Article
J. Braz. Chem. Soc., Vol. 22, No. 8, 1439-1445, 2011. Printed in Brazil - ©2011 Sociedade Brasileira de Química
0103 - 5053 $6.00+0.00
A
*e-mail: [email protected]
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides
Hélio A. Stefani,*,a Jesus M. Pena,a Julio Zukerman-Schpectorb and
Edward R. T. Tiekinkc
aFaculdade de Ciências Farmacêuticas, Universidade de São Paulo, São Paulo-SP, Brazil
bDepartamento de Química, Universidade Federal de São Carlos, 13565-905 São Carlos-SP, Brazil cDepartment of Chemistry, University of Malaya, Kuala Lumpur 50603, Malaysia
Neste artigo é descrita a síntese de sistemas diínicos conjugados contendo substituintes elétron-atratores e elétron-doadores via a deteluração catalizada por paládio de bis-(ariletinil)teluretos e bis-(alquiletinil)teluretos. Este procedimento foi realizado sob condições atmosféricas em DMF usando Pd(Oac)2 como catalisador e AgOAc como um aditivo na presença de trietilamina. Esta
rota oferece acesso eiciente a sistemas diínicos conjugados em um curto período de tempo. A estrutura cristalográica por difração de raios X do telureto de bis(p-toluiletinila) e a conformação no estado sólido mostram uma cadeia supramolecular alinhada ao longo do eixo b, sustentada por interações CH…π.
The synthesis of symmetric conjugated diyne systems with withdrawing or electron-donating substituents via a palladium-catalyzed detelluration of bis(arylethynyl)tellurides and bis(alkylethynyl)tellurides is described. This procedure is effected under atmospheric conditions in DMF using Pd(OAc)2 as a catalyst and AgOAc as an additive in the presence of triethylamine.
This route offers eficient access to conjugated diyne systems in short reaction time. X-ray crystallographic structure and solid-state conformation of bis(p-tolylethynyl)telluride show a supramolecular chain aligned along the b axis, sustained by C-H…π interactions.
Keywords: tellurium, bis(phenylethynyl)tellurides, bis(alkynylethynyl)tellurides, detelluration, diynes, palladium
Introduction
Organotellurium compounds play an important role in organic synthesis, and they have received considerable attention because of their potential availability and useful biological activity.1 As reported by Bergman and co-workers2 more than three decades ago, aryltelluriums undergo detelluration upon treatment with degassed Raney nickel to afford biaryl compounds. Although the reaction is interesting and synthetically useful, the necessity of more than a stoichiometric amount of the required metal is still a serious drawback. However, attempted transition metal-catalyzed detellurations have been unsuccessful to date.2,3 Compounds containing chains of conjugated triple bonds4,5 are of paramount importance as versatile and useful building blocks in organic synthesis. Among these compounds, 1,3-butadiynes6 have been prominently
utilized as substructures in the formation of valuable intermediates for natural products7 and pharmaceuticals such as antitumor,8 antibacterial,9 anti-inlammatory,10 and antifungal agents.11
These conjugated diynes also serve as the core functional group in organic molecular materials such as linearly σ-conjugated acetylenic oligomers and polymers,12 macrocycles13 (Figure 1), and supramolecular scaffolds.14 Oxidative dimerization of sp-hybridized terminal alkynes mediated by Cu(I) or Cu(II) salts under either catalytic or stoichiometric conditions is the most commonly used synthetic methodology for obtaining symmetrically substituted 1,3-butadiyne.
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc. 1440
Results and Discussion
Herein, we describe a convenient protocol for the synthesis of symmetrical conjugated diynes through the palladium-catalyzed detelluration of functionalized bis(arylethynyl)tellurides and bis(alkylethynyl) tellurides at room temperature in presence of air (Scheme 1).
The approach to preparing symmetrical conjugated diynes 2a-j was based on a palladium-catalyzed detelluration reaction of functionalized bis(phenylethynyl)tellurides and bis(alkylethynyl) tellurides 1a-j. The parent precursors bis(arylethynyl)telluride 1a-j were conveniently prepared in good to moderate yields according to the procedure described by Engman and Stern.19
We initially optimized the conditions for the detelluration of functionalized bis(phenylethynyl)telluride 1. To ind optimal conditions for the detelluration reaction, bis(phenylethynyl) telluride 1a was selected as a model substrate, and a variety of catalysts were screened as described in Table 1. All reactions were monitored by TLC and GC/MS.
We initially surveyed palladium catalysts for use in this detelluration reaction. A comparison of different palladium catalysts such as PdCl2(dppf)•CH2Cl2, Pd(OAc)2, PdCl2PEPPSI-iPr, PdCl2, and PdCl2(PhCN)2 (72%, 52%, 50%, 31% and 46% yields, respectively). Although the best yield (72% yield) was obtained using PdCl2(dppf). CH2Cl2, we choose Pd(OAc)2 as catalyst due its availability in our laboratory. No product formation was observed using catalysts such as Fe(acac)2, CuCl and NiCl2(dppe) (entries 8-10). The reaction also did not proceed in the absence of a Pd catalyst (Table 1, entry 1).
One equiv. of AgOAc was used as an additive, along with 4 equiv. of triethylamine, and MeOH as solvent.
After the determination of the optimal catalyst for this transformation, we then studied the inluence of the base. In our initial attempts, we used triethylamine, and the desired compound was obtained in 72% yield. We also attempted the same reaction with some other organic and inorganic bases such as NaOAc, Cs2CO3, K2CO3, DIPEA, pyridine and cyclohexylamine, obtaining the detelluration product in yields ranging from 27% to 67%.
To further determine the optimal conditions for the detelluration reaction, we performed the model reaction in various solvents. When MeOH, 1,4-dioxane, toluene and CH3CN were employed, the reaction yields were poor or moderate (52%, 44%, 38% and 58%, respectively). While
Figure 1. Structure of diyne macrocycles. t-Bu t-Bu Dec Dec Dec Dec
Scheme 1. Synthesis of conjugated diynes.
R Te R Pd, AgOAc R R
base, solvent air, r.t.
1a 2a
Table 1. Effect of a catalyst on the detelluration reaction of bis(phenylethynyl)telluride 1a
Te
Catalyst 10 mol %, AgOAc
Et3N, MeOH, r.t.
1a 2a
entry Catalysta Yield / (%)b
1 - nr
2 PdCl2(dppf).CH2Cl2 72
3 PdCl2PEPPSI-iPr 50
4 Pd(OAc)2 52
5 PdCl2 31
6 PdCl2(PhCN)2 46
7 Fe(acac)2 nr
8 CuCl nr
9 NiCl2(dppf) nr
Reactionconditions: bis(phenylethynyl)telluride 1a (1 equiv.), catalyst (10 mol %), Et3N (4 equiv.), AgOAc (1.0 equiv.), MeOH (5 mL), r.t. a10 mol % of
Stefani et al. 1441 Vol. 22, No. 8, 2011
using DMSO as solvent provided a good yield (72%), DMF provided the best yield of the desired product (86%).
From these studies, it was determined that a reaction mixture containing 1.0 equiv. of bis(phenylethynyl)telluride 1a, 4 equiv. of Et3N, 1 equiv. of AgOAc, and 10 mol % of Pd(OAc)2 in 5 mL of DMF at room temperature stirred under atmospheric conditions for 60 min provided the best conditions for the synthesis of conjugated diyne 2a. To demonstrate the eficiency of this detelluration reaction, we then explored its generality with a variety of bis(arylethynyl)tellurides and bis(alkylethynyl)tellurides. The results are summarized in Table 2.
After optimizing the conditions for the synthesis of symmetrical conjugated diyne 2a, it was synthesized a series of conjugated diynes (2a-j) in 21-86% yields (see Table 2 and Experimental). The reaction was carried out at room temperature. The reaction proceeded with electron-withdrawing substituents attached to the alkynyltelluride and with electron-donation substituents. All of the obtained products provided 1H and 13C NMR spectra that were in full agreement with their assigned structures.
On the basis of available literature20 we propose a possible catalytic cycle for the detelluration reaction of
Table 2. Detelluration reaction of bis(arylethynyl)tellurides and bis(alkylethynyl)tellurides
Te
Pd(OAc)2 10 mol %, 1.0 equiv. AgOAc
4 equiv. Et3N, DMF, air, r.t., 60 min
R R R R
1a-j 2a-j
Bis(alkynyl)tellurides Diynes Yielda / (%)
1a Te 2a 86 1b Te F F
F Te F
F F
F F
F 2b F
F F 60 Te F F 1c F F 2c 65
Te CH3
H3C
1d
CH3 H3C
2d 38 Te F F 1e F F 2e 52 Te S S 1f S S 2f 44
Te OCH3
H3CO
1g
OCH3 H3CO
2g
39
Te (CH2)4CH3 H3C(H2C)4
1h (CH2)4CH3 H3C(H2C)4 2h 47 Te O O
1i O 2i O
65
Te
1j
2j
89b
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc. 1442
bis(arylethynyl)tellurides and bis(alkylethynyl)tellurides as described in Figure 2.
According to this cycle the reaction proceeds by the formation of Pd(II) complex with acetylene followed by the conversion of this intermediate into another palladium species B, which leads the formation of conjugated diyne C along the reduction of the Pd(II) complex to Pd(0). The palladium species is later oxidized in the presence of O2 to give the initial Pd(II) species completing the cycle.
Due to our ongoing interest in tellurium structures, especially those involving π-interactions, the crystal and molecular structure of the bis-(p-tolylethynyl)telluride starting material was determined.21 The tellurium atom is located on a crystallographic twofold axis with the C-Te-C angle being 92.23 (15)°. The dihedral angle formed between the phenyl rings is 87.27 (7)° (Figure 3).
In the crystal structure, the telluride molecules are connected into supramolecular chains along the b axis via C-H...π interactions, as shown in Figure 4.
Conclusions
In summary, we demonstrated the synthesis of functionalized symmetrical 1,3-diyne systems through the palladium-catalyzed detelluration reaction of bis(arylethynyl) tellurides and bis(alkylethynyl)tellurides. The use of this methodology for the synthesis of more complex polyacetylenic compounds is currently under study in our laboratory.
Experimental
Proton nuclear magnetic resonance spectra (1H NMR) were obtained at 300 MHz. Spectra were recorded in CDCl3 solutions. Chemical shifts are reported in ppm, referenced to the solvent peak of CDCl3 or tetramethylsilane (TMS) as the external reference. Data are reported as follows: chemical shift (d), multiplicity, coupling constant (J) in Hertz and integrated intensity. Carbon-13 nuclear magnetic resonance spectra (13C NMR) were obtained at 75 MHz. Spectra were
Figure 2. Possible catalytic cycle of the homocoupling reaction of alkynylditellurides.
LnPd(OAc)2
L2Pd
Ar
Ar Pd(0)
L2Pd(II) OAc Ar Ar Te Ag 2 AcOTe Ar AcOTe Ar
(AcO)2Te Ag + Ar Ar O2 Pd O O 2HOAc
H2O2 Pd H2O + O2
deteluration product transmetalation
reductive elimination A B C D transmetalation
Figure 3. Molecular structure of bis(p-tolylethynyl)telluride showing numbering scheme and displacement ellipsoids at the 70% probability level. Symmetry operation i: -x, y, 3/2-z.
Stefani et al. 1443 Vol. 22, No. 8, 2011
recorded in CDCl3 solutions. Chemical shifts are reported in ppm, referenced to the solvent peak of CDCl3. Abbreviations to denote the multiplicity of a particular signal are s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sex (sextet) and m (multiplet). Column chromatography was performed using silica gel (230-400 mesh) following the methods described by Still et al.22 Thin layer chromatography (TLC) was performed using silica gel F254, 0.25 mm thickness from Merck. For visualization, TLC plates were either placed under ultraviolet light, or stained with iodine vapor, or acidic vanillin. The following solvents were dried and puriied by distillation from the reagents indicated: tetrahydrofuran from sodium with a benzophenone ketyl indicator. All other solvents were ACS or HPLC grade unless otherwise noted. Air and moisture-sensitive reactions were conducted in lame-dried or oven dried glassware equipped with tightly itted rubber septa and under a positive atmosphere of dry nitrogen or argon. Reagents and solvents were handled using standard syringe techniques. Temperatures above room temperature were maintained by use of a mineral oil bath with an electrically heated coil connected to a controller.
General experimental procedure for preparing conjugated diyne compounds 2a-j
A suspension of bis(phenylethynyl)telluride (1a) (0.0824 g, 0.25 mmol), PdOAc2 (5.6 mg, 10% mol), triethylamine (0.101 g, 1 mmol) and silver(I) acetate (0.041 g, 0.25 mmol) in 5 mL of DMF was stirred at room temperature under air for 60 min. The reaction mixture was then diluted with ethyl acetate (25 mL), and the organic layer was washed with a saturated solution of NH4Cl (2 × 10 mL) and water (2 × 10 mL), dried over MgSO4 and concentrated under vacuum. The crude product was puriied by lash silica column chromatography using hexane as the eluent and subsequently characterized.
Bis(phenylethynyl)telluride (1a)19
Yield 86%; 1H NMR(300 MHz, CDCl
3) d (ppm) 7.30-7.38 (m, 6H), 7.47-7.56 (m, 4H). 13C NMR (CDCl
3, 75.5 MHz,) d(ppm) 73.7; 81.3; 121.6; 128.4; 129.5; 132.3; EM m/z (%) 202 (100), 200 (24), 150 (8), 101 (13), 88 (10).
1,4-Bis(2,5-luorophenylethynyl)telluride (1b)
Red solid; mp 187-188 °C. Yield 60%; 1H NMR(CDCl 3, 300 MHz) d(ppm)6.84 (dd, 1H, J 5.2 Hz, J 13.0 Hz) 7.45 (dd, 1H, J 8.0 Hz, J 15.3 Hz). 13C NMR (CDCl
3, 75.5 MHz) d(ppm)165.4 (d, J 11.9 Hz, C-F), 161.8 (d, J 46.4 Hz, C-F), 135.1 (d, J 2.3 Hz, C-F), 1 1.76 (d, J 3.7 Hz, C-F), 108.0 (d, J 4.2 Hz, C-F),104.8 (d, J 19 Hz, C-F), 48.6 (2C), 29.4 (2C).125Te NMR (CDCl
3, 95 MHz) d(ppm)374.1.
EM m/z (%) 274.2 (100), 275.2 (16), 138.2 (15), 223.2 (6). IR (KBr) ν
max/cm
-1:3440,3099, 2139, 1613, 1586, 1142,
1096, 967, 855, 779, 729, 512, 498, 485. HRMS (ESI) calc. for C16H6F4TeNa
+ 426.9365; found 426.9368.
1,3-Bis(m-luorophenylethynyl)telluride (1c)
White solid; mp 129-130 °C. Yield 58%; 1H NMR(CDCl 3, 300 MHz) d(ppm)6.96-7.14 (m, 8H). 13C NMR (CDCl
3, 75.5 MHz ) d(ppm)44.5 (2C); 111.5 (2C); 116.4 (d, J1 21.1 Hz, C-F); 118.8 (d, J2 22.8 Hz, C-F); 124.4 (d, J3 9.36 Hz, C-F); 129.9 (d, J4 3.03 Hz, C-F); 129.8 (d, J5 8.64 Hz, C-F); 162.2 (d, J 6 245.5 Hz, C-F). 125Te NMR(CDCl
3, 95 MHz) d(ppm) 374.1. EM m/z (%) 238.1 (100), 236.1 (13), 218.1 (6), 217.2 (5), 168.2 (4), 119.2 (16). IR (KBr) ν
max/cm -1: 3440,
3067, 2143, 1604, 1581, 1484, 1423, 1266, 1136, 941, 872, 784, 678, 571, 517, 457. HRMS (ESI) calc. for C16H8F2TeNa
+
390.9553; found 390.9538.
1,4-Bis(p-tolylphenylethynyl)telluride (1d)23
Yield 52%; 1H NMR(CDCl3, 300 MHz) d (ppm) 2.35 (s, 6H); 7.40 (d, J 7.8 Hz, 4H); 7.12 (d, J 7.8 Hz, 4H); 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 21.6 (2C); 73.5 (2C); 81.6 (2C); 118.8 (2C); 129.2 (4C); 132.4 (4C); 139.5 (2C); EM m/z (%) 230 (100); 215 (17); 115 (17); 101 (15).
1,4-Bis(p-luorophenylethynyl)telluride (1e)
White solid, mp 129-130 °C. Yield 51%; 1H NMR (CDCl3, 300 MHz) d (ppm) 6.91 (t, J
1 and J2 8.52 Hz,
4H); 7.45 (dd, J1and J2 5.37 Hz, 4H). 13C NMR (CDCl 3, 75.5 MHz) d (ppm) 42.7 (2C); 111.6 (2C); 115.6 (d, J1 22.0 Hz, C-F); 118.9 (d, J2 3.5 Hz, C-F); 134.2 (d, J3 8.5 Hz, C-F); 162.9 (d, J4 249.3 Hz, C-F). 125Te NMR(CDCl
3, 95 MHz) d (ppm) 364.5. EM m/z (%) 264.2 (100), 265.1 (33), 249.2 (32), 233.6 (6), 231.4 (6), 221.1 (41), 220,2 (95), 201.5 (5.75), 200,4 (6) 132.3 (13), 117.1(5), 116.1 (8). IR (KBr) ν
max/cm
-1: 3443, 3099, 3080, 2143, 1595, 1503,
1480, 1224, 1209, 1150, 1096, 838, 749, 533. HRMS (ESI) calc. for C16H8F2TeNa
+ 390.9553, found 390.9538.
1,4-Bis(3-thyenyletynyl)telluride (1f)
White solid, mp 100-101°C. Yield 67%, 1H NMR (CDCl3, 300 MHz) d (ppm) 7.14 (dd, J
1and J2 1.14 Hz,
2H); 7.25 (t, J 3.0 Hz, 2H); 7.57 (dd, J1 1.2 Hz and J2 1.14 Hz, 2H). 13C NMR (CDCl
3, 75.5 MHz) d (ppm) 42.7 (2C); 107.6 (2C); 122.1 (2C); 125.2 (2C); 130.1 (2C); 130.8 (2C). 125Te NMR (CDCl
3, 95 MHz) d (ppm) 364.4. EM m/z (%) 214.1 (100), 169,2 (19), 156.2 (4), 144.2 (6), 126.2 (4), 107.2 (11), 69.1 (6), 45.0. IR (KBr) ν
max/cm -1:
3431,3099, 2129, 1352, 1075, 940, 933, 865, 825, 783, 692, 670, 621, 582. HRMS (ESI) calc. for C12H6S2TeNa
+
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc. 1444
1,4-Bis(4-methoxyphenylethynyl)telluride (1g)23
Yield: 58%1H NMR (CDCl
3, 300 MHz) d (ppm) 7.37 (d, J 8.5 Hz, 4H); 6.67 (dd, J1 2.6 Hz and J2 8.5 Hz, 4H); 3.79 (s, 6H). 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 20.9 (2C), 55.2 (2C), 111.2 (2C), 115.0 (4C), 129,8 (2C), 134.0 (4C), 143.1 (2C), 160.1 (2C), EM m/z (%) 264.2 (100), 220.2 (95), 221.1 (41), 249.2 (32), 110.2 (21), 218.2 (10). IR (KBr) ν
max/cm
-1:3437,3005, 2984, 2958, 2936, 2903,
2891, 2838, 2824, 2154, 1463, 1440, 1432, 1427, 1348, 1273, 1187, 1082, 1069, 995, 976, 899, 880, 606, 590. HRMS (ESI) calc. for C18H14O2TeNa+, found 414.9851.
1,4-Bis(p-pentylphenylethynyl)telluride (1h)
Yellow solid; mp 57-58 °C. Yield: 50%. 1H NMR (CDCl3, 300 MHz) d(ppm) 0.89 (t, J 6.6 Hz, 6H); 1.33 (m, 8H); 1.60 (m, 4H); 2.61 (m, 4H); 7.14 (d, J 8.0 Hz, 4H), 7.39 (d, J 7.8 Hz, 4H). 13C NMR (CDCl
3, 75.5 MHz) d (ppm) 14.0 (2C); 22.5 (2C); 30.96 (2C); 35.9 (2C); 42.1(2C); 112.8 (2C); 120.1 (2C); 128.4 (2C); 132.1 (2C); 144.3 (2C). 125Te NMR(CDCl
3, 95 MHz) d (ppm) 357.6. EM m/z (%)285.3 (100), 342.3 (92), 228.1 (61), 343.3 (25), 226.1 (12), 41.1 (9). IR (KBr) ν
max/cm
-1:3435,3024,
2955, 2929, 2855, 2141, 1503, 1466, 842, 825, 815, 723, 572, 530. HRMS (ESI) calc. for C26H30TeNa
+ 495.1307;
found 495.1286.
1,4-Bis(methoxyethynyl)telluride (1i)
Yellow solid, mp 44-45 °C. Yield 78%. 1H NMR (CDCl3, 300 MHz) d(ppm) 4.32 (s, 4H); 3.39 (s, 6H). 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 40.2 (2C); 57.4 (2C); 60.6 (2C); 109.9 (2C). EM m/z (%)69.0 (100), 77.1 (14), 78.1(14), 63.1 (13), 51.1 (11), 53.0 (8).
1,4-Bis(butylethynyl)telluride (1j)24
Yellow oil; 1H NMR (CDCl
3,300 MHz) d (ppm) 0.90 (t, J 7.2 Hz, 6H); 1.36-1.57 (m, 8H); 2.50 (t, J 6.9 Hz, 4H).
13C NMR (CDCl
3, 75.5 MHz) d (ppm) 13.6 (2C); 20.7 (2C); 21.9 (2C); 30.8 (4C); 114.3 (2C).
General procedure to homocoupling reaction
A suspension of bis(phenylethynyl)telluride (1a) (0.0824 g; 0.25 mmol), Pd(OAc)2 (0.0056 g, 10% mol), triethylamine (0.101 g; 1 mmol) and silver acetate (0.041 g, 0.5 mmol) in 5 mL of DMF was stirred at room temperature, under air atmospheric by 60 min. The reaction mixture was diluted with ethyl acetate (25 mL), and the organic layer washed with NH4Cl (2 × 10 mL), dried over MgSO4 and concentrated under reduced pressure. The crude product was puriied by lash silica column chromatography using hexane as eluent and subsequently characterized.
1,4-Bis(phenylbuta-1,3-diyne (2a)25
Yield 86%. 1H NMR(CDCl
3, 300 MHz) d (ppm) 7.30-7.38 (m, 6H), 7.47-7.56 (m, 4H). 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 73.7; 81.3; 121.6; 128.4; 129.5; 132.3.
1,4-Bis(2,4-diluorophenyl)buta-1,3-diyne (2b)
White solid; mp 187-188 °C. Yield 60%. 1H NMR (CDCl3, 300 MHz) d (ppm) 6.83-6.90 (m, 4H); 7.54-7.46 (m, 2H). 13C NMR (CDCl
3, 75.5 MHz) d (ppm) 29.7 (2C); 74.8 (2C); 104.6 (d, J 1.3 Hz, C-F); 111.9 (d, J 25.7, C-F); 135.3 (d, J 9.9 Hz, C-F); 160.1 (d, J 196.0 Hz, C-F); 165.6 (d, J 53.8 Hz, C-F). IR (KBr) ν
max/cm
-1: 3439, 3101, 2955,
2924, 2854, 1730, 1609, 1582, 1496, 1468, 1435, 1299, 1268, 1143, 1098, 965, 857, 852, 812, 741, 710, 612, 601, 508, 475, 429.Microanalysis Calc. 70.25% C, 2.46% H; found 70.44% C; 2.54% H.
1,3-Bis(m-luorophenyl)buta-1,3-diyne (2c)26
Yield 65%; 1H NMR(CDCl
3, 300 MHz) d (ppm) 7.22-7.29 (m, 8H). 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 74.4 (2C); 80.6 (2C); 116.9 (d, J1 21.0 Hz, C-F); 119.2 (d, J2 22.9 Hz, C-F); 123.4 (d, J3 9.5 Hz, C-F); 128.4 (d, J 4 3.1 Hz, C-F); 130.1 (d, J 5 8.6 Hz, C-F); 162.2 (d, J 6 245.8 Hz, C-F).
1,4-Bis(p-tolyl)buta-1,3-diyne (2d)27
Yield 52%. 1H NMR(CDCl
3, 300 MHz,) d (ppm) 2.35 (s, 6H); 7.40 (d, J 7.8 Hz, 4H); 7.12 (d, J 7.8 Hz, 4H); 13C NMR (CDCl
3, 75.5 MHz,) d (ppm) 21.6 (2C); 73.5 (2C); 81.6 (2C); 118.8 (2C); 129.2 (4C); 132.4 (4C); 139.5 (2C).
1,4-Bis(p-luorophenyl)buta-1,3-diyne (2e)28
Yield 47%. 1H NMR(CDCl
3, 300 MHz) d(ppm) 7.03 (dd, 1H, J 7.8 Hz, J 9.7 Hz); 7.51 (ddd, J 2.2 Hz, J 5.3 Hz, J 6.9 Hz). 13C NMR (CDCl
3,75.5 MHz) d (ppm) 73.5 (2C); 80.4 (2C); 115.9 (d, J1 22.1 Hz, C-F); 117.8 (d, J2 3.56 Hz, C-F); 134.5 (d, J3 8.50 Hz, C-F); 163.0 (d, J4 250.1 Hz, C-F).
1,3-Bis(3-thyenyl)buta-1,3-diyne (2f)28
Yield 44%;1H NMR(CDCl
3, 300 MHz,) d(ppm) 7.12 (d, J 5.0 Hz, 2H); 7.25-7.28 (m, 2H); 7.58 (d, J 2.9 Hz, 2H).
13C NMR (CDCl
3, 75.5 MHz) d(ppm) 73.6 (2C); 121.0 (2C); 125.6 (4C); 131.2 (4C).
1,4-Bis(p-methoxyphenyl)buta-1,3-diyne (2g)28
Yield 39%; 1H NMR (CDCl
3, 300 MHz) d(ppm) 7.42 (d, J 8.5 Hz, 4H); 6.69 (dd, J1 2.5 Hz and J2 8.5 Hz, 4H); 3.80 (s, 6H). 13C NMR (CDCl
Stefani et al. 1445 Vol. 22, No. 8, 2011
1,4-Bis(p-penthylphenyl)buta-1,3-diyne (2h)29
Yield 38%; 1H NMR(CDCl
3, 300 MHz) d (ppm) 0.87 (m, 6H); 1.30 (m, 8H); 1.59 (t, J 7.0 Hz, 4H); 2.59 (m, 4H); 7.12 (dd, J1 3.8 Hz and J2 7.9 Hz, 4H); 7.40 (t, J 8.6 Hz, 4H). 13C NMR (CDCl
3, 75.5 MHz) d (ppm) 13.9 (2C); 22.5 (2C); 31.4 (4C); 35.9 (2C); 73.5 (2C); 83.9 (2C); 119.0 (2C); 128.5 (4C); 132.4 (4C); 144.4 (2C).
1,6-Dimethoxy-2,4-hexadiyne (2i)29
Yield 21%; 1H NMR(CDCl
3, 300 MHz) d(ppm) 3.39 (s, 6H); 4.18 (s, 4H). 13C NMR (CDCl
3, 75.5 MHz) d (ppm) 57.4 (2C); 59.8 (2C); 70.1 (2C); 75.1 (2C).
Supplementary Information
Supplementary data are available free of charge at http://jbcs.sbq.org.br as PDF ile.
Acknowledgments
The authors thank FAPESP (2007/59404-2) and CNPq for fellowships to H. A. S. and J. M. P. (300.613/2007-5 and 142.741/2008, respectively) for inancial support.
References
1. Patai, S.; Rappoport, Z.; The Chemistry of Organic Selenium and Tellurium Compounds, Wiley: Chichester, 1986, vol. 1; Irgolic, K. Y. In Houben-Weyl Methoden der Organischen Chemie; Müller, E., ed., 4th ed., vol. E12b, G. Thieme: Stuttgart,
1990; Petragnani, N.; Stefani, H. A. Tetrahedron2005, 61, 1613; Petragnani, N.; Stefani, H. A. In Tellurium in Organic Chemistry;Second, Updated and Enlarged Edition, Academic Press, Elsevier: Amsterdam, 2007.
2. Bergman, J.; Carlsson, R.; Sjöberg, B.; Org. Synth. 1977, 57, 18; Bergman, J.; Tetrahedron 1972, 28, 3323; Bergman, J.; Engman, L.; Tetrahedron 1980, 36, 1275.
3. Barton, D. H. R.; Ozbalik, N.; Ramesh, M.; Tetrahedron Lett. 1988, 29, 3533.
4. Stang, P. J.; Diederich, F.; Modern Acetylene Chemistry, VCH: Weinheim, Germany, 1995, p. 506; Patai, S.; The Chemistry of the Carbon-Carbon Triple Bond, Wiley-Interscience: London, 1978.
5. Damle, S. V.; Seomoon, D.; Lee, P. H.; J. Org. Chem. 2003, 68, 7085.
6. For general reviews on coupling reactions between sp-carbon centers, see: Sonogashira, K. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., eds.; Pergamon: Oxford,
1991, Vol. 3, pp. 551-561; Siemsen, P.; Livingston, R. C.; Diederich, F.; Angew. Chem., Int. Ed. 2000, 39, 2632. 7. Bohlmann, F.; Burkhardt, T.; Zdero, C.; Naturally Occurring
Acetylenes, Academic Press: London, 1973, p. 548.
8. Mayer, S. F.; Steinreiber, A.; Orru, R. V. A.; Faber, K.; J. Org. Chem. 2002, 67, 9115.
9. Stefani, H. A.; Costa, I. M.; Zeni, G.; Tetrahedron Lett. 1999, 40, 9215.
10. Zeni, G.; Panatieri, R. B.; Lissner, E.; Menezes, P. H.; Braga, A. L.; Stefani, H. A.; Org. Lett. 2001, 3, 819.
11. Stüts, A.; Angew. Chem., Int. Ed. 1987, 26, 320.
12. Diederich, F.; Rubin, Y.; Angew. Chem., Int. Ed. 1992, 31, 1101; Tour, J. M.; Chem. Rev. 1996, 96, 537; Martin, R. E.; Diederich, F.; Angew. Chem., Int. Ed. 1999, 38, 1350.
13. Kimball, D. B.; Haley, M. M.; Mitchell, R. H.; Ward, T. J.; Bandyopadhyay, S.; Williams, R. V; Armantrout, J. A.; J. Org. Chem. 2002, 67, 8798.
14. O’Connor, M. J.; Haley, M. M.; Org. Lett. 2004, 6, 2385. 15. Stefani,H. A.; Guarezemini, A. S.; Cella, R.; Tetrahedron 2010,
66, 7871.
16. Eglinton, G.; Galbraith, A. R.; J. Chem. Soc. 1959, 889. 17. Hay, A. S.; J. Org. Chem. 1962, 27, 3320.
18. Taylor, R. J. K.; Organocopper Reagents, Oxford University Press: New York, 1994, p. 352.
19. Engman, L.; Stern, D.; Organometallics 1993, 12, 1445. 20. Punniyamurthy, T.; Velusamy, S.; Iqbal, J.; Chem. Rev. 2005,
105, 2329.
21. Caracelli, I.; Zukerman-Schpector, J.; Pena, J. M.; Stefani, H. A.; Tiekink, E. R. T.; Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, o685.
22. Still, W.C.; Kahn, M.; Mitra, A.; J. Org. Chem. 1978, 43, 2923. 23. Murai, T.; Imaeda, K.; Kajita, S.; Kimura, K.; Ishihara, K.;
Kato, S.; Phosphorus, Sulfur Silicon Relat. Elem. 1992, 67, 239. 24. Citeau,G. A.; Giolando, D. M.; J. Organomet. Chem. 2001, 625,
23.
25. Coste, A.; Couty, F.; Evano, G.; Synthesis 2010, 9, 1500. 26. Jiang, H-F.; Liu, H-L.; Zhan, H-Y.; Zhou, L.; Chin. J. Chem.
2007, 25, 1413.
27. Chen, S-N.; Wu, W-Y.; Tsai, F-Y.; Green Chem. 2009, 11, 269. 28. Wang, D.; Li, J.; Li, N.; Gao, T.; Hou, S.; Chen, B.; Green
Chem. 2010, 12, 45.
29. Stefani, H. A.; Singh, F. V.; Amaral, M. F. Z. J.; Tetrahedron Lett. 2009, 50, 2636.
Submitted: November 11, 2010 Published online: April 5, 2011
Supplementary Information
S
I
J. Braz. Chem. Soc., Vol. 22, No. 8, S1-S20, 2011. Printed in Brazil - ©2011 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
*e-mail: [email protected]
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides
Hélio A. Stefani,*,a Jesus M. Pena,a Julio Zuckerman-Schpectorb and Edward R. T. Tiekinkc
aFaculdade de Ciências Farmacêuticas, Universidade de São Paulo, São Paulo-SP, Brazil
bDepartamento de Química, Universidade Federal de São Carlos, 13565-905 São Carlos-SP, Brazil
cDepartment of Chemistry, University of Malaya, Kuala Lumpur 50603, Malaysia
Table S1. Chemical shifts for 127Te
Tellurides 125Te NMR (d ppm, CDCl 3)
S
Te S
364.48
Te 332.57
Te
F
F
F
F
387.55
Te
F F 364.52
Te
F F
374.18
Te
O O 348.06
Te
C5H11 C5H11 357.60
Te 357.83
Te 362.78
All spectra were obtained using diphenyl ditelluride as internal standard. Te2Ph2
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S2
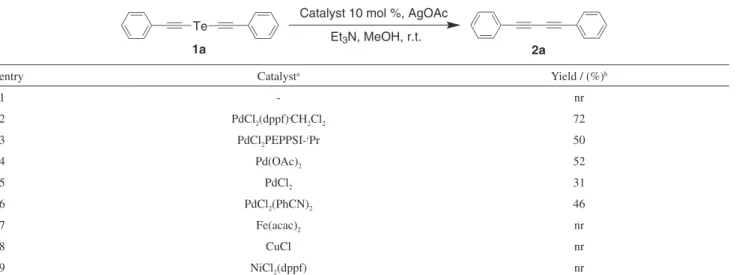
Figure S1. 1H NMR (CDCl
3,300 MHz) of 1a.
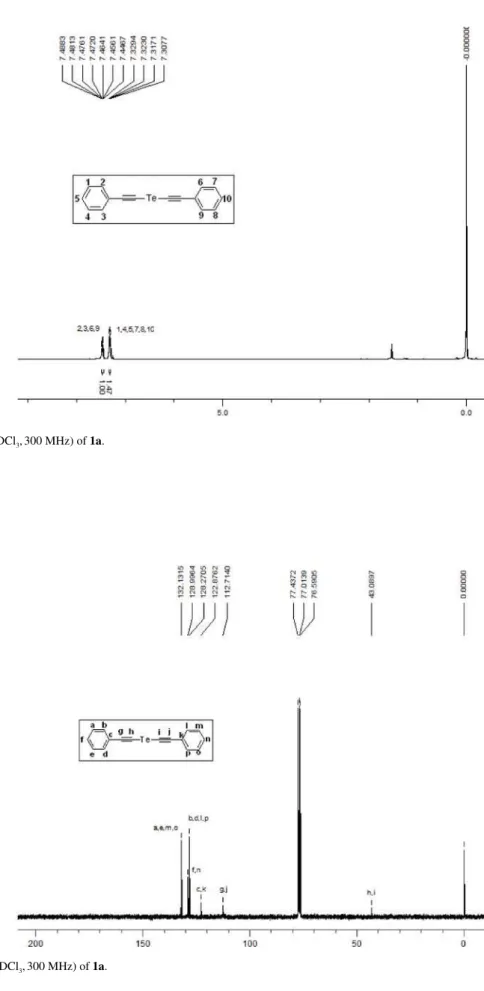
Figure S2. 13C NMR (CDCl
Stefani et al. S3 Vol. 22, No. 8, 2011
Figure S3. 1H NMR (CDCl
3,300 MHz) of 1b.
Figure S4. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S4
Figure S5. 1H NMR (CDCl
3,300 MHz) of 1c.
Figure S6. 13C NMR (CDCl
Stefani et al. S5 Vol. 22, No. 8, 2011
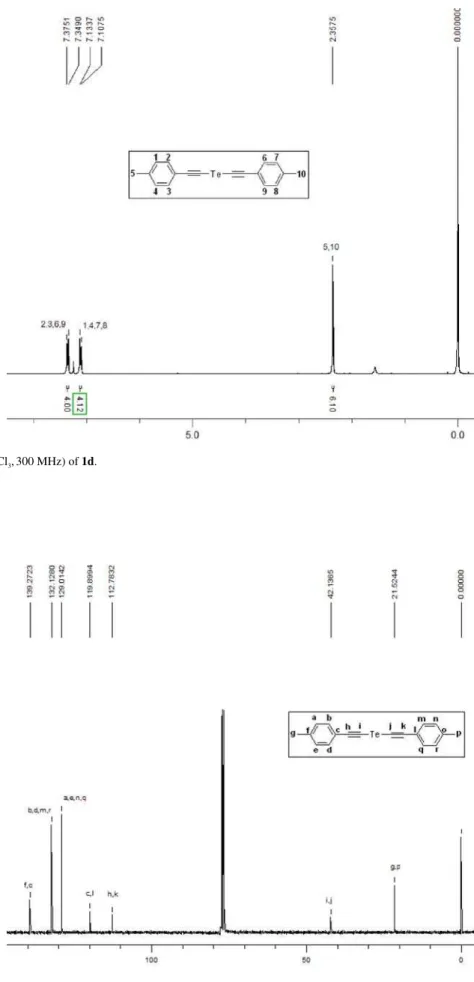
Figure S7. 1H NMR (CDCl
3,300 MHz) of 1d.
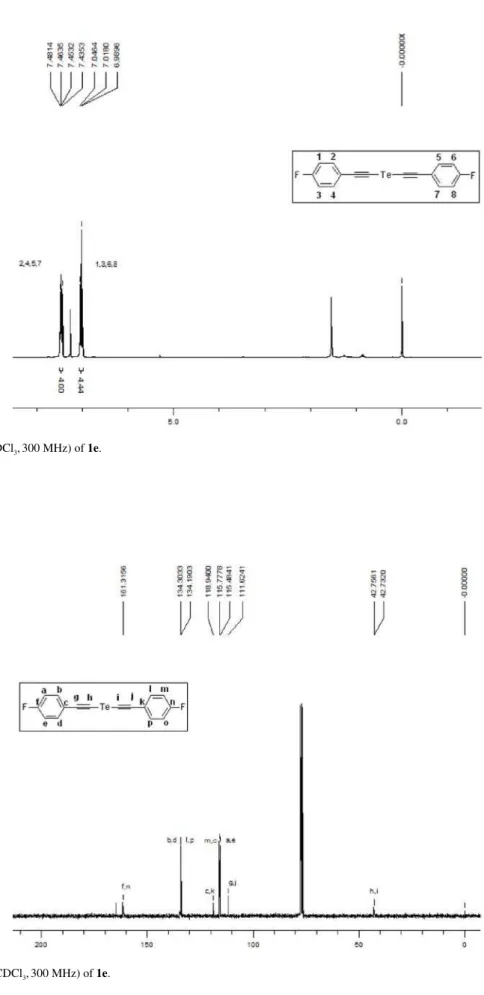
Figure S8. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S6
Figure S9. 1H NMR (CDCl
3,300 MHz) of 1e.
Figure S10. 13C NMR (CDCl
Stefani et al. S7 Vol. 22, No. 8, 2011
Figure S11. 1H NMR (CDCl
3,300 MHz) of 1f.
Figure S12. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S8
Figure S13. 1H NMR (CDCl
3,300 MHz) of 1g.
Figure S14. 13C NMR (CDCl
Stefani et al. S9 Vol. 22, No. 8, 2011
Figure S15. 1H NMR (CDCl
3,300 MHz) of 1h.
Figure S16. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S10
Figure S17. 1H NMR (CDCl
3,300 MHz) of 1i.
Figure S18. 13C NMR (CDCl
Stefani et al. S11 Vol. 22, No. 8, 2011
Figure S19. 1H NMR (CDCl
3,300 MHz) of 1j.
Figure S20. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S12
Figure S21. 1H NMR (CDCl
3, 300 MHz) of 2a.
Figure S22. 13C NMR (CDCl
Stefani et al. S13 Vol. 22, No. 8, 2011
Figure S23. 1H NMR (CDCl
3, 300 MHz) of 2b.
Figure S24. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S14
Figure S25. 1H NMR (CDCl
3,300 MHz) of 2c.
Figure S26. 13C NMR (CDCl
Stefani et al. S15 Vol. 22, No. 8, 2011
Figure S27. 1H NMR (CDCl
3,300 MHz) of 2d.
Figure S28. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S16
Figure S29. 1H NMR (CDCl
3, 300 MHz) of 2e.
Figure S30. 13C NMR (CDCl
Stefani et al. S17 Vol. 22, No. 8, 2011
Figure S31. 1H NMR (CDCl
3,300 MHz) of 2f.
Figure S32. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S18
Figure S33. 1H NMR (CDCl
3,300 MHz) of 2g.
Figure S34. 13C NMR (CDCl
Stefani et al. S19 Vol. 22, No. 8, 2011
Figure S35. 1H NMR (CDCl
3, 300 MHz) of 2h.
Figure S36. 13C NMR (CDCl
Synthesis of 1,3-Diynes via Detelluration of Bis(ethynyl)tellurides J. Braz. Chem. Soc.
S20
Figure S37. 1H NMR (CDCl
3,300 MHz) of 2i.
Figure S38. 13C NMR (CDCl