock-Out
KOS - Kagami-Ogata Syndrome LNA - Locked Nucleic Acids lncRNA –long noncoding RNA MEG - Maternally Expressed Gene mESC – mouse Embryonic Stem Cell
miPSC – mouse Induced Pluripotent Stem Cell miRNAs - microRNAs
P/S – Penicillin/Streptavidin PBS – Phosphate-Buffered Saline PEG - Paternally Expressed Gene PFA – Paraformaldehyde
PRC2 - Polycomb repressive complex-2 PWS – Prader-Willi Syndrome
RNA FISH - RNA Fluorescent in situ hybridization RNAi - RNA interference
rRNA - ribosomal RNA
snoRNA – small nucleolar RNA
SNP – Single Nucleotide Polymorphism snRNA - spliceosomal RNA
SRS - Silver-Russell syndrome TAE – Tris-acetate-EDTA TFs – Transcription Factors TH - thyroid hormones TS – Temple Syndrome UPD – Uniparental Disomy
1
1. Introduction
Stem Cells – from Embryonic to Induced Pluripotent Stem Cells
The discovery that human embryonic stem cells (hESCs) could be derived from preimplantation embryos and be indefinitely expanded in vitro led to high expectations for these cells as a great source for regenerative medicine and disease modelling 1. This is because, hESCs have self-renewal
characteristics (i.e., potential to endlessly divide while maintaining an undifferentiated state) and pluripotent plasticity (that is the ability to generate any cell type belonging to the three germ layers of the embryo: ectoderm, mesoderm, or endoderm) 2. However, ethical considerations associated to the
use of human embryos and risk of immune rejection after transplantation are some of problems for the use of hESCs for their downstream applications.
The solution to overcome these limitations appeared to be the generation of pluripotent cells by reprogramming patient-derived somatic cells. Indeed in 2006, Takahashi and Yamanaka described for the first time the generation of these cells which were named induced Pluripotent Stem Cells (iPSCs) 3.
First iPSCs were obtained from mouse embryonic and adult fibroblasts by retrovirus-mediated transfection of Yamanaka factors under ESC culture conditions. These cells exhibited the morphology and the pluripotent properties of ESCs 3. It was later described the generation of reprogrammed from
human adult fibroblast using the same Yamanaka factors. Once again, these hiPSCs seemed to be similar to hESCs in morphology and functional features, including in the ability to differentiate into cell type of three germ layers 4,5.
These hiPSCs opened the prospect to reprogram patient-derived cells, helping to elucidate novel pathological mechanisms underlying human diseases and to reveal new therapeutic drugs (Figure 1.1).
Furthermore, clinical trials involving iPSCs for regenerative medicine have also been initiated 6. Despite
their relative success, concerns about the safety of the use of these cells have been raised. One of the first problems was related to the use of retroviruses or lentiviruses for generating iPSC, which might cause insertional mutagenesis. For that reason, approaches using non-insertional methods have been reported using plasmids, adenovirus, Sendai virus, proteins or synthesized RNAs, however the efficiency of reprogramming remained lower when compared to insertional approaches 7. Another
2 major point of discussion regarding iPSCs was whether they are really equivalent to ESCs and, if not, whether differences are functionally relevant. Several studies indicated that effectively subtle differences exist between ESCs and iPSCs, namely, at the level of gene expression, epigenetic modifications, specifically on DNA methylation and on pluripotency capacity 8. In particular, the fact
that we currently lack the ability to control the epigenetic state of iPSCs fully, especially at the epigenetic level, is a major limitation to downstream applications, such as disease modelling. However, since epigenetic states can be modulated, there is a window of opportunity to fix this.
Epigenetics
The term Epigenetic refers to the study of functionally relevant changes in gene activity that do not involve alterations in the DNA sequence and which are heritable, at least from cell to cell, but could potentially be reversible. These changes can occur at different periods in life and be induced by environmental factors 9.
Epigenetic control act on three main levels: DNA, histones, and nucleosomes. At DNA level, the most relevant known modification occurs through methylation of the cytosine residue in context of CpG dinucleotides. At the histones level, it occurs through diverse post-translation modifications (e.g., methylation, acetylation, ubiquitination, sumoylation and phosphorylation and others) at specific amino acids mostly at the protruding tails of histones. At the nucleosome level by remodeling 9,10. The most
studied and understood is CpG DNA methylation that consist in a covalent addition, by de novo DNA methyltransferases (DNMTs), of a methyl group to the cytosine residue in CpG dinucleotides 11. DNA
methylation is linked with gene repression when occurs at CpG-rich regions located at promoters. However, when methylation occurs in the sparse CpG dinucleotides on the gene body has a positive correlation with gene expression 9.
The epigenetic machinery is involved in many important biological processes, including genomic imprinting, X-chromosome inactivation, heterochromatin formation and transcriptional regulation 9. Epigenetics is important for the maintenance of transcriptional landscape which confers
cell identity, being one of the main obstacles to the success and efficiency of somatic reprogramming techniques. Indeed, reprogramming of somatic cells requires drastic changes in the epigenetic profile of a cell, which could cause undesirable mistakes. Several sources of epigenetic errors have been described in iPSCs, like the failure to reset the epigenetic memory of donor cell and acquisition of stochastic errors during reprogramming 12. Moreover, iPSCs could also gain epigenetic aberrations through adaptation to
long-term in vitro culture conditions. All these defects create epigenetic heterogeneity between the iPSCs. Epigenetic processes, such as genomic imprinting, provides a good read-out to study this epigenetic heterogeneity in iPSCs.
Genomic imprinting
Genomic imprinting is an epigenetic mechanism of gene regulation that causes monoallelic expression of a small subset of genes depending on their parental origin in mammalian cells 13.
Mammalian cells are characterized by having two sets of chromosomes, one inherited from the mother and another from the father. Thus, typically both copies of each gene have the same potential to be expressed or repressed according to the cell type. However, genes regulated by genomic imprinting defies this rule, with only one of the two chromosomes being expressed in a parental-specific manner
11. These two parentally inherited chromosomes have identical genetic sequences and, therefore genetics
is unlikely to explain the differential expression of imprinted genes 11,14,15. Genomic imprinting is a
3 a maternally expressed imprinted it will be expressed on the maternal chromosome and repressed on the paternal homolog in all female and male progeny 11.
So far 100 genes in the human and 124 in the mouse genomes were identified as being imprinted genes 16. Interestingly, while the majority of these genes are imprinted in whole organism and placenta,
some of them are only imprinted in specific tissues and/or during specific developmental phases 16.
Many are grouped in clusters suggesting the presence of long-range cis-acting regulatory elements that can control the parental-origin specific expression 11,14,17. Indeed, for each of these clusters a specific
region designated as Imprinting Control Region (ICR), has been identified as the master-regulator of imprinting regulation for all the genes within a cluster 13,14.
Deletions of ICRs in mouse and human cause the alteration of imprinting pattern of the whole domain, proving their essential role in regulation of the imprinted expression. The ICR sequences are not the same in all clusters, but they are highly dense in CpG dinucleotides 16. All ICRs are characterized
as being differentially DNA methylated between the two parental alleles and this feature is essential for the establishment imprinting regulation 13. This is the reason why genomic imprinting is an epigenetic
phenomenon since it is dependent on parental-specific DNA methylation patterns. This type of imprinting marks which distinguishes the parental alleles, are initially erased in primordial germ cells and then established during later phases of gametogenesis in a sex-specific manner. These marks are then maintained stable in all future developmental stages, thus forming the so-called imprinting life cycle, ensuring correct gene expression in all somatic cells 18.
Beyond the ICRs, imprinted clusters typically contain other Differentially Methylated Regions (DMRs), whose methylation is established after fertilization, known as somatic DMRs. They can act as tissue-specific regulatory elements, promoters or silencers 11,16. It is thought that these DMRs depend
hierarchically on the methylation pattern of ICR (also named gametic DMRs). This idea is supported by the fact that deletions of somatic DMRs only affects expression of the adjacent genes without causing any impact on the imprinting expression of the remaining genes in the cluster 11.
Furthermore, most of imprinted clusters contain at least one long non-coding RNA (lncRNA), defined as a non-coding transcript with more than 200 nucleotides, that display specific temporal and spatial expression patterns. Generally, in an imprinted cluster, protein-coding genes are expressed from the same parental chromosome, while lncRNA(s) are expressed from the opposite chromosome. This suggest that lncRNAs may have a regulatory role in a silencing of protein-coding genes in cis, which consequently determines their imprinting expression 11,16.
Studies that led to the identification of the imprinted genes allowed at the same time predict what their functions might be, namely, the first experiments using nuclear transfer technology showed that embryos reconstituted from two paternal (androgenetic embryos) or two maternal (gynogenetic embryos) pronuclei failed to developed properly and died, while embryos reconstituted from one paternal and one maternal pronuclei produced fertile and viable offspring 19,20. At the time of death,
androgenetic embryos revealed defects in embryonic tissues and gynogenetic embryos in extraembryonic tissues. These strikingly different results demonstrated the existence of parental-specific differences and that both parental contributions are essential for correct embryogenesis. Imprinted genes are associated with roles in growth and development of the embryo, placenta and neonate 11.
Accordingly, deletions or mutations in imprinted genes that cause disruption of the correct expression are linked with several growth, neurodevelopmental and metabolic defects and with several human imprinting syndromes. For example, failure in imprinting of the SNRPN-UBE3A domain are associated with Prader-Willi (PWS) and Angelman Syndromes (AS) and in DLK1-DIO3 domain with Temple (TS) and Kagami-Ogata Syndromes (KOS) (see below). Imprinting defects are also related with cell transformation and cancer 21.
4
DLK1-DIO3 domain
Several studies indicate that imprinted regions are very susceptible to epigenetic errors during the reprogramming process. Among many imprinted domains, the DLK1-DIO3 locus has been highlighted with one of the most affected in both mouse (miPSCs) and human (hiPSCs) iPSCs 8,22,23.
The DLK1-DIO3 domain, is an imprinted domain located on human chromosome 14q32 and distal mouse chromosome 12 24. Comparative sequence analyses demonstrated that organization and
imprinting of this cluster is highly conserved between mammals 25.
The first clear evidence for the existence of imprinting involving genes on mouse chromosome 12 occurred in 1993, when Cattanach and Rasberry intercrossed mice heterozygous for balanced chromosomal translocations and generated mice with maternal chromosome 12 duplication and paternal chromosome 12 deficiency and vice-versa. Both mice exhibited prenatal lethality, growth abnormalities and developmental defects in several organ systems, what suggested the requirement of both parental copies 14. Around the same time, in humans, a growing number of conditions associated with uniparental
disomy (UPD) of the chromosome 14 were also reported 26,27. Interestingly, patients shared notable
phenotypic similarities with mice with uniparental disomies for the chromosome 12, although they were viable at birth 14. All these indications led to the subsequent search for the imprinted genes on the mouse
chromosome 12/human 14 28,29,30,31.
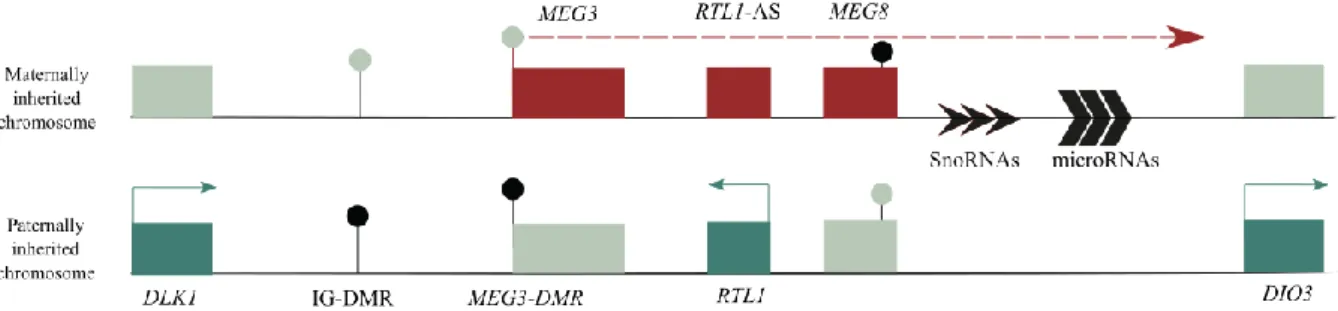
The DLK1-DIO3 imprinted cluster contains three protein-coding genes, Delta like non-canonical Notch ligand 1 (DLK1), Retrotransposon-like 1 (RTL1, also identified as Peg11) and type III iodothyronine deiodinase gene (DIO3), expressed from the paternally inherited chromosome. The maternal homolog expresses several noncoding RNA genes, including the MEG3 lncRNAs (previously known as Gtl2), RTL1-antisense (RTL1-as) and Maternal expressed 8 (MEG8) and several small non-coding RNAs such as, C/D-box snoRNAs and microRNAs 14 (Figure 1.2).
Figure 1.2 - Schematic representation of the DLK1-DIO3 domain. Blue rectangles represent imprinted protein-coding genes that are paternally expressed, red rectangles represent imprinted noncoding RNAs transcripts that are maternally expressed, and the grey rectangles represent the repressed alleles of imprinted genes. The DMRs are represented by circles: the grey circles represent unmethylated regions while black circles represent the methylated regions. IG-DMR is the DLK1-DIO3 imprinting center.
Paternally Expressed Genes, PEGs
DLK1 was the first paternally expressed gene (PEG) identified in this cluster 28. It encodes a
transmembrane glycoprotein that comprises six epidermal growth factor (EGF)-like motifs in the extracellular domain, a transmembrane domain and a short intracellular tail 14,32,33. These structural
features and the high grade of homology with proteins encoded by homeotic genes like Delta, Serrate and Notch, and therefore DLK1 is considered as a member of the EGF-like homeotic protein family 33.
Notch receptors and their ligands are also part of this family, however in contrast to them, DLK1 do not contain the Delta:Serrate:-Lin-12 (DSL) domain, that is responsible for the activation and interaction
5 with NOTCH receptor 14,33,34. Despite this lack, several studies suggest that DLK1 can interacts with
NOTCH, more precisely acting as NOTCH antagonist 14,34,35,36. In mice, the Dlk1 knock-out (KO) results
ingrowth retardation and accelerated adiposity 37.This is consistent with known roles of DLK1 in the
differentiation of some tissues 33,37,38,39 and as inhibitor of adipogenesis 37,40. This gene is mainly
expressed during embryonic stages and is dosage-sensitive, so when it is overexpressed cause phenotypes like defects in prenatal lethality and postnatal energy homeostasis 39,41,42. Postnatally,
expression is retained only in a few tissues, for example in adult brain the expression is restricted to neurons of some areas 39.
RTL1 is a retrotransposon-like gene that shares homology with the Ty3/gypsy retrotransposons
family, but without the ability to autonomously retrotranspose 14. RTL1 is principally expressed at
late-fetal stage in a subset of tissues and in the placenta. Indeed, Rtl1 KO mice indicate that Rtl1 is essential for correct placental development since placentas of Rtl1 KO mice (deletion on the paternal allele only) demonstrated severe abnormalities in the fetal capillaries where the feto-maternal interaction occurs. Additionally, most of mice with no Rtl1 expression showed late-fetal or neonatal lethality 43.
In contrast to DLK1 and RTL1, DIO3 imprinting is less obvious, since the maternal allele is not completely silenced, and only a bias towards paternal expression is observed 44,45. In human, there is no
evidence that DIO3 is, in fact, an imprinted gene. DIO3, located ~800 kb downstream of DLK1, encodes Type 3 iodothyronine deiodinase (D3), a conserved selenocysteine-containing enzyme that belongs to iodothyronine deiodinase family 14,46 . Enzymes of this family are responsible for metabolize thyroid
hormones (TH) and the interaction between them allows to maintain the TH levels within the physiological range 46,44. In the particular case of the D3, the high levels of activity in placenta, uterus
and fetal tissues suggest that have a role in regulation of TH availability during development, thereby ensuring the protection of the developing fetus against excessive amounts of this active hormone 46,47.
Indeed, this function is proved with Dio3 KO mice that exhibit neonatal mortality, growth retardation and abnormal thyroid status 48. In the developing and adult brain, Dio3 is also very expressed, once
again it is suggested an important role in TH levels regulation in order to prevent neurological abnormalities 49.
Maternally Expressed Genes, MEGs
The maternally expressed gene (MEG) MEG3, previously known as Gene trap locus 2 (Gtl2), expresses a noncoding poly-adenylated transcript with multiple alternatively spliced forms without apparent open reading frame 50,28,30. MEG3 plays an important role in embryonic development.
According to this, two studies with Meg3 KO mice with deletions upstream and comprising Meg3 were generated. In both cases the mice died, but while in one of the studies maternal inherited deletion of
Meg3 resulted in mice with skeletal muscle defects and perinatal death in the other, mice no Meg3 were
born with an apparent normal phenotype but died within 4 weeks after birth, possibly due to severely hypoplastic pulmonary alveoli and hepatocellular necrosis 51,52. In humans, it can act as a tumor
suppressor and as a regulator of others tumor suppressors, for example p53 24. It is also involved in
neural developmental 53.
Beyond MEG3, other non-coding RNAs are, as previously mentioned, expressed from the maternally inherited chromosome, namely, the microRNAs (miRNAs) and the C/D small nucleolar RNAs genes (snoRNAs).
miRNAs are an abundant class of small non-coding RNAs (21-25 nucleotides) processed from longer hairpin RNA precursors. miRNAs can interact with mRNA and trigger them either for degradation through RNA interference (RNAi) or translation repression, according to the grade of complementarity 14,54. Several imprinted miRNAs genes are expressed at the DLK1-DIO3 locus 55. The
6 majority are contained within recognized host transcripts. One example is RTL1-AS transcript, which contains seven miRNAs processed from five hairpins, two of these miRNAs are perfectly complementary to RTL1 and are all transcribed in an antisense orientation to this paternally expressed coding gene. Thus, the miRNAs generated from Rtl1-as can target Rtl1 mRNA and mediate its degradation through RNAi-mediated machinery as demonstrated in mouse studies 14,54. Beyond this
important role, it was suggested that one of the miRNAs processed from Rtl1-as might have a role in tumorigenesis 56. Another example is the larger transcript, which in the mouse is named Mirg, that
contains 40 miRNAs clustered downstream of the snoRNAs 14. Some of these miRNAs are associates
with functions in mitochondrial metabolism and in control of neonatal metabolic adaptation 57,58. All the
miRNAs are expressed in placenta, developing embryo and adult, where their expression is mostly restricted to the brain 55.
At last, this imprinted cluster contains tandemly repeated C/D snoRNA genes 59. C/D snoRNAs
are typically involved in 2’-O-methylation of spliceosomal RNAs (snRNAs) and/or ribosomal RNA (rRNAs) 60. It has also been reported that they can act in regulation of pre-mRNA alternative splicing,
mRNA abundance, activation of enzymes and be processed into shorter ncRNAs 61. These repeated
intron-encoded C/D snoRNAs belong to a transcription unit and most of them are arranged into two tandem arrays of 31 and 9 copies of related sequences. This unique transcription unit includes an enigmatic lncRNA named MEG8 lncRNA of unknown function and understudied 59. They are mainly
expressed in the brain and their function is not yet known 14.59.
Several indications suggested that MEG3 and the remaining maternal non-coding RNAs are part of a large polycistronic transcription unit initiated at the transcriptional start site of the MEG3 and then processed post-transcriptionally to generate individual intergenic transcripts 14. Consistent with this
notion, no typical promoter region beyond MEG3 promoter has so far been defined in this region and all these non-coding RNAs show a strikingly similar tissue-specific profile of expression 62.
Furthermore, deletion involving MEG3 promoter results in loss of all the others non-coding transcripts expression 57.
Imprinting regulation of DLK1-DIO3 domain
The main element involved in imprinting regulation at the DLK1-DIO3 domain is the intergenic DMR (IG-DMR), that act as an ICR and ensures the monoallelic expression of all genes in the domain. IG-DMR inherits the methylation mark in the paternal chromosome while the maternal chromosome is inherited in an unmethylated state (Figure 1.2) 63,64. A study by Lin et al. (2003) demonstrated that, in
mice, deletion of IG-DMR from the maternally inherited chromosome cause a maternal to paternal epigenotype switch, resulting in biallelic expression of paternally expressed imprinted genes and repression of the genes usually expressed from the maternal chromosome. In contrast, deletion of the paternal methylated IG-DMR, resulted in no significant alterations on imprinting expression 64 and has
been proposed to be the default state 64. How the maternal unmethylated IG-DMR regulates imprinting
at this locus remains to be fully understood. One possibility is that maternal IG-DMR displays enhancer features important for the activation of the MEG3 lncRNA during preimplantation development 57, and
this lncRNA could then be important for the inactivation of protein-coding genes (DLK1, RTL1 and
DIO3) in the maternal allele (discussed below).
In addition, others regulatory somatic DMRs are part of this domain. MEG3-DMR, whose methylation are hierarchically regulated by the IG-DMR, is an important example (Figure 1.2) 64. This
DMR spans the MEG3 promoter and its first exon and intron 14 and its methylation status dictates
whether MEG3 gene (and the other MEGs) are expressed: methylated MEG3-DMR results in silencing of MEG3, while unmethylated MEG3-DMR results in MEG3 expression . Another example is MEG8-DMR, which is located at intron 2 of the MEG8 gene (Figure1.2) 65,66. The function is yet unknown,
7 since there is no evidence that it can influence the monoallelic expression of the MEG8 gene 65. In
contrast to the other DMRs previously described, this DMR is unmethylated on the paternal inherited chromosome and methylated on the maternal homolog 66,65.
Although most of the different DMRs have been identified in this cluster, the precise mechanism(s) of imprinting regulation at the DLK1-DIO3 domain remains not fully understood.
Role of MEG3 in imprinting regulation and gene regulation in general
Based on the role of lncRNAs in other imprinting domains, it has already been postulated that one or more of the non-coding genes of DLK1-DIO3 domain could be the responsible for regulation of imprinting expression of the protein-coding genes. MEG3 lncRNA has been pointed out as possible candidate 14. Indeed, two Meg3 KO mice studies are congruent with this hypothesis since in both cases,
the deletion of Meg3 affects the expression of the neighboring imprinted genes 51,52. Furthermore, Sanli
et al., in a recent study of Meg3 KO deletions in mouse ESCs, suggested that maternal Dlk1 repression is controlled through Meg3 expression because these deletions caused Dlk1 loss of imprinting upon neuronal differentiation. Although all these results suggest a potential role for MEG3 lncRNA as a critical regulatory cis-element necessary for the silencing of protein-coding genes on the maternal allele, direct evidence for a role of the lncRNA molecule itself remains to be proved.
Beyond the possible MEG3 function as cis-regulator of imprinting, this lncRNA may also have some trans effects. In agreement with this idea, Mondal et al., previously showed that MEG3 can, through RNA-DNA triplex structures, bind to other genomic loci and regulate expression of those genes, many of which belongs to the TGF-β pathway. Curiously, MEG3 binding sites colocalize with bindings sites of components of the Polycomb repressive complex 2 (PRC2) 67, which is normally associated with
the stable maintenance of silencing states. Additionally, it was also described that, in vitro and in vivo,
MEG3 can interacts with components of PRC2, as lysine methyltransferase (KMT) Ezh2 68,69 and this
interaction might assist or mediated MEG3 lncRNA trans effects . However, how MEG3 acts mechanistically to execute its functions remains still to be deeply investigated.
Imprinting disorders associated with DLK1-DIO3 domain
In 1991, Temple et al. and Wang et al. described for the first time cases of Maternal Uniparental Disomy (UPD(14)mat) and Paternal Uniparental Disomy (UPD(14)pat) for chromosome 14, respectively 26,27. UPD consist on the inheritance of two homologous chromosomes or chromosomal
region from only one parent 70, caused by balanced Robertsonian translocations 71. Because of that, these
conditions were initially designated as UPD(14)mat and UPD(14)pat syndromes. However, more recently, other molecular causes such as epimutations (i.e., methylation anomalies) or microdeletions that affect the 14q32 imprinted region lead to similar UPD(14) phenotypes. Consequently, the names Temple syndrome (TS14) (OMIM 616222) and Kagami-Ogata syndrome (KOS14) (OMIM 608149) have been proposed and approved by the European Network for Human Congenital Imprinting Disorders (EUCID) 72,73. These syndromes result from opposite molecular changes involving, at least,
the IG-DMR and MEG3-DMR 65,70. Study of both syndromes has allowed to decipher regulation
mechanisms involved in the 14q32 imprinted region.
Temple Syndrome (TS14)
TS14 is characterized by pre- and postnatal growth retardation, muscular hypotonia, truncal obesity, feeding difficulties in early childhood and premature puberty. Facial characteristics include a
8 broad forehead and short nose with a wide nasal tip. Most patients have also small hands and feet and short stature 72. Until 2017, only 65 patients have been reported with this condition. These patients show
clinical features that overlap with other imprinted syndromes such as PWS and Silver-Russell syndrome (SRS). The majority of the patients were initially suspected to have one of these two syndromes instead of TS, thus many other cases might be underdiagnosed in clinical practice. Due to this, the real prevalence of TS14 in the general population remains unknown 74. It is therefore important that diagnosis
be based mainly on genetic analysis rather than clinical findings.
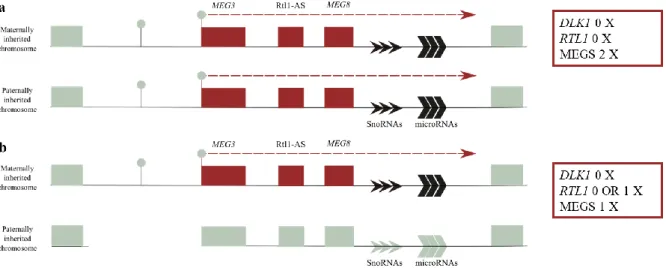
UPD(14)mat is the most frequently recognized cause of TS, accounting for 72% of the cases (Figure 1.3a), followed by epimutations (cause by hypomethylation) (19%) (Figure 1.3a) and by microdeletions (9%) (Figure 1.3b)74. Genotype–phenotype analysis in patients with microdeletions and
with TS14 phenotypes, including growth retardation, PWS/SRS-like features in infancy, and precocious puberty suggest that these characteristics are caused principally by absence of DLK1 expression.
Moreover, genome-wide studies and custom-genotyping arrays revealed that DLK1 is related with determination of menarchial age 75. Many TS phenotypes are also present in Dlk1 KO mice, which show
pre- and postnatal growth retardation and obesity 37, supporting a major role for DLK1 loss in TS.
Furthermore, in a few patients metabolic abnormalities were observed in agreement with previously described function of DLK1 as inhibitor of adipogenesis 74. RTL1 may also have a role for the
development of the TS phenotype judged by the defects on Rtl1 KO mice characterized growth retardation and placental hypoplasia 43. Nevertheless, it was described one case of a microdeletion
including only RTL1 but not the DMRs, which revealed an apparently normal phenotype, so further studies are essential to elucidate the importance of RTL1 for the TS14 phenotype 74.
Kagami-Ogata Syndrome (KOS14)
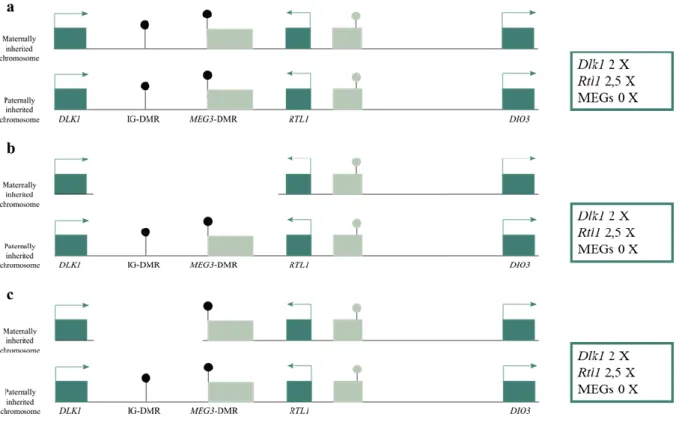
KOS14 patients exhibit a much more severe phenotype, characterized by polyhydramnios, placentomegaly, excessive birth weight, facial abnormalities, a characteristic small bell-shaped thorax, abdominal wall defects, variable developmental delay and/or intellectual disability, poor sucking usually requiring gastric tube feeding, hepatoblastoma and a mortality rate of 20–25 % in the neonatal period. This unique set of clinical features are shared between patients with UPD(14)pat (~65%), with epimutations (hypermethylations) affecting IG-DMR and/or MEG3-DMR (~15%) (Figure 1.4a) or
Figure 1.3 - Schematic representation of defects at the DLK1-DIO3 locus in TS14. Red rectangles represent imprinted noncoding RNAs transcripts and the grey rectangles represent the repressed alleles of imprinted genes. The DMRs are represented by circles: the grey circles represent unmethylated regions while black circles represent the methylated regions. a) UPD(14)mat or epimutations; b) Microdeletions - Rtl1 expression varies according to the location of microdeletion.
9 with microdeletions (~20%) (Figure 1.4b and c)73. This causes silencing of all the MEGs and
overexpression (twice or more) from PEGs.
It is also postulated that, cases of epimutations (hypermethylations) of maternal IG-DMR also results in maternal to paternal epigenotypic change (Figure 1.4a) 76.
Additionally, in KOS patients the RTL1 expression level becomes ~ 2.5 times increased in the absence of maternal functional RTL1-as, that act as a trans-acting repressor for RTL1. Furthermore, correlations between clinical features and deleted parts evidence that the excessive RTL1 expression seems to be a major role in development of the KOS(14) phenotype.
Interestingly, contrary to the expected given the function of DIO3, none of the patients has described as having hypothyroidism, which suggest that DIO3 is not an imprinted gene in humans 76.
Imprinting defects of DLK1-DIO3 domain in iPSCs and impact on pluripotency
As previously described, imprinting patterns are very well maintained in somatic cells, except in exceptional contexts such as, during cancer progression. In contrast during iPSC reprogramming, imprints have been shown to very sensitive to the massive epigenetic rewiring necessary for a differentiated cell to turn into a stem-like cell. Indeed, many imprinting defects, due to hypermethylation of the unmethylated parental alleles or hypomethylation of the methylated parental one, have been described in both mouse and human iPSCs 8,22,77,78, 79. In particular, the DLK1-DIO3 region have been
shown to be especially sensitive to imprinting errors, due to hypermethylation of the maternal allele in both mouse and human iPSCs 8,22,79 . In a 2010 paper, Stadtfeld et al. demonstrated that hypermethylation
of the IG-DMR was very frequent in miPSCs and causes abnormal silencing of the imprinted non-coding transcripts such as Meg3 and miRNAs expressed on the maternal mouse chromosome 12 8. Furthermore,
Figure 1.4 - Schematic representation of defects at the DLK1-DIO3 locus in KO14. Blue rectangles represent imprinted protein-coding genes and the grey rectangles represent the repressed alleles of imprinted genes. The DMRs are represented by circles: the grey circles represent unmethylated regions while black circles represent the methylated regions. a) UPD(14)pat or epimutations; b) Microdeletions involving the IG-DMR and the MEG-DMR; c) Microdeletions involving the IG-DMR, but not the MEG3-DMR
10 the authors demonstrated that dysregulation of imprinting at this region affected the developmental potential of miPSCs. Indeed, miPSCs hypermethylated for the IG-DMR with repressed Meg3 contributed poorly to formation of chimeras and failed to support the development of entirely iPSC-derived animals (or all-iPSC mice), in contrast to iPSCs with normal imprinted expression at the
Dlk1-Dio3 region 8. Therefore, correct imprinting regulation of the DLK1-DIO3 is directly associated with
pluripotency potential in the mouse. Likewise, a study comparing human ESCs with normal or hypermethylated IG-DMR demonstrated that many genes related to nervous system development were downregulated upon dysregulation of imprinting at the DLK1-DIO3 locus. As a consequence, cells derived from hESCs with hypermethylated IG-DMR showed lower expression levels of stage-specific neural linage markers and reduce neurite formation when compared with cells with normal imprinting at this region 80. Therefore, correct imprinting of the DLK1-DIO3 region is important for correct
neuronal differentiation, at least, in vitro. Hypermethylation of the IG-DMR causes biallelic expression of protein-coding genes and complete silencing of the MEGs. The specific role played by each of these transcriptional alterations for these phenotypes of reduced pluripotency and differentiation potential is unknown. It will be interesting to understand whether, for example, silencing of MEG3, which has been proposed to regulate imprinting of the protein-coding genes, is the driving force of these problems found in miPSCs and hESCs.
Aims of the study
Previous studies indicate that MEG3 lncRNA, like other lncRNAs in imprinted domains, can be the responsible for imprinting regulation of gene expression, such as, the specific silencing of DLK1 on the maternal chromosome 68. However, all these studies were based on the use of KO approaches, with
other DNA sequences being deleted in addition to MEG3 gene, which does not allow to clarify the role of MEG3 transcript in imprinting regulation. Thus, the aim of this study is to determine directly the role of MEG3 lncRNA on imprinting of DLK1-DIO3 domain, through modulation of MEG3 RNA specifically, without affecting its genomic locus in human cells, including hiPSCs. Furthermore, dysfunction of imprinting regulation at the DLK1-DIO3 region is known to affect pluripotency , and the lack of MEG3 has been suggested to play a role in it 8,80. Therefore, in this dissertation we also want to
evaluate the impact of MEG3 on the pluripotency capacity of iPSCs.
11
2. Materials and Methods
Biological Samples and tissue culture
Fibroblasts
Dermal fibroblasts from punch-skin biopsies of a 30 years old woman were kindly provided by Dr. Sofia Duarte (iMM/Centro Hospitalar de Lisboa Central), the biopsies were collected in accordance with European and National ethical regulation and approved by the Ethical Committee of Instituto de Medicina Molecular João Lobo Antunes (iMM) and Hospital de Santa Maria. Written informed consent was obtained from fibroblasts donors.
Dermal fibroblasts cryopreserved in liquid nitrogen and in vials were thawed from for 1-2 minutes (min) in a water bath at 37ºC and transferred to a previously prepared Falcon tube (Sarstedt) containing 5ml of fibroblast medium: DMEM (Life Technologies) supplemented with 10% of Fetal Bovine Serum (FBS; Life Technologies), 1mM of L-Glutamine (Life Technologies), and 1% of Penicillin/Streptavidin (P/S; Life Technologies). Then the Falcon tube was centrifuged at 1000 rpm for 3 minutes and the supernatant was discarded. Cells were then resuspended in 5ml of fibroblast medium and seeded in T25 flasks (Thermo Fisher Scientific), previously coated with 0.1% gelatine (Sigma-Aldrich).
When the fibroblasts reached 80-90% confluency, they were passed using TrypLE™ Express (recombinant cell-dissociation enzymes); Life Technologies) solution. For passassing, the culture medium was removed from the T25 flasks and cells were washed once with phosphate-buffered saline (PBS, Sigma-Aldrich), followed by incubation for 3-5 minutes with TrypLE™ Express solution at 37ºC. Cells were then resuspended by adding fibroblast medium and seeded in a gelatine-coated T75 flask (Sarstedt). When the cells reached 80-90% confluency, they were again passed using the same procedure or were collected for DNA and RNA extraction.
For freezing the cells were dissociated from the flask surface with TrypLE™ Express using the same strategy as described as above. Then cells were resuspended by adding fibroblast medium and transferred into a Falcon tube which was posteriorly centrifuge at 1000 rpm for 3 min. The supernatant was discarded, and the cells were resuspended in Fibroblast Freezing Medium: 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich) in FBS. Resuspended cells were after transferred to a cryovial (Nunc) (two cryovials per one T75 flask) and placed in a cooler at -80°C over-night. After this, cells can be stored in liquid nitrogen.
hiPSC
iPSC reprogramming of fibroblasts was performed by Isabel Onofre and Dr. Ana Rita Álvaro in the laboratory of Professor Luis Pereira de Almeida at CNC/UC, according a previously published protocol 81. Subsequently, several independent isogenic iPSC lines from fibroblasts were obtained and
isolated and, in this study, we used four of them named: iPSCs B, C, D and E.
To thaw these cells from cryopreserved vials, a Falcon tube containing 10 ml washing medium: DMEM-F12 (Life Technologies) supplemented with 10% of KnockOut Serum Replacement (Life Technologies, 1% of nonessential aminoacids, 1mM of L-Glutamine, 0.1mM of β-Mercaptoethanol (Life Technologies) and 1% of P/S (Life Technologies) was pre-warmed to 37ºC. Pre-warmed washing medium was added drop-wise to the cryovial containing the cells and defrosting cell suspension was transfer for a second falcon tube which was then centrifuged at 1000 rpm for 3 min at room temperature (RT): The supernatant was discarded and the cell pellet was resuspended in 1.5 ml of warm mTeSR1
12 medium [mTeSR™1 (STEMCELL Technologies) supplemented with 0.5% of P/S] and plated in one well of 6-well plate (TPP), previously coated with matrigel (Corning). For matrigel coating, matrigel was diluted in DMEM/F12 (1:30) and then used to coat the wells of a 6-well plate which was incubated at RT for at least 2 hours before use. This matrigel solution was removed before to cell seeding. mTeSR1 medium was changed daily.
When colonies reached ~75% confluency, cells were passed using an enzyme-free method: cells were washed with 1.5 ml of PBS and then incubated with 1 ml PBS-EDTA (1.0 ml of 0.5mM EDTA (VWR) in PBS) for 3 min to allow cells to become less attached to the surface of the well. PBS-EDTA was then removed and mTSER1 medium was added to the cells. Cells were scraped from the well using a cell scraper but taking care not to get single-cell suspensions and transferred to a Falcon tube with additional mTeSR1 medium according to the desired dilution (typically 1 in 3 or 1 in 4). Cells were then seeded in the wells of a 6-well plates, also previously coated with matrigel and mTeSR1 medium was changed daily.
For cryopreservation, a non-enzymatic EDTA-based procedure to detach the cells was also used. After incubation and removal of PBS-EDTA, the cells were scraped from the well with 1.5 ml of Washing Medium [DMEM-F12 supplemented with 10% of KnockOut Serum Replacement, 1% of nonessential aminoacids, 1mM of L-Glutamine, 0.1mM of β-Mercaptoethanol (Life Technologies) and 1% of P/S] to collect cells. Then the cell suspension was transferred to a Falcon tube and centrifuged at 1000 rpm for 3 min. The supernatant was discarded, and the cell pellet was carefully resuspended in iPSC Freezing Medium (10% DMSO (Sigma-Aldrich) in Knockout Serum Replacement) to avoid formation of single cells. Resuspended cells were then transferred to a cryovial and placed in a cooler at -80ºC over-night. After this, cells were stored in liquid nitrogen.
Antisense LNA GapmeRs Transfections
For loss-of-function studies of MEG3 lncRNA was used Antisense Locked Nucleic Acids (LNA) GapmeRs, which are an efficient tool for silencing of nuclear lncRNAs. The custom design of specific LNAs against exon 3 of MEG3 and control (Table 2.1), was generated by an online LNA GapmeRs design program for an optimal performance.
Table 2.1- Antisense LNAs GapmeRs for MEG3
Antisense LNA GapmeR MEG3 Sequence
LNA Meg1 (QIAGEN, 339511 LG00197781-DDA) GTAAGACAAGCAAGAG
LNA Meg2 (QIAGEN, 339511 LG00197782-DDA) CTCACTAGGGCATTGG
LNA Control (QIAGEN, 339511 LG00000002-DDA) AACACGTCTATACGC
For LNA transfection, we use RNAiMAX kit (Invitrogen) and followed the manufacturer’s instructions. Briefly, fibroblasts and iPSCs were cultured to 60-80% confluency on gelatine and Matrigel coated 6-well plates, respectively.
The mix solutions A (Optimem + lipofectamine RNAiMax) and B (Optimem + 25µM LNA GapmeRs) were prepared and then were incubated for 5 min at RT. After incubation, the solution A was gently transferred to solution B and then the solution A+B was incubated again for 20 min at RT. The solution A+B was added to the cells that were incubated overnight at 37º. In the second day, the medium of the cells was changed, and this procedure was repeated. On the third, cells were harvested for future RNA extraction and/or FISH.
13
Molecular Biology Techniques
Allelic Specific Imprinting Assessment
DNA extraction
For genomic DNA extraction, iPSCs B, C and D were pelleted after detachment through cell scrapping or TrypLE™ Express. Cells were centrifuged for 3 min at 1000 rpm and supernatant was discarded. Then the cells were resuspended in 1ml of PBS, transferred to an Eppendorf tube and centrifuged again for 3 minutes at 1000 rpm. The supernatant was discarded, and the pellet was stored at -80ºC, if not used immediately.
Then, cell pellet was resuspended in 500μl of lysis buffer [100mM NaCl (NZYTech), 10mM Tris pH8.0 (Sigma-Aldrich), 25 mM EDTA pH8.0 and 0.5% SDS (NZYTech) with freshly added 0.2 μg/μl of Proteinase K (Bioline)] and incubated overnight at 56ºC. On the next day was added 55μl of 3M sodium acetate pH5.2 (Merck Millipore) and 500μl of UltraPure™ Phenol:Cloroform:Isoamyl Alcohol (25:24:1, v/v; Invitrogen). The samples were mixed and centrifuged for 10min at maximum speed at RT and the top phase was carefully transferred to a new Eppendorf tube. Then was added 500μl of chloroform (Merck Millipore) and strongly shaked. Samples were centrifuged once more with same conditions and the top phase was again transferred to a new eppendorf tube. Subsequently was added 450μl of isopropanol (Merck Millipore) to the samples which was next mixed by inverting and incubated at -20ºC for at least 1h. After incubation, samples were centrifuged for 30min at maximum speed at RT. The supernatant was discarded and 700μl of 70% ethanol was gently added to the DNA pellet formed after centrifugation (Merck Millipore). Samples were then centrifuged for 5min at maximum speed at RT and the supernatant was discarded. A short spin was performed to remove the remaining supernatant and the pellets were open air dried for 20 min at RT. The pellets were resuspended in 200μl of DNase/RNase-free water and incubated at 37ºC for 3h for the DNA pellet to be well resuspended and quantification and purity evaluation were measured through spectrophotometer Nanodrop2000 (ThermoScientific).
DNA Methylation Analysis
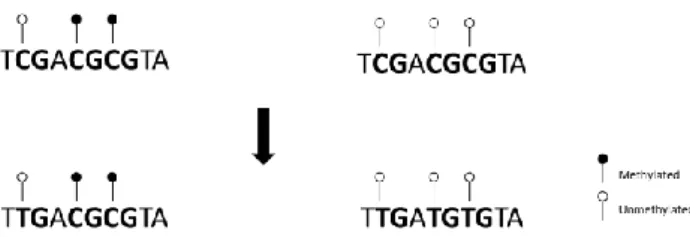
In order to analyze DNA methylation, genomic DNA was treated with sodium bisulfite, which converts all unmethylated cytosines into uracils, while leaves methylated cytosines residues intact (Figure 2.1). Bisulfite treatment was performed with the EZ DNA Methylation-Gold™ kit (Zymo Research), according the manufacturers’ instructions. This procedure consists in a conversion step in a thermal cycler, followed by use of spin columns to clean and desulphonate DNA. In the end, 11 μL of Bisulfite treated DNA were obtained for each sample. These treated DNA samples were then amplified by nested PCR reactions designed for the specific regions.
Figure 2.1 – Bisulfite conversion process. Bisulfite treatment converts all unmethylated cytosines into uracils, which are detected as thymines after PCR reaction. Methylated cytosines remain intact.