Universidade de Aveiro Departamento deEletr´onica, Telecomunica¸c˜oes e Inform´atica, 2013
Nuno Miguel Pereira
Mogas da Silva
Sistema computacional para an´
alise e redesenho de
genes
Computational system for gene analysis and
redesign
Universidade de Aveiro Departamento deEletr´onica, Telecomunica¸c˜oes e Inform´atica, 2013
Nuno Miguel Pereira
Mogas da Silva
Sistema computacional para an´
alise e redesenho de
genes
Computational system for gene analysis and
redesign
Disserta¸c˜ao apresentada `a Universidade de Aveiro para cumprimento dos requisitos necess´arios `a obten¸c˜ao do grau de Mestre em Engenharia de Com-putadores e Telem´atica, realizada sob a orienta¸c˜ao cient´ıfica do Dr. Jos´e Lu´ıs Oliveira (Professor Associado da Universidade de Aveiro e Investigador no IEETA) e da Dr.aGabriela Moura (Investigadora Auxiliar, Departamento de Biologia e CICECO, Universidade de Aveiro).
o j´uri / the jury
presidente / president Armando Jos´e Formoso de Pinho
Professor Associado com Agrega¸c˜ao da Universidade de Aveiro
vogais / examiners committee Jos´e Lu´ıs Oliveira
Professor Associado da Universidade de Aveiro (orientador)
Rui Pedro Lopes
Professor Coordenador do Departamento de Inform´atica e Comunica¸c˜oes do Instituto Polit´ecnico de Bragan¸ca
agradecimentos / acknowledgements
Agrade¸co em primeiro lugar ao meu professor, e orientador, Jos´e Lu´ıs Oliveira, pelo acompanhamento, disponibilidade e ajuda.
`
A minha co-orientador Gabriela Moura, pela disponibilidade e ideias que apresentou e, principalmente, por todas as d´uvidas esclarecidas relacionadas com gen´etica.
Agrade¸co em especial ao Paulo Gaspar, membro do grupo de bioinform´atica do IEETA, pelo constante acompanhamento, disponibilidade, troca de ideias, orienta¸c˜ao e ajuda durante todo este trabalho.
Agrade¸co a todo o grupo de bioinform´atica do IEETA.
Agrade¸co a todos os meus colegas e amigos de curso, pelo apoio e con-vivˆencia ao longo destes anos.
Agrade¸co `a Ana Rosa por todo incentivo e apoio momentos de maior desˆanimo.
Um agradecimento em especial aos meus pais, pela oportunidade que me deram de poder estar aqui, por todos os valores que me ensinaram e pelo suporte e apoio incondicional.
Resumo A evolu¸c˜ao das tecnologias permitiu ao homem explorar diversas ´areas de forma mais eficiente e r´apida. Uma das ´areas onde a inform´atica e a com-puta¸c˜ao tˆem um grande impacto ´e a biologia, permitindo aos investigadores resolverem tarefas de forma mais eficiente, e em tempo ´util, sem recorrer `
a experimenta¸c˜ao pr´atica em laborat´orio. Em biologia molecular, in´umeros m´etodos computacionais s˜ao usados, como por exemplo, na sequencia¸c˜ao e anota¸c˜ao de genomas e tamb´em em m´etodos de redesenho de genes. Por sua vez, com o crescente poder computacional, procura-se realizar o maior n´umero de tarefas no menor tempo poss´ıvel, recorrendo-se para isso a diversas metodologias de otimiza¸c˜ao. Estas podem ser apresentadas de diversas formas, desde o recurso a mais mem´oria e melhor desempenho do hardware bem como a algoritmos mais eficientes.
Esta tese procura avaliar de que forma distintos fatores associados `as car-acter´ısticas de cada gene ajudam a explicar a evolu¸c˜ao destes para o seu estado atual. Os padr˜oes evolutivos potenciados por este estudo podem igualmente ser utilizados como motivos principais no redesenho de genes. Estas tarefas s˜ao computacionalmente dispendiosas e os tempos de execu¸c˜ao elevados devido `as muitas combina¸c˜oes de diferentes m´etodos que s˜ao re-alizadas. Para minorar este problema, esta tese apresenta tamb´em algumas solu¸c˜oes para otimizar os m´etodos de redesenho de genes, de forma a que estes obtenham os mesmos resultados num menor tempo poss´ıvel.
Abstract Technology evolution has allowed man to explore different areas more effi-ciently and quickly. One of the areas where informatics and computation have a major impact is biology, allowing researchers to solve tasks more efficiently, and on time, without resorting to laboratory experimentation. In the field of molecular biology, several computational methods are used, for instance, genome sequencing and annotation and also in gene redesign methods.
Meanwhile, with the growing computational power, there is a need to make the greatest number of tasks in the shortest time possible, using for it several optimization methodologies. Those can be presented in different forms, from the usage of more memory and better performance provided by the hardware as well as more efficient algorithms.
This thesis tries to explore how distinct factors related to each gene charac-teristics can aid to explain how each gene evolved to its current state. The evolutionary patterns enhanced by this study can also be used as the main reasons in gene redesign. These tasks are computationally expensive and their execution times are high due to the various combinations performed with different methods. To mitigate this problem, this thesis also presents some solutions to improve the gene redesign methods performance, in a way that they can achieve the same results in a shorter time.
Contents
Contents i
List of Figures iii
List of Tables v
1 Introduction 1
1.1 Motivation . . . 1
1.2 Objectives . . . 1
1.3 Thesis outline . . . 2
2 Genetics background and EuGene 5 2.1 Protein synthesis . . . 5
2.2 Synonymous codons . . . 7
2.3 Codon optimization . . . 8
2.3.1 Rare codons . . . 9
2.3.2 Codon correlation effect . . . 9
2.3.3 Ramp effect . . . 10
2.4 EuGene - Gene Redesign Software . . . 11
2.4.1 Brief description . . . 11
2.4.2 Redesign methods . . . 12
2.5 Summary . . . 14
3 Requirements and Architecture 15 3.1 User requirements . . . 15
3.1.1 General objectives . . . 15
3.1.2 User interface . . . 16
3.2 Functional requirements . . . 17
3.2.1 Combinatorial system using gene redesign methods . . . 18
3.3 Non-functional requirements . . . 19
3.3.1 Portability . . . 19
3.4 Summary . . . 20
4 Optimization 21 4.1 Finding the bottleneck . . . 21
4.2 Simulated Annealing . . . 22
4.4 Plugins optimizations . . . 26 4.4.1 Codon Usage . . . 28 4.4.2 Repeats Removal . . . 31 4.4.3 GC Content . . . 32 4.4.4 Codon Context . . . 33 4.4.5 Site Removal . . . 34
4.4.6 Hidden Stop Codons . . . 35
4.4.7 UnModified tRNAs . . . 37
4.4.8 RNA Secondary Structure . . . 37
4.4.9 Codon Correlation Effect . . . 38
4.5 Summary . . . 39
5 GEA - Gene Evolution Analysis 41 5.1 Plugins parameters . . . 41 5.2 Random Genes . . . 43 5.3 Similarity score . . . 44 5.4 Cross-validation . . . 45 5.5 Plugin weighting . . . 47 5.6 Data storage . . . 47 5.7 Evolution system . . . 48 5.7.1 User interface . . . 51 5.7.2 Automation script . . . 52 5.8 Summary . . . 52 6 Optimization Results 55 6.1 Plugins improvement . . . 55 6.1.1 Codon Usage . . . 55 6.1.2 Repeats Removal . . . 56 6.1.3 GC Content . . . 57 6.1.4 Codon Context . . . 58 6.1.5 Site Removal . . . 59
6.1.6 Hidden Stop Codons . . . 59
6.1.7 UnModified tRNAs . . . 60
6.1.8 RNA Secondary Structure . . . 61
6.1.9 Codon Correlation Effect . . . 62
6.2 Summary . . . 63
7 Conclusions 65 7.1 Future Work . . . 66
Bibliography 67
List of Figures
2.1 DNA molecules with double-helix form . . . 5
2.2 Portion of DNA: gene. DNA is transcripted to RNA . . . 6
2.3 Resulting amino acid chain provided by the translation process . . . 6
2.4 Substitution of a codon (CUU) by a synonymous one (CUC) . . . 8
2.5 Gene translation process . . . 8
2.6 Codon Correlation Effect process . . . 10
2.7 EuGene quick information about a single gene . . . 11
2.8 EuGene overall operation mode schema . . . 12
3.1 Basic user interface mock-up . . . 16
3.2 Quick evolution parameters analysis mock-up . . . 17
3.3 Optimization system draft . . . 19
4.1 Simplified version of the Simulated Annealing algorithm flowchart . . . 22
4.2 Plugins execution time of two distinct genes with 300 codons length . . . 24
4.3 Plugins execution time of two distinct genes with 600 codons length . . . 25
4.4 Interface that a plugin must respect in order to be used in EuGene . . . 28
4.5 Codon Usage algorithm sample . . . 29
4.6 Changes done in getCodonUsageRSCU() method . . . 30
4.7 Changes done in getCodonRelativeAdaptiveness() method . . . 30
4.8 Changes done in Repeats Removal plugin . . . 31
4.9 Codon sequence highlighting how GC content is measured . . . 32
4.10 Old GC Content plugin algorithm . . . 32
4.11 New version of the GC Content plugin algorithm . . . 33
4.12 New version of the Codon Context algorithm . . . 34
4.13 Site Removal : sequences that can be avoided . . . 35
4.14 Site Removal : iterations effort . . . 35
4.15 Out-of-Frame stop codon - Solution to overcome this issue . . . 36
4.16 RNA Secondary Structure algorithm - block 1 . . . 37
4.17 RNA Secondary Structure algorithm - block 2 . . . 38
4.18 Codon Correlation Effect internal table structures . . . 39
5.1 Redesign method parameter illustration . . . 42
5.2 Synonymous genes generation problem . . . 44
5.3 Hamming Distance between two words . . . 44
5.4 Cross-Validation to check redesign methods parameters veracity . . . 46
5.6 Gene evolution activity diagram . . . 50
5.7 GEA application interface . . . 51
5.8 GEA application automation script . . . 52
6.1 Codon Usage performance improvement . . . 56
6.2 Repeats Removal performance improvement . . . 57
6.3 GC Content performance improvement . . . 57
6.4 Codon Context performance improvement . . . 58
6.5 Site Removal performance improvement . . . 59
6.6 Hidden Stop Codons performance improvement . . . 60
6.7 UnModified tRNAs performance improvement . . . 61
6.8 RNA Secondary Structure performance improvement . . . 62
List of Tables
2.1 Standard Genetic Code table. . . 7
2.2 Gene optimization methods available in EuGene . . . 13
3.1 List of main requirements . . . 18
4.1 Block times using random plugins . . . 21
4.2 Difference between RNA Secondary Structure and Codon Condext . . . 25
4.3 Hidden Stop Codon table strategy . . . 36
Chapter 1
Introduction
1.1
Motivation
Every living being is built according to a genetic library, called genome, which is made of Deoxyribonucleic Acid (DNA). From the ancient times, genomes have suffered successive changes that lead to the evolution of the organisms. Some of those mutations are caused by errors during DNA duplication (replication), a crucial step so that a new copy of the genome can pass to the next generation. Other mutations can also be caused by the environment where the organism live. In other words, evolution is driven by mutations of the DNA.
Genes are nothing more than portions of an organism’s genome, that carry the code used for building the molecular structures that are key for living organisms. Therefore, under-standing how a gene originates is still a field that requires special attention from researchers. To do so, they often need to express those genes in laboratory, using a host species. Hence, several tools aid researchers investigating and better understanding the process of synthesiz-ing proteins in host species in a quick and accurate faction, based on several known algorithms that redesign the DNA sequence to improve the protein yield and quality.
On the other hand, today’s society want results faster and with better quality. In com-puter science this means that a program, must be efficient, accurate and fast to execute its goal. Studying and developing new algorithms and optimization techniques, that can reduce execution time, is a major challenge and a motivation for computer science enthusiasts.
This thesis explores two distinct fields: gene for heterologous expression optimization algorithms and algorithmic optimization. Using distinct gene design algorithms, this work tries to determine what factors influenced a gene to mutate into its current state. Also, this normally needs a huge computing time, since it explores millions of possibilities that can explain a gene’s evolution. Also, optimization techniques should be applied to the known gene optimization methods in order to reduce their overall execution time. Furthermore, combining all this achievements into a single package should result in a dataset that can be used by researchers to explain what biological processes a gene went through until it reaches its current state. This also served as a motivational purpose for the development of this thesis.
1.2
Objectives
The set of informatics solutions for gene sequence redesign grows everyday. Also, there are an evergrowing spring of genetic digital data that needs to be evaluated to explain genes
evolution.
EuGene1 a former software package for multivariate gene optimization for heterologous expression, already processes and analyzes some information about genes, using biological concepts like codon usage, GC content (guanine-cytosine content) or hidden stop codons. Hence, the goal of this thesis is not only to explore further approaches for gene sequence redesign and optimization for heterologous expression but also try to combine those methods to explain how a gene may have evolved to its current state. This project objectives included the following:
• Study and implement additional gene redesign algorithms into EuGene software, such as:
– Rare Codons
– Codon Correlation Effect – Ramp Effect
• Study and implement performance optimization techniques for the gene redesign pro-cess.
• Enhance the global optimization block used in EuGene (Simulated Annealing algorithm) to decrease its execution time.
• Study and implement an architecture that can combine all the gene redesign methods in order to find the best input parameters that conducted a gene to its present form. From an engineering point of view, creating a system that explores all gene redesign methods in an exhaustive way is a tenacious job, being the reason why this thesis outline resulted in an exploratory work where once the results are achieved, they are stored for future investigation. Thus, this analysis should be performed once for every gene of a genome, and the results should be made available for later use by biology researchers. Also, a major goal of this thesis is to decrease every gene redesign method execution time as much as possible to decrease the computation time of each analysis.
1.3
Thesis outline
This thesis is organized in seven distinct chapters, being the remaining six briefly described bellow:
Chapter 2 presents some biological background needed to support this thesis goal. Here it is presented the basic principals regarding genetic information as well as some common biological concepts. This includes the process behind protein synthesis, what are synonymous codons and why they are important, and some gene redesign methods available in the lit-erature. Moreover, the software that served as core for this thesis is presented showing its features and how they were explored.
Chapter 3 explains the main requirements and the architecture of the project. Here the user requirements are detailed, enhancing why this project is important for researchers. Also, from the engineering point of view, the functional and non-functional requirements are listed. This includes the architecture behind the software and how it will work and aid the researchers.
Chapter 4 shows the gene redesign methods performance and how they can be optimized. Here it is analyzed where the system overall performance can be improved. Moreover, it details every gene redesign method and presents some methodologies used to improve their global performance, reducing the time they take to achieve an optimal result.
Chapter 5 shows how the redesign methods were used to achieve this thesis goals. Also, it presents how gene redesign methods parameters were chosen, how random genes were generated and also how to calculate a similarity score between gene sequences. Moreover, the structure behind the data storage is presented and how it can be used by researchers.
Chapter 6 discusses the improvements achieved with the decisions taken in chapter 4. It presents all individual gene redesign analyzes, showing how faster the new plugins version are, when compared directly with their older version. A quick summary with the improvements achieved for each redesign method is also presented.
Chapter 7 shows the conclusions of this work. It explains how the optimizations were important, and a validation of how the redesign methods can be used for other purposes regarding EuGene. Finally this chapter also points out some research lines for future work.
Chapter 2
Genetics background and EuGene
2.1
Protein synthesis
When we think about genetics, most of the times we associate this concept with DNA (deoxyribonucleic acid). Nevertheless, genetics can be described as the science which studies the heredity, variation, molecular structure and function of genes in living organisms.
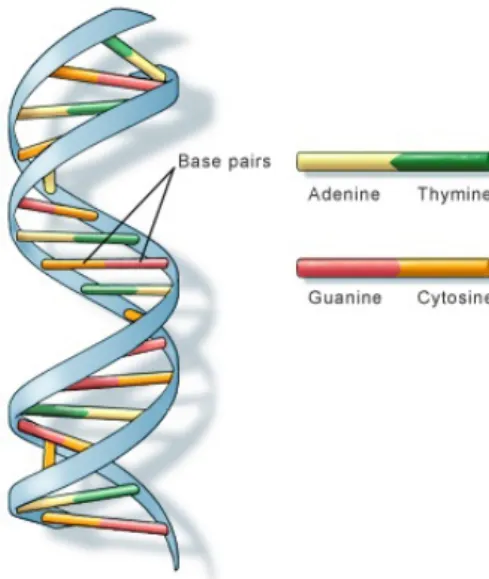
The DNA molecules are present in almost every cell of a person’s body and the same is true in almost all other organisms. Hence, it is important to know what kind of information DNA carries. DNA properties allows it to function as a very efficient vehicle to store information [1]. DNA stores information, encoded as a sequence of four chemical bases: adenine (A), guanine (G), cytosine (C), and thymine (T). The order in which these bases appear determines the information available for building and maintaining an organism, similar to how we use the alphabet to build different combinations of letters in order to build words and sentences. Most DNA molecules are structured as a double-helix and the four nucleotides (bases) are located as shown in the figure 2.1:
Figure 2.1: DNA molecules with double-helix form. All four different nucleotides are presented with different colors.
it in smaller sequences of interest, called genes. This division is important because each gene can define an organism characteristic, for instance, in human DNA we can find genes that determine one’s height, eye color, etc.
This genetic information is used through a process that copies the nucleotide sequence of the gene and produces another nucleic acid, similar to DNA, called RNA. The process of synthesizing RNA from DNA is called a transcription [2].
Although RNA can be seen as a copy of DNA, it differs in certain ways, for instance: while DNA is formed by a double-stranded chain, RNA is formed by a single-stranded chain; RNA has the bases Adenine (A), Uracil (U), Cytosine (C) and Guanine (G), i.e, the thymine base is replaced by Uracil.
A RNA transcript is composed by exons and introns. Exons are the nucleotide sequence that remains present in the final messenger RNA (mRNA) sequence, as presented in figure 2.2, while introns are the sequences that are discarded during mRNA maturation.
Figure 2.2: Portion of DNA: gene. DNA is transcripted to RNA and the resulting nucleotide triples, that code for protein synthesis, are called codons. Introns are the portion of nucleotides that doesn’t code for protein synthesis.
After the transcript process, the resulting sequence,the mature mRNA, is decoded by the Ribosome, in a process called translation. The goal of this process is to produce a specific amino acid chain, the polypeptide. The translation process start always with a start codon (most common start codon is AUG) and is not complete while the formed chain does not face a stop codon (UAA, UAG or UGA) (figure 2.3).
Figure 2.3: Example of the resulting amino acid chain provided by the translation process. As shown, this sequence always starts with a start codon and ends with a stop codon.
Note that a codon is nothing more than a sequence of three consecutive nucleotides from the mRNA sequence. Hence, the complete sequence must have always a multiple of three nucleotides length. After the translation process is complete, the resulting polypeptide chain will fold into an active protein.
2.2
Synonymous codons
In the previous section it was given a brief overview of the translation process. That process involves a set of twenty amino acids and each amino acid is encoded by a codon (sequence of three nucleotides). It was stated before that there are four types of nucleotides (adenine (A), guanine (G), cytosine (C), and thymine (T)) meaning that we can have up to sixty four different codons. Because there are 64 possible codons and only 20 amino acids (plus stop codons) some codons may encode for the same amino acid, being called synonymous codons. This characteristic is often referred to as the redundancy of the genetic code. Synonymous codons are easily found in the “Genetic Code Table”, that displays the information about each amino acid and the associated list of codons who encode it. The table 2.1 represents the standard genetic code:
U C A G
U
UUU - Phe UCU - Ser UAU - Tyr UGU - Cys U UUC - Phe UCC - Ser UAC - Tyr UGC - Cys C UUA - Leu UCA - Ser UAA - Stop UGA - Stop A UUG - Leu UCG - Ser UAG - Stop UGG - Trp G
C
CUU - Leu CCU - Pro CAU - His CGU - Arg U CUC - Leu CCC - Pro CAC - His CGC - Arg C CUA - Leu CCA - Pro CAA - Gln CGA - Arg A CUG - Leu CCG - Pro CAG - Gln CGG - Arg G
A
AUU - Ile ACU - Thr AAU - Asn AGU - Ser U AUC - Ile ACC - Thr AAC - Asn AGC - Ser C AUA - Ile ACA - Thr AAA - Lys AGA - Arg A AUG - Start/Met ACG - Thr AAG - Lys AGG - Arg G
G
GUU - Val GCU - Ala GAU - Asp GGU - Gly U GUC - Val GCC - Ala GAC - Asp GGC - Gly C GUA - Val GCA - Ala GAA - Glu GGA - Gly A GUG - Val GCG - Ala GAG - Glu GGG - Gly G
Table 2.1: Standard Genetic Code table.
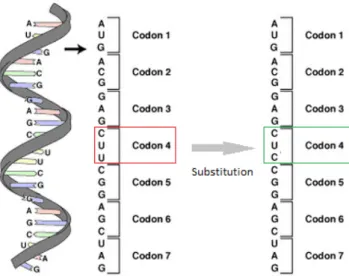
One of the main objectives of optimizing genes for heterologous expression, is to achieve an improved nucleotide sequence. This improvement results from a process called synonymous substitution, where codons can be replaced by other equivalent codons (see table 2.1), without changing the resulting protein sequence. When a replacement occurs, the change is generally neutral. This means that these changes will not affect the protein that is produced. A simple example of the this process is shown in figure 2.4.
Figure 2.4: Substitution of a codon (CUU) by a synonymous one (CUC), both coding for the amino acid leucine, that does not change the resulting protein sequence.
2.3
Codon optimization
In section 2.1, a brief description regarding the translation process was presented. How-ever, this process is a lot more complex. To fully understand the codon optimization tech-niques, it is necessary to know more about the translation process, especially the role of tRNA (transfer ribonucleic acid). The tRNA can be described as a linkage bridge between nucleotide sequence and the amino acid sequence of proteins. Hence, it can be described as a carrier that carry the correct amino acid to the mRNA on the ribosome during the translation process as shown in figure 2.5:
Figure 2.5: Gene translation process where the mRNA sequence is decoded by the ribosome to synthesize a protein.
This information is important for the following sections because codons take a major role in proteins improvement (gene redesign) as described in section 2.2. Next, several characteristics of the mRNA with relevance for gene optimization will be presented.
2.3.1 Rare codons
A low-usage codon is defined as a codon that is used rarely or infrequently in the genome. A rare codon is not only used rarely in a genome, since codon usage and tRNA abundance are usually well correlated, but is also decoded by a low-abundant tRNA [8]. This brings problems to the translation process. The translation rate for a rare codon is much slower than the one for a more abundant codon since the tRNA availability is lower for rare codons. One approach to tackle this effect, is to firstly identify the rare codons. Thus, it is assumed that a codon can be considered rare if it appears less then 5 times out of 1000 in a ORFeome of an organism. Note that this threshold to consider what is a rare codon can be higher or lower.
However, despite a codon being rare, it can be required for the gene expression and thus should be kept. One way to determine if this is the case is to do an orthologous comparison because, if the rare codon is required for a given position of a gene, that position should be filled by that rare codon, not only in the gene of that organism, but also in its orthologs. Hence, if the codon is considered rare and appears in the same position of the gene orthologs, or this positions are occupied by other rare codons, it should be kept, otherwise, it should be replaced by a more frequent codon.
2.3.2 Codon correlation effect
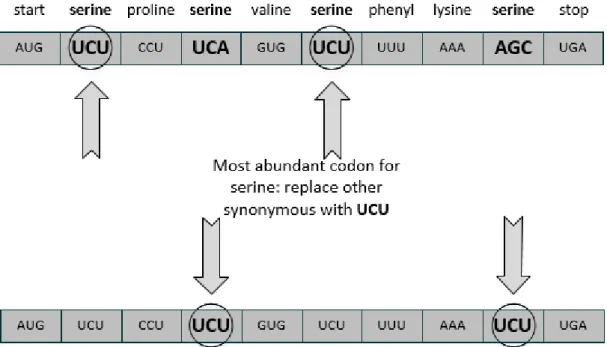
In the previous section, was stated that numerous tRNAs can compete with each other, at the acceptor site of ribosomes, until the correct tRNA is selected. This competition can make the process slow, reducing its efficiency. Thus, as well as in the “ramp effect”, multiple tRNAs can encode more than one synonymous codon so reducing the number of needed tRNAs [5]. Hence, the speed of translation can be improved as well as the complexity behind all this process. This process can be applied to the entire gene, trying to avoid as much as possible the switch of tRNAs, by using the same synonymous codon along the gene sequence. The translation speed can be greatly improved since the tRNAs do not need to disperse in order to get charged again without leaving the ribosome vicinity (figure 2.6).
This effect can be more important if a high level of expression is required. Nevertheless, one should be aware that synonymous codons substitutions that change codon usage frequencies from infrequent to frequent, in regions with slow mRNAs translations, can deleteriously affect the protein quality, mostly because this can exhaust the tRNA pool of the cell, causing an overall imbalance between codons and their cognate tRNAs [6]. One possible solution to override this problem would be knowing how many tRNAs are available. As a consequence, codons decoded by abundant tRNAs need to be more frequent than their synonymous [7].
Figure 2.6: Codon Correlation Effect process - The gene is evaluated choosing the codon who appears often in the gene for each amino acid. After that all other codons for the same amino acid are replaced with the codon who appears more. This process will force the use of the same tRNA increasing the translation process
2.3.3 Ramp effect
The translation process is not in any way linear. mRNA translation evolves multiple stages and many mechanisms that are complex and hard to detail. Furthermore they go out of the outline of this thesis. However, the translation process is critical for gene expression and it must be as much as possible efficient. The abundance of charged tRNAs that correspond to the different codons that encode a protein, was suggested to determine the speed and accuracy of translation [3]. This means that codons have a unique role in translation process and the abundance, or stringency, of tRNAs determines the high and low efficiency respectively. In other words, we can specify that abundant codons are decoded with high efficiency, because the are better adapted to the tRNA pool. With this information, we can define two different types of translation during the translation process. The first, that will be named slow region, complies the codons where we have different tRNAs to encode the same amino acid. In this case synonyms codons are avoided resulting on a slow translation since every codon needs to wait for a new tRNA. The second type, that will be named fast region, is where we can encode the same amino acid with the same tRNA, if it is a synonym. Thus, this process relates the abundance of tRNA and the amino acid translation speed and accuracy [4].
Codons can be translated at different speeds as stated before and this is because the frequency of codons is directly correlated with tRNA abundance. Thus, if there are several rare codons at the beginning of a gene sequence, they will be translated at slow speed since rare codons are usually decoded by rare tRNAs [3]. Therefore, the redesign efficiency of the translation process can be optimized using this knowledge. The main idea is to use rare synonymous codons at the beginning of the gene, so each codon is translated by less abundant
tRNAs which takes more time. Furthermore, the area of effect of this “ramp effect” can be defined, for instance, for the first 30 codons. After this region, the codons should be translated normally and the ribosome congestion should ever be avoided, or partially avoided.
2.4
EuGene - Gene Redesign Software
In the previous sections, several processes that can improve a protein expression were discussed. Those processes are connected to the translation process, which is quite sensitive, and they all try to achieve a common goal which is to synthesize a protein faster, without changing its quality and functionality. As a result, several informatics solutions have been developed that have a huge role aiding this processes. These solutions are widely used in different fields. For instance, improving the protein production can be very important in the development of new vaccines as well as in their manufacture [9].
In this section, a specific gene optimization system is explained. Next, will be presented the main characteristics of the system as well as how it can be explored even further, using new algorithms as well as a new application for those gene redesign methods. Furthermore, a deep analysis is made regarding the performance of the overall system and where it can be optimized.
2.4.1 Brief description
As described above, EuGene explores expert algorithms to redesign genes for heterolo-gous expression. Thus, besides redesign methods, EuGene display several informations about a specific selected gene. Furthermore, to identify a gene, EuGene can use FASTA1 and Gen-Bank2 formats to extract any database identifiers. Those are then used to access NBCI and obtain gene and genome names and also the resulting protein sequence [10]. After the gene selection, EuGene displays in an intuitive way the gene sequence as well as the amino-acid sequence and relevant information about that gene (figure 2.7.
Figure 2.7: EuGene quick information about a single gene - CAI, RSCU, codon pair bias and some other useful information
1
FASTA format is a text-based format for nucleotide or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes.
2GenBank format (GenBank Flat File Format) consists of an annotation section and a sequence section.
The annotation section has information about the gene, like organism, definition etc and the sequence sequence has the gene nucleotide sequence.
In a seamless automatic form, EuGene calculates the “CAI” (Codon Adaptation Index), “RSCU” (Relative Synonymous Codon Usage), “CPB” (Codon Pair Bias, for codon context), number of codons and also the “GC Content” (quantity of Guanine and Cytosine pairs) of the opened gene. This pre-calculated information is an important attribute for the redesign methods, so each method do not need to re-calculate this information, resulting also in a performance tweak.
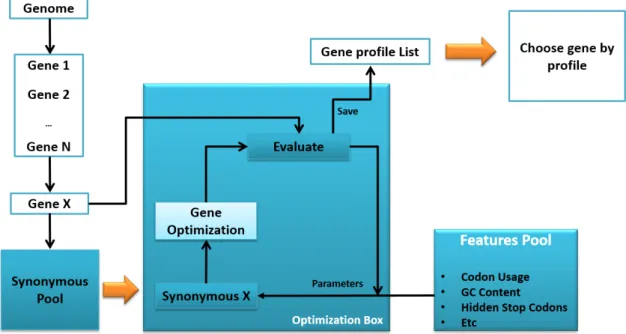
Hence, EuGene has a set of functionalities that cooperate between them in order to achieve a good performance and result, and also a display of the set of relevant actions that are rather transparent to the user. Figure 2.8 presents an overview of the system.
Figure 2.8: EuGene overall operation mode schema: 1 - Possibility to load FASTA and GenBank file formats to retrieve information about a genome and its genes; 2 - Displays information about a specific selected gene (CAI, RSCU...); 3 - Selection of several optimization methods to improve the protein expression (subsection 2.4.2); 4 - Observe results from the optimization methods, including the new codon sequence as well as information about the score achieved by each method.
In conclusion, this section presents some features of EuGene in a summarized way, opening ways to the next big feature, the redesigning methods, that will be detailed in subsection 2.4.2.
2.4.2 Redesign methods
The main feature presented by EuGene is the capability to optimize codon sequences using a combination of several different approaches. Those comprehend a set of algorithms that allows the customization of a gene following specific redesign algorithms. Table 2.2 shows which codon optimization methods are currently available to use in EuGene. Note that this table does not try to explain in detail how every method implemented in EuGene works, but it gives a brief overview about their propose in the context of gene redesign.
Optimization redesign method Brief description
Codon Usage
Allows to maximize or minimize the codon usage. This is done by counting the number of times each of the 64
codons appears; Repeats Removal
Replaces zones with repeated nucleotides with synonymous codons breaking the repeated sequence
chain; GC Content
Calculates the percentage of GC nucleotides. For instance to maximize this effect every codon is replaced
by a synonymous with higher GC amount;
Codon Context
Calculates the number of times a pair of consecutive nucleotides appears. Thus, optimizing by codon context is
finding the synonymous codons that maximize the frequencies of those consecutive pairs; Site Removal
Remove specific nucleotide sequences, for instance Kozak sequences (GCCACCAUGG), by replacing
the codons within it by synonymous codons; Hidden Stop Codons
Replaces out-of-frame stop codons, for instance, CCUAAC, by replacing two codons by synonymous
in order to eliminate out-of-frame stop codons; Unmodified tRNAs
Avoid a specific set of codons for Eukaryote or Bacteria. These codons
are decoded by less efficient tRNAs; RNA Secondary Structure
RNA secondary structure prediction in order to eliminate double strand
occurrences;
Table 2.2: Gene optimization methods available in EuGene. A set of different approaches are
presented as well as some brief information about each method.
To use this redesign methods, in order to achieve a good optimized sequence, EuGene uses two distinct optimization techniques: Genetic Algorithm3 and Simulated Annealing4. These two methods are used in distinct situation, depending if the user wants quicker results or deeper results. Simulated Annealing is a faster algorithm that tries to find an optimal solution quicker but with less precision. On the other hand, Genetic Algorithm search deeper, making it slower, not only for one optimal solution but for a list of possible best equivalent solutions, allowing selection of the solution that offers the best trade-off between the selected redesign methods [10].
3
The Genetic Algorithm is an adaptive strategy and an global optimization technique. It is an Evolutionary Algorithm and belongs to the broader field of Evolutionary Computation [11]
4
Simulated Annealing is a global optimization algorithm that belongs to the field of Stochastic Optimization and Metaheuristics [11]
2.5
Summary
This chapter described a brief introduction to the biological process behind the protein synthesis. Moreover, a definition of DNA was presented as well as its contents, genes, that are used in the protein synthesis. This process is described in all its steps, since how a gene is copied from DNA until a chain of amino acids is formed and after becoming a protein.
Furthermore, it is explained how genes can be redesigned by substituting codons by equiv-alent ones that code for the same amino acid. This flexibility allows to explore several redesign methods with the purpose of increase the proteins qualities, also some of this methods were detailed. Finally, a software (EuGene) that is capable of join several gene redesign methods in a unique tool was presented. This software acts as the root for this thesis since its foundation rise from EuGene features.
Chapter 3
Requirements and Architecture
EuGene is a powerful tool that implements several redesign algorithms, as stated in chap-ter 2, and some of its features can be reused for another purpose. Therefore, this chapchap-ter presents how those features can be reused and also further detailed information about the requirements and the architecture that led to this project implementation.
3.1
User requirements
With a tool such as EuGene, that accommodate several tools in one single application, researchers notice that the evolution of genes could maybe be explained, if they could predict what kind of mutations led to that evolution [12]. The redesign methods available in EuGene, can hence be combined in such way that allow explaining how a gene evolved to a synonymous one.
Overall, using such redesign methods, become clear that a new tool could be developed to evaluate how a gene evolved to its current state. This was the main goal of this project, i.e, to combine several redesign methods, and their parameters, and try to achieve as much as possible a new gene with high similarity to its current state. This computational process takes time to complete because several redesign methods needs to be combined, and evaluated, until an optimum solution is found. Moreover, computing time increase as the gene length increases, and therefore, the performance provided by the system should be optimized to mitigate high execution times. As extra requirements, some new features will be implemented to enhance EuGene functionality.
The following sub-sections describe the most important user requirement, as well as the architecture behind the application.
3.1.1 General objectives
Gathering a complete set of requirements is the most important step at the beginning of any software project. Thus, the most important requirement relies on the ability to generate random synonymous genes from a specific gene and then apply several combined gene redesign methods to each. After all those methods have been applied, it is necessary to evaluate the score of the resulting sequence, for each synonymous, in comparison with the original one. These scores can be obtained by using known techniques that can tell how different the sequences are, for instance providing a similarity percentage, like Hamming Distance. The
whole process is detailed in chapter 5. Finally, the optimization of the execution time is a second major requirement.
3.1.2 User interface
When humans interact with a machine, an important role relies on the user interface. A good interface should provide a “user friendly” experience allowing users to completely understand how they can use the application.
With this in mind, a first interface draft was needed, which layout should accommodate most of the required features such as plugins selection, genes and results display. Based on EuGene application, a new interface mock-up was built resulting as shown in figure 3.1:
Figure 3.1: Basic user interface mock-up to accommodate the main features
The main reason behind this similar interface is that the resources used by this project application are the same used by EuGene, i.e. gene redesign methods that can either be used in this project application. Thus, in this illustration, numbers show the main parts of the application layout and the ones which need further detail.
The first part (number one), represents the menu bar. This bar should have four distinct menus: File, Edit, View, Gene Pool. The File menu is intended to provide functionalities such as create a new project or load past projects. Loading a saved gene is important to check what kind of mutations are responsible for gene evolution. On the other hand, the Edit menu provides a set of settings to improve the algorithm accuracy. Those include the number of random synonymous genes to be generated as well as the maximum number of iterations the algorithm can take to converge. The View menu is an extra option just to enhance user interaction experience, by showing or hiding panels. Finally, the Gene Pool Menu is intended to load genes from files or even insert a gene manually.
as well as the option to select the desire ones. Here it should also be possible to see what kind of parameters each method use. This zone also include the Run button that allows that application to start evolve the optimization methods to achieve a final result.
In the project area we can see two distinct zones. One regarding the gene representation (3), where the user can see the nucleotide sequence as well as the amino acid sequence of the loaded gene. The second zone, represented by number 4, shows the final results achieved. Here the user should be able to see information such as how many random genes where used as well as the best parameters combination used to achieve the final result. Also the final similarity score obtained using those methods and parameters should be shown, as presented in panel marked with number 5. This zone also has the option to make a quick validation. This enables the user to test the achieved parameters with new random genes or even insert synonymous gene sequence as shown in figure 3.2:
Figure 3.2: Quick evolution parameters analysis mock-up
This mock-ups represents a quick draft of how the basic application functionalities should be represented. The final results may change due to new specifications or even performance issues.
3.2
Functional requirements
From the software engineering point of view, functional requirements are defined by the objective/function of the software system, and hence, they should describe what data the system should complies and how it is used. Moreover, functional requirements are supported by non-functional requirements (section 3.3) which impose some constrains like portability. Therefore, there were identified some crucial requirements that the system should have.
Firstly it is necessary to be able to manipulate gene sequences, namely opening and parsing a genome file and display the gene sequence (codon and protein structures). This requirement is inherited from EuGene since it has a well defined module called Gene Pool which takes care of the genome load and manipulation.
Other requirement is the capability of use the developed gene redesign methods that EuGene currently supports and also use new redesign methods, Keep Rare Codons, Codon Correlation Effect and Ramp Effect.
Finally, the system should be able to explore all redesign methods applying them to random synonymous genes generated from a wild type gene (original gene), in order to achieve the best set of parameters that lead the synonymous to evolve, as much as possible, in a new
sequence similar to the wild type. This can be defined as one of the main requirement of this thesis. Moreover, having such tool that can produce this results in lesser time is crucial and therefore the redesign methods performance should be tuned as well as the global optimization process, detailed in chapter 4.
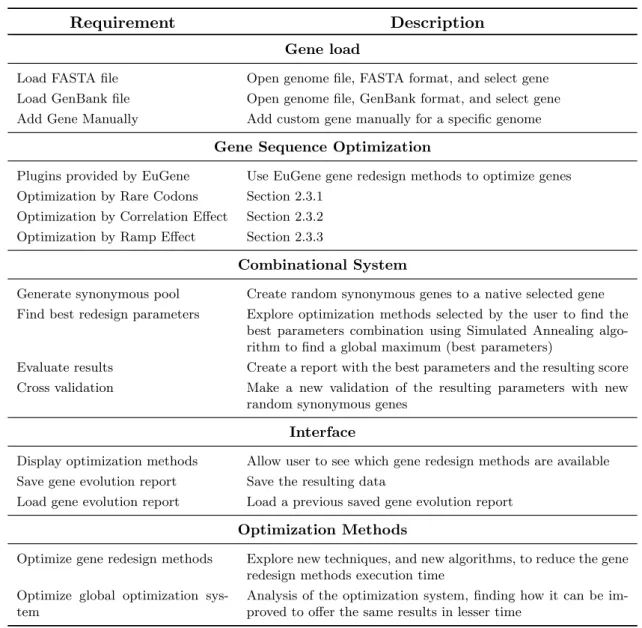
This main requirements, as well as the interface needs are presented in table 3.1.
Requirement Description
Gene load
Load FASTA file Open genome file, FASTA format, and select gene Load GenBank file Open genome file, GenBank format, and select gene Add Gene Manually Add custom gene manually for a specific genome
Gene Sequence Optimization
Plugins provided by EuGene Use EuGene gene redesign methods to optimize genes Optimization by Rare Codons Section 2.3.1
Optimization by Correlation Effect Section 2.3.2 Optimization by Ramp Effect Section 2.3.3
Combinational System
Generate synonymous pool Create random synonymous genes to a native selected gene Find best redesign parameters Explore optimization methods selected by the user to find the
best parameters combination using Simulated Annealing algo-rithm to find a global maximum (best parameters)
Evaluate results Create a report with the best parameters and the resulting score Cross validation Make a new validation of the resulting parameters with new
random synonymous genes
Interface
Display optimization methods Allow user to see which gene redesign methods are available Save gene evolution report Save the resulting data
Load gene evolution report Load a previous saved gene evolution report
Optimization Methods
Optimize gene redesign methods Explore new techniques, and new algorithms, to reduce the gene redesign methods execution time
Optimize global optimization sys-tem
Analysis of the optimization system, finding how it can be im-proved to offer the same results in lesser time
Table 3.1: List of main requirements that the application should support
With all these requirements, it is noticeable that exists a crucial requirement: combination of several redesign methods. The next sub-section provide additional information about it.
3.2.1 Combinatorial system using gene redesign methods
The main feature of the system relies on the combinatorial system. This system can be seen as a box that receives a gene and tries to give hints of how this gene evolved. That process encompasses generating a pool of synonymous and then applies to each synonym
several redesign methods. Each of these methods should try to achieve the highest score as possible, i.e, increase the protein quality as much as possible. Though, each method have a set of parameters, for instance, the codon correlation effect have the possibility to increase the correlation between codons or even decrease this correlation, as explained in section 2.3.2. These parameters should be tested in different combinations for all redesign methods in order to achieve a final result - maximum similarity of each synonymous and the original gene. This process can help explaining what factors led a gene to evolved to its current state.
Note that due to the different possibilities of combining the redesign methods parameters, this process is exhaustive, and therefore it takes long time to complete. On the other hand, once a gene is evaluated, it no longer requires further evaluation since the set of parameters achieved by each redesign methods is final. A simple draft of how this system works can be seen in the figure 3.3:
Figure 3.3: Optimization system draft displaying the system flow. First a genome is chosen and from it is selected a gene. After, a pool filled with synonymous genes is generated, and for each synonymous a set of redesign methods are applied, iteratively, until an optimal result is found
3.3
Non-functional requirements
3.3.1 Portability
Nowadays, one of the biggest bottleneck in software applications is the portability. This means that independently of the operating system, processor or machine, the application should run in all environments. Hence, using Java programming language solves this issue given its portability for different operating systems. Thus, this project software should run without any problem in any environment with Java, for instance, Linux, Windows or Mac OS.
3.4
Summary
In this chapter, the main requirements for this thesis were presented. After reading this chapter it should be possible to understand the need to generate random genes from a native one, and process them in order to achieve a similar gene to the native. This process is done by applying several redesign methods with random parameters and then applied a metric like Hamming Distance to evaluate the similarity. Moreover, an interface scratch is presented that shows how all the requirements should fit in the application. This application, inherit from EuGene most of its visual contents as well as new features to support the functional require-ments. These requirements were presented in a compact table allowing a quick understanding of what work needs to be done to accomplish this thesis goal. Furthermore, the need to have an application that can run in distinct operating systems was presented in order to ensure portability.
Chapter 4
Optimization
Chapter 3 presented main requirements to achieve this project goal. Thus, being an ex-ploratory work, the performance of the existent redesign methods, as well as the optimization process, must the tackled. Those methods, implemented in EuGene, makes use of a simple approach where the execution time does not matter a lot. For this project, where those meth-ods are going to be used repeatedly, a more deep analysis was performed in order to see how their execution time can be decreased, or at least for most of them. Moreover, this chapter also explores the Simulated Annealing process, presented in EuGene, and a possible solution to improve its performance.
4.1
Finding the bottleneck
In any computer program, there are some code zones that are executed more frequently than others. Despite being more frequently executed, does not entirely means that they are slowing down an entire process and thus, finding the critical spot, bottleneck, is a meticulous task. Therefore, the optimization process presented in EuGene, was carefully analyzed and divided into three distinct blocks - Simulated Annealing entire process, codon neigh-bour generation and plugins exectution . The goal of this division is to find where the optimization process can be improved. Thus flowchart ( 4.1) shows how this division was performed. Note that these are the most important blocks in the SA algorithm. For the previous blocks presented, an average execution time was measured resulting in the following table 4.1:
Number plugins Total Time Plugins execution Neighbour generation
3 85588 84123 147
5 67721 66183 172
7 120415 118672 179
Table 4.1: Block times using random plugins - This results were achieved using distinct plugins in each case and with a gene with 1000 codons length. Other tests were made using genes with distinct lengths and the results were similar. Also, times are presented are in milliseconds.
As observed, it is clearly that the bottleneck remains in the plugins execution environment. They use almost all the time of the Simulated Annealing algorithm and, therefore, is the perfect spot to perform a deeper analysis (section 4.3). These results showed that the
Two main blocks Has
converge ?
Get random codon synonymous
Run plugins and get score
End Start
yes no
Figure 4.1: Simplified version of the SA flowchart showing the most important blocks
average consumption time of the plugins execution took around 98% off the entire Simulated Annealing process.
4.2
Simulated Annealing
The redesign methods execution takes almost all the computational time of the Simu-lated Annealing algorithm. Hence, before checking how this algorithm can be improved it is important to understand how it works and what it tries to achieve.
Over the last decades, many optimization heuristics have been developed. Those have the objective to find the optimal solution for a given problem. Therefore, improvement heuristics start with an arbitrary configuration for a given problem, and tries to improve its solution iteratively by changing small pieces of the original configuration and evaluating the new one. A simple and well known heuristic is the Greedy Algorithm. In short, it move pieces of the initial configuration, randomly, and only accept the changes made by the move if the new configuration is better than the previous one. On the other hand, if the configuration is worst, it is discarded and one stays at the “previous best” configuration.
However, using such approach can lead to stuck situations where the solution found is a local maxima but not a global one. When this happens, the system can not improve the solution. To overcome this problem, it is necessary to work with more acceptance functions and not only choosing pieces randomly. Note that more acceptance functions do not free the system to get stuck in local maximas, but reduce the probability of that to happen[13]. One used optimization algorithm that follows this approach is the Simulated Annealing.
Every redesign method that is present in EuGene returns a score for each sequence, according to the redesign algorithm objective. This allows Simulated Annealing to perform an aggregation of all the redesign methods scores and therefore perform an evaluation by checking if the global score achieved is better than the previous one. Note that in the gene context, each synonymous sequence of the original gene sequence represents a possible solution.
Hence, Simulated Annealing works according to the choose of several candidate solutions, even if its worst than the current, and evaluate its final score. As the iterations goes by, Simulated Annealing choose only the best solutions (it does not guarantee the optimum solution). This process can be referred as slower cooling since initially Simulated Annealing
will find good solutions, and, with time, it will become harder to find better solutions than the previous obtained[14].
The original implementation of this algorithm (in EuGene), uses Strings to manipulate and create new gene sequences. Most of the effort of the algorithm, besides the effort used by the gene redesign methods, is used in obtaining a new random neighbour (codon) and apply it to the current best sequence. In order to improve this solution, every gene sequence was converted to an Array of Integers, where each position uses an ID that identifies a specific codon. This allows to manipulate the gene sequence by just using array references. Using this approach, not only makes the Simulated Annealing algorithm to generate new sequences faster, but also aid the gene redesign methods in the sequence manipulation, since most of those methods operates at codon level.
A small test was made to prove how using an array of integers can improve the overall performance. Using Java programing language, it was created a StringBuilder with 4725 characters and an array of integers with 4725 positions initialized with random integers. Then, it were made 100000 iterations over those variables according to the following:
• sequence1.replace(randomIndex, randomIndex, ”A”); for the StringBuilder • sequence2[randomIndex] = 1; for the integer array
The measured time was at milliseconds scale with the results of 453 and 9 respectively. Despite being a small test it shows how faster it is to attribute a value to an array than replacing a specific Character in a String sequence.
This approach can certainly improve the overall Simulated Annealing algorithm as well as similar operations in the other gene redesign methods.
4.3
Plugins Analysis
Manipulating several data structures can be a hard task since each one have a unique behavior. However, due to the plugin structure adapted in EuGene, each redesign method can be tackled independently. From this problem point of view, each redesign method uses a unique algorithm and therefore their behavior is distinct.
Achieving an optimal result takes time and the algorithm complexity plays a major roll that needs to be tackled. Hence, as first step, each plugin was analyzed individually. The purpose of this analysis is to measure the average time each plugin takes to achieve the best result and therefore know how the system overall performance is affected. For this test scenario a couple of tests were made, using two distinct genes lengths. Those lengths may define the convergence time of each plugin, however, other factors like the gene codon sequence, may fit better in one plugin algorithm than other, and therefore the algorithm does not need to put the same effort as it should in a more “scrambled” sequence. In other words, if for instance a hypothetical redesign method had the objective of removing the base A, and the codon sequence does not have any A, then a single iteration would be necessary to perform such task and the optimal solution is easily reached. However, if a sequence has several A bases, it would be necessary to remove each appearance, which would add more complexity to the algorithm by removing codons and replacing them by synonymous ones that does not have A bases. Hence, the gene length may not be the only factor affecting the algorithm performance.
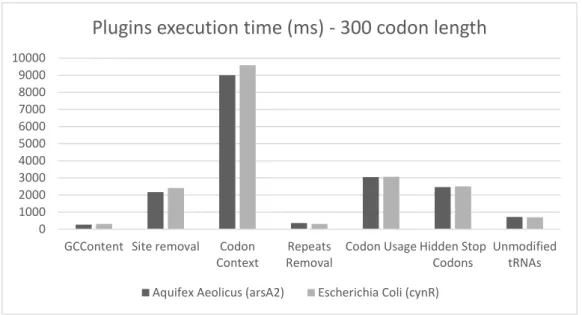
In these initial tests, and to measure the time each plugin takes, only the gene length was taken in consideration. This is enough to find which plugins are performing bad in a general case scenario. Hence, four distinct genes were tackled from two different genomes -Escherichia Coli and Aquifex Aeolicus. From each genome, two genes were randomly chosen, one with a codon length of 200 and another with 600. Also, each plugin was run five times for each gene and an average execution time was taken. The purpose of taking a mean time is to avoid an inefficiently convergence of the Simulated Annealing algorithm that can sometimes not achieve the optimal solution. The following execution times, in milliseconds, were observed: 0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000
GCContent Site removal Codon Context
Repeats Removal
Codon Usage Hidden Stop Codons
Unmodified tRNAs
Plugins execution time (ms) - 300 codon length
Aquifex Aeolicus (arsA2) Escherichia Coli (cynR)
Figure 4.2: Plugins execution time of two distinct genes with 300 codons length
Chart 4.2, shows that at least four plugins have a major impact in the global performance. For a small gene, with 300 codons, the redesign method Codon Context can take up to approximately 9,5 seconds which is pretty high. For instance, if the combinatorial system presented in chapter 3 took up to 500 iterations to find the optimal parameters, meaning that it would need to evaluate 500 different parameters for this plugin, then the required time for completion would be around: 9, 5 ∗ 500 = 4750s, which, for a single plugin, would result in a global execution time of approximately 80 minutes. Other plugins, like Site Removal, Codon Usage and Hidden Stop Codons also take a considerable amount of time to complete as we can see in the previous chart. Chart 4.3, presents similar results for a bigger gene. It is noticeable that the same plugins have a higher execution time, proving that they are a bottleneck.
From the available plugins provided by EuGene, one is missing in this tests - RNA Secondary Structure prediction redesign method. This one has a much higher execution time being that the reason why is not presented along with the other plugins. The chart would not be easily read since it would present a huge discrepancy between the RNA Secondary Structures and the other plugins. However, table 4.2 shows a direct comparison between this plugin and the worst one presented before, Codon Context.
0 10000 20000 30000 40000 50000 60000 70000
GCContent Site removal Codon Context
Repeats Removal
Codon Usage Hidden Stop Codons
Unmodified tRNAs
Plugins execution time (ms) - 600 codons length
Aquifex aeolicus (lepA) Escherichia Coli (lepA)
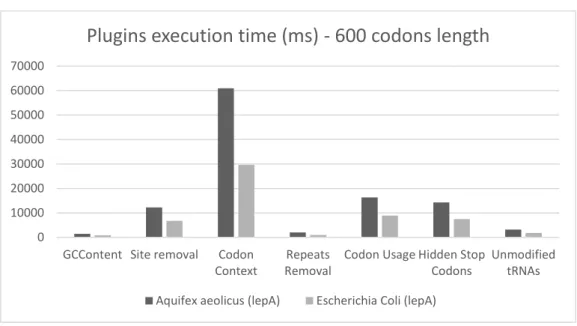
Figure 4.3: Plugins execution time of two distinct genes with 600 codons length
RNA Sec. Struct. Codon Context Time Difference
Genes with 300 codon length
Aquifex Aeolicus (arsA2) 98043 ms 9002 ms ∼11x more time Escherichia Coli (cynR) 101716 ms 9578 ms ∼10x more time
Genes with 600 codon length
Aquifex Aeolicus (lepA) 1061091 ms 60970 ms ∼17x more time Escherichia Coli (lepA) 607737 ms 29638 ms ∼21x more time
Table 4.2: Difference between RNA Secondary Structure and Codon Condext - The Time Difference column shows how many times the Codon Context plugin could run until the RNA Secondary Structure plugin achieve the best result.
execute than Codon Context being this the main reason why it is not present in the charts. It would not be possible to see clearly the average time that all plugins takes to execute and, therefore, the need to make improvements at this level. Despite being the plugin that takes more time to execute, it does not make it the unique plugin that needs a performance boost. Every other plugin may be, and must, be improved.
Looking at RNA Secondary Structure execution time, it is noticeable how long it takes to execute. Note that this results were taken for a single plugin at a time, and if for instance, we run four plugins at once, they would need even more iterations to converge. The conjugation of several plugins can force some plugins to scramble the results achieved by other plugin, for instance, if one plugin was intent to replace all A bases by C and another plugin was intent to replace all C bases by A. This results in an even greater time needed to achieve the best result.
The execution time for a single iteration is long, and for multiple iterations, in a com-binatorial environment, it could take weeks to achieve an optimal result. Since every plugin will be tested several times, with distinct parameters, every second that can be improved in the algorithms execution is important and therefore it should be the next step taken before advancing to the development final application (chapter 5).
4.4
Plugins optimizations
Section 4.3 showed how important it is to make optimizations. Hence, several distinct strategies can be applied to each plugin according to its behavior. Previously, it was shown that EuGene is written using Java programming language and, being this language an Object Oriented one, objects cost time and CPU on their creation. One approach to improve plugins execution time could be holding some objects in memory since managing them in memory is much faster than processing them over and over again for getting the same result. This can free CPU to do another tasks while objects are being kept in memory. Moreover, avoiding unnecessary temporary objects, which takes time to create, can also affect an application performance. Note that every second that can be spare is important to achieve this thesis goals. On the other hand, some plugins may also be implemented using new algorithms that can be more efficient than the previous ones.
Using memory, there are some good programing practices that should be used. Memory leaks are possible in Java, like in any other programming language. For instance, it is possible to have memory leaks by holding on to objects without releasing their references. This usage stops the Java Garbage Collector from reclaiming those objects and therefore increasing the usage of memory being used [15]. This could lead to an inefficient usage of objects and therefore to an excessive memory consumption. Hence, next is presented a few guidelines that lead to a good programing and also to produce more efficient code:
• Avoid objects replication. This is important in routines where objects are used fre-quently. The over creation of the same object is unnecessary as well as inefficiently and also adds an overhead that can be avoided;
• Use static variables when multiple classes need access to the same object. It is preferable to use a static variable than have each class instance holding a separate reference for the same object. This also creates less variables since each class will use the same object without the need of creating one for each class;
• Avoid String exhaustive manipulation. On solution could be using integer IDs to iden-tify strings. Integers are much less complex to manipulate and to compare;
• In a Thread environment, some methods must be synchronized. This synchronization should be applied to the method block that needs to prevent concurrent access and not the entire method;
• Using Threads can vastly improve system performance since it allows parallel compu-tation;
These are just some examples of good practices that can lead to a more efficient and optimized code. More examples could be named but this list would be too extensive. However, one more example of a good practice should be referenced - data structures usage. Choosing the correct data structure to hold data can sometimes improve the system performance greatly, for instance, choosing between a Vector or an ArrayList. Despite being considered deprecated the Vector class may be suitable in some situations where concurrent modification matters. Vector has its methods synchronized granting concurrent security (mutual exclusion) while ArrayList does not. On the other hand, since ArrayList does not have synchronized methods, offers better performance and is more suitable for most applications. Note that it is possible to force an ArrayList to be synchronized using Collections - Collections.synchronizedList(List). This small details can make difference in the overall system performance.
Likewise Vector and ArrayList, one often common problem, relies on the choose of the appropriate Map structure. HashMap does not have any method synchronized and hence does not offer object thread safe while HashTable offers synchronization in all methods. Also, being a direct consequence, HashMap presents higher performance while HashTable is slower. In short, choosing the right data structure for a given application can vastly improve the system overall performance and therefore is a task that needs to be carefully thought depending of the system needs. With some of this ideas present, some modifications can be performed to the plugins algorithms but, before analyze each plugin individually a common modification was done.
These plugins were developed for EuGene and, curiously, EuGene avoids the usage of ArrayLists. Being a multi-thread application, specially using Swing1, that is not thread-safe,
the usage of Vector class was a good choice that could prevent most of the deadlocks that might appear. Note that Swing use a specific thread to manipulate his components EDT -Event Dispatch Thread. Hence, for data manipulation, where there is a need to insert and remove values, the Vector class offers thread safety, although, it may not be necessary if the concurrent access is well controlled, or even if it does not exists. Most of the operations used by the plugins, that use a Vector data structure, are gets (operation of retrieve a value from the vector). This kind of operation does not modify the vector data and therefore being in a Vector data structure provides an unnecessary overhead due to the synchronization of the class. Hence, plugins Vectors data structures were replaced by ArrayList to avoid this overhead. This substitution despite offers a performance improvement, also provides a better programing methodology since Vector class is already deprecated.
Besides this change at the plugin level, it was also made in all EuGene application classes. The usage of only Vector classes could bring down system overall performance and not only
1
Swing is the primary Graphical User Interface provided by Java. Since Swing components are fully imple-mented in Java they are plataform-independent and therefore a good choice for use in distinct environments
at the plugins runtime. This change was carefully done since some vectors really needed to be synchronized - it was used the Collections.synchronizedList(List) to overcome this synchro-nization problems. Despite being out of this thesis outline, it was a simple task that enhance EuGene overall performance.
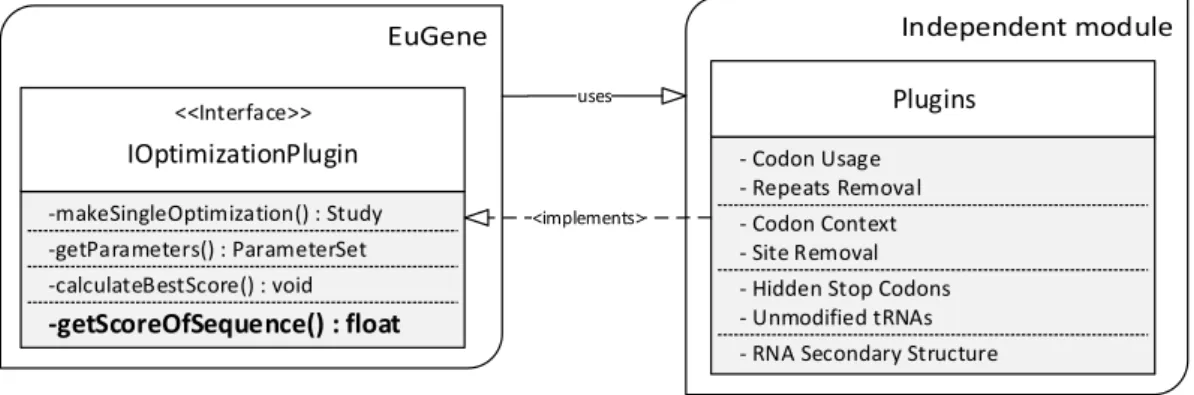
Finally, an individual analysis was performed to each plugin resulting in some changes that enhance their performance. Before those changes are presented, it is relevant to known how plugins are implemented generally. In order to develop a gene redesign method that can fit in EuGene, from the software developing point of view, it needs to respect a contract -interface. This interface forces the implementation of several methods that need to exists in the new plugin so it can be integrated by EuGene. Figure 4.4 enhance some of the methods that a plugin should implement. Relatively to performance, the most relevant method is the
<<Interface>> IOptimizationPlugin -makeSingleOptimization() : Study -getParameters() : ParameterSet -getScoreOfSequence() : float -calculateBestScore() : void Plugins - Codon Usage - Codon Context - Unmodified tRNAs - Hidden Stop Codons - Repeats Removal
- Site Removal
- RNA Secondary Structure <implements>
uses
Figure 4.4: Interface that a plugin must respect in order to be used in EuGene
getScoreOfSequence. Its objective is to evaluate a given input codon sequence and return a score from 0 to 100 where 100 is the optimum score. This result is obtained by checking how much the input sequence matches the plugin best possible score. Hence, it is noticeable that all the effort done by the plugins occur in this method since it is the one who will evaluate every new input sequence provided by the evolutionary algorithm.
Therefore it is now possible to understand the importance of this method and why it needs to be improved, even in a slighter way. The next subsections show what modification were done at each gene redesign method individually that can toggle their performance.
4.4.1 Codon Usage
The Codon Usage plugin has a set of distinct parameters like maximize or minimize the codon usage for a given gene. Also, it allows the gene customization using RSCU (Relative Synonymous Codon Usage) and CAI (Codon Adaptation Index) approaches. The main goal is to count the number of times each of the 64 distinct codons appear using one of the previous approaches. For instance, if the chosen parameter is “Maximize”, than this plugin objective is to replace each codon in the gene sequence by the synonymous one that appears often in the whole genome.
The bottleneck of each plugin relies in the getScoreOfSequence method, and hence, fig-ure 4.5 shows a simple version of the algorithm used by this plugin.
This algorithm is quite simple, where a unique iteration over all the codon sequence is enough to get the codon usage value. However, the get methods used to obtain the RSCU