Integrative Analysis of the Developing Postnatal Mouse Heart Transcriptome.
Texto
Imagem

Documentos relacionados
From the two different developmental stages (myce- lium and fruiting body), we identified differentially expressed genes by transcriptome analysis and found that numerous
Gene ontology (GO) term enrichment analysis of the genes with differential H3K4me3 peaks, revealed statistically significantly enriched GO terms only in the genes with
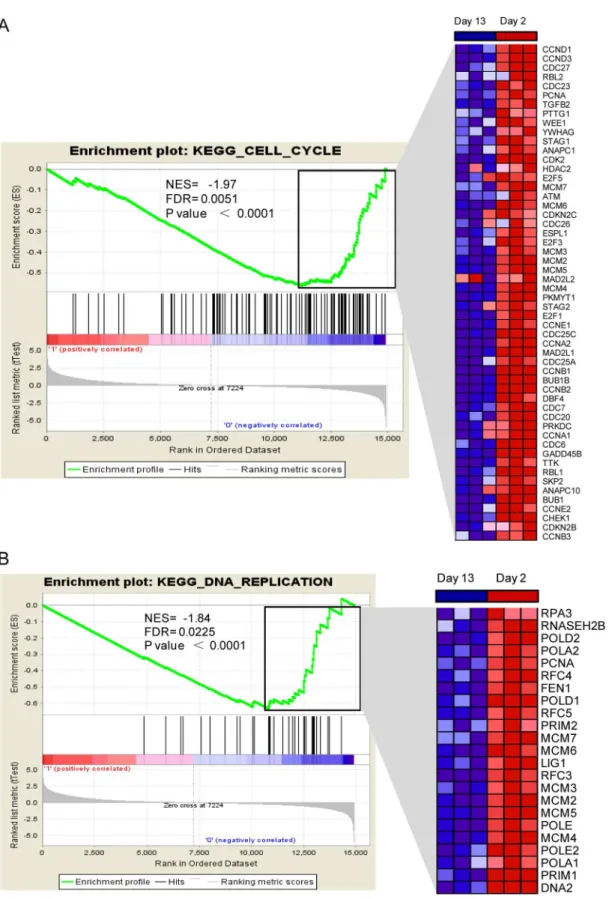
Gene Ontology (GO) and pathway enrichment analyses of DEGs were performed, followed by protein-protein interaction (PPI) network, transcriptional regulatory network as well
Gene ontology (GO) enrichment analysis for the differentially expressed target genes of selected miRNAs in esophageal adenocarcinoma (EAC) and squamous-cell carcinoma (ESCC)..
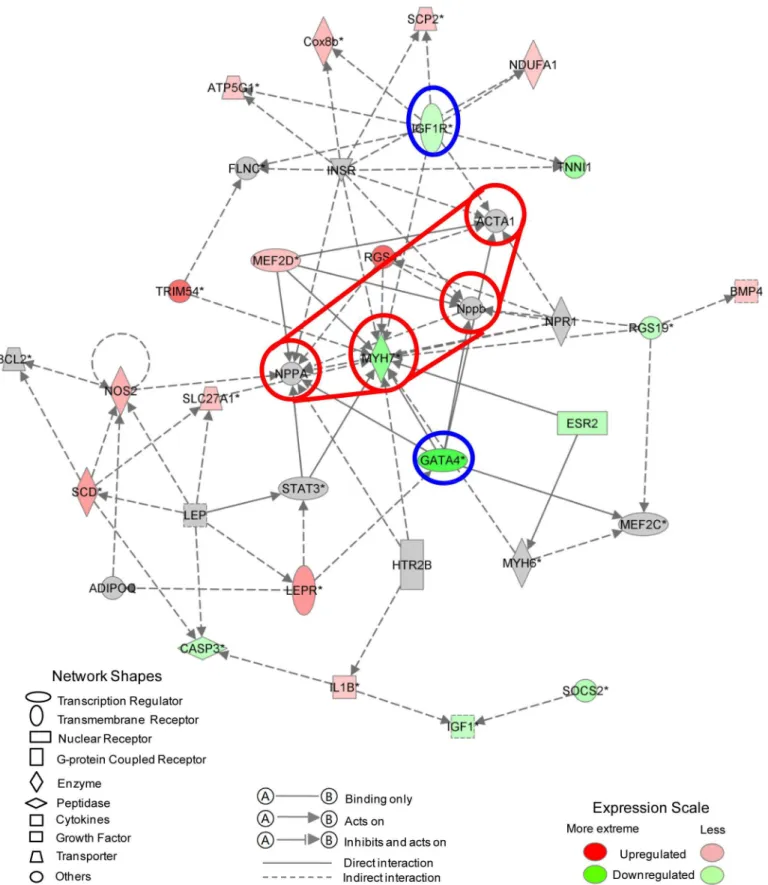
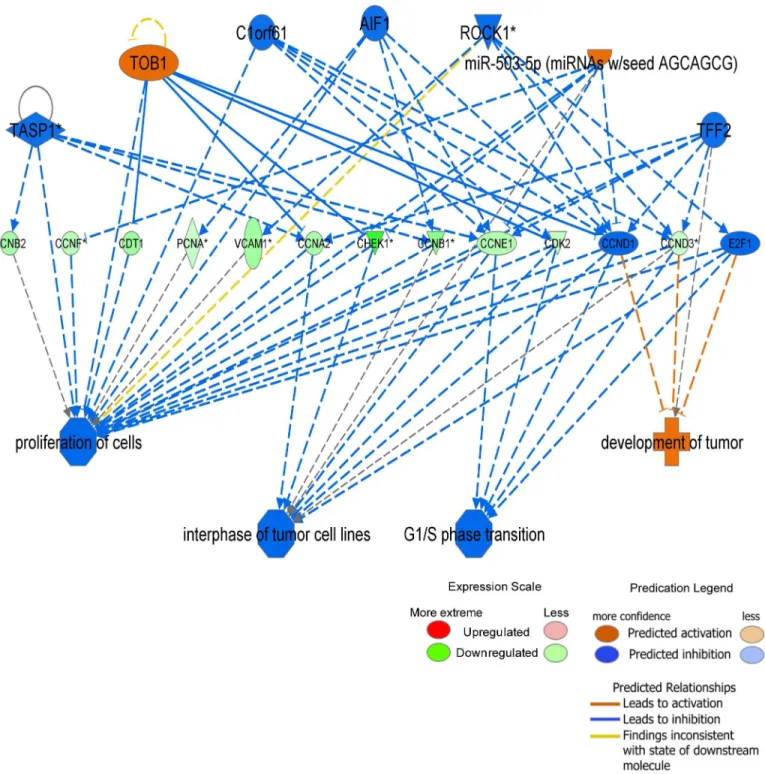
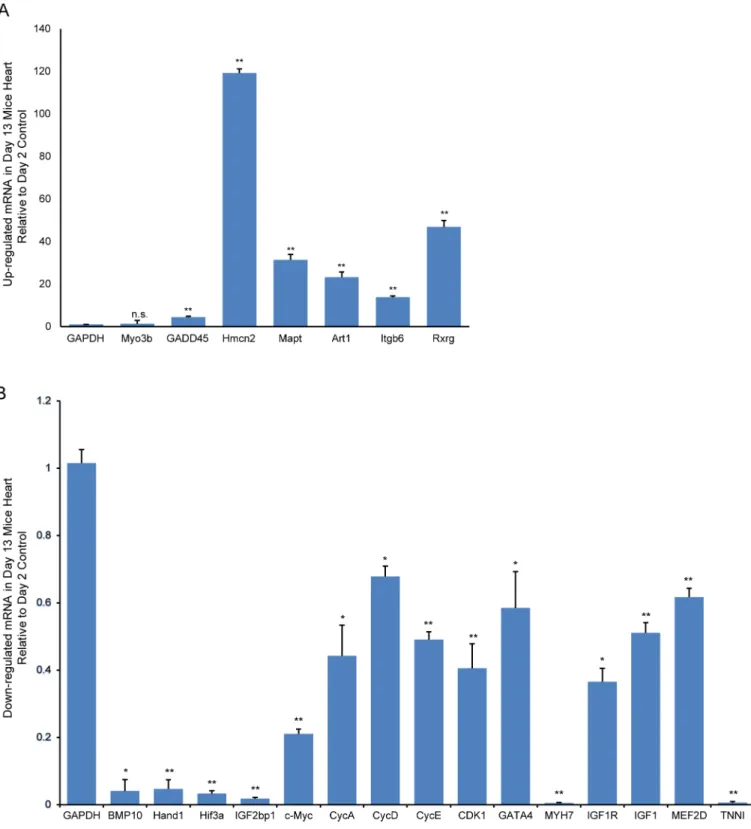
Functional identification of gene networks was performed using Ingenuity Pathway Analysis program as previously described [39].The tables representing the differentially
Furthermore, comparison of yak and cattle ovary transcriptome data revealed that 1307 genes were significantly and differentially expressed between the two libraries, wherein 661
Gene ontology (GO) enrichment of differentially expressed genes revealed protein folding, cell morphogenesis and cellular component morphogenesis as the top three functional terms
Interestingly, the central nodes were more informative for pathway enrichment analysis compared to all network nodes and also 49 di ff erentially expressed genes.. In conclusion,
