UNIVERSIDADE FEDERAL DE OURO PRETO
DEPARTAMENTO DE FÍSICAMESTRADO EM CIÊNCIAS: ÊNFASE EM FÍSICA DE MATERIAIS
Efeitos Optoeletrônicos em Copolímeros do Fluoreno
Márcio Marques da Silva
Ouro Preto - MG
Márcio Marques da Silva
Efeitos Optoeletrônicos em Copolímeros do Fluoreno
Dissertação apresentada ao Departamento de Física como requisito parcial para obtenção do título de Mestre em Ciências.
Orientador(a): Profa. Dra. Melissa F. S. Savedra
Ouro Preto - MG
Catalogação: sisbin@sisbin.ufop.br
S586e Silva, Márcio Marques da.
Efeitos optoeletrônicos em copolímeros do fluoreno [manuscrito] / Márcio Marques da. – 2014.
54f.: il. color., grafs.
Orientadora: Profa. Dra. Melissa Siqueira.
Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas. Departamento de Física.
Área de concentração: Física de Materiais
1. Estrutura eletrônica - Teses. 2. Detectores nucleares de semicondutor - Teses. 3. Polímeros - Teses. I. Siqueira, Melissa. II. Universidade Federal de Ouro Preto. III. Título.
Agradecimentos
À minha Orientadora, Profa. Dra. Melissa Siqueira pela oportunidade de sempre aprender, pelo fundamental estímulo a pesquisa científica e pela liberdade intelectual permitida que estou certo ter comtribuído bastante para meu desenvolvimento profissional.
À minha família, em especial minha noiva Fátima, pela paciência e companheirismo. Estes apostaram em minha formação mesmo sem entenderem muitas vezes o que eu fazia.
Aos amigos do curso, sempre divertidos e ao mesmo tempo muito comprometidos com seus trabalhos, mas sempre boas companhias para um café.
Aos amigos João Paulo e a Nathália Yoshioka pela amizade e acaloradas discussões compartilhadas.
Ao Prof. Marcello Savedra pelas boas discussões e contribuições para o trabalho. E aos nobres companheiros dos seminário da Melissa: Carlos Eduardo e Lucas Correia.
Aos amigos de velhos “carnavais”, Sérgio Borba, Márcio Ribeiro, Renata Silveira, Fernanda Karoline(a maga), Thiago Menotti, Valfrido Furtado (Fenômeno), ao velho Roberto Eli (Barraca) e o nobre Paulo “Batata”.
Ao CNPq e a FAPEMIG que forneceram o suporte financeiro para esta pesquisa.
Resumo
Nesta dissertação de mestrado foram realizadas caracterizações ópticas, elétricas e es-truturais de dois copolímeros do fluoreno a partir de cálculos baseados na Teoria do Funcional da Densidade (DFT) e semi-empíricos. As estruturas eletrônica, molecular, e a relação conformacional com algumas propriedades optoeletrônicas e espectroscópicas dos copolímeros baseados em fluoreno, poli[(9,9-dioctil-2,7-divinilenofluorenileno)-alt-co-(2-metoxi-5-(2-etilexiloxi)-1,4-fenileno)] e poli[(9,9-diexilfluorenil-2,7-diil)-alt-co-(2-metoxi-5-2-etilexiloxi-1,4-fenileno)], renomeados CF108 e CF136 respectivamente, foram analisadas iniciando por estruturas constituídas por uma unidade monomérica, e então expandidas até onze unidades. Tal procedimento foi realizado para investigar como o crescimento das cadeia oligoméricass (e consequente aumento da conjugação) influencia os espectros UV-Vis e as energias dos orbitais de fronteira, onde foi observado deslocamento batocrômicos dos espectros e um decréscimo linear da diferença energética dos orbitais HOMO-LUMO. As distribuições HOMO e LUMO, energia total, momento de dipolo total foram investigados para cada molécula e fazem parte da caracterização optoeletrônica, onde uma análise detalhada das excitações do espectro de absorção eletrônico calculado teoricamente dos sistemas citados acima foi realizada, e verificou-se as transições envolvidas nos estados excitados característicos. A caracterização estrutual para os dois sistemas consistiu em analisar os efeitos da inclusão do grupo funcional vinil entre o fluoreno do monômero da molécula CF108, bem como o feito da variação dos ângulos diédros nas propriedades optoeletrônicas de ambos os sistemas. Observa-se que o vinil parece contribuir para a esta-bilidade energetica do primeiro sistema supracitado em relação ao segundo. A magnitude do momento de dipolo total dos oligômeros CF136, em geral, é menor em comparação com os correspondentes do outro material. Contudo, as informações concernentes as correlações expostas acima tem papel central para a caracterização e otimização das propriedades de sistemas conjugados, o que pode ser denominado engenharia molecular.
Lista de ilustrações
Figura 1 – Previsão de crescimento do consumo global de painéis OLED, estimada para o período de 2013 a 2018, segundo pesquisa de mercado realizada pela iSuppli [6]. . . 14 Figura 2 – Exemplos de aplicações e dispositivos que utilizam sistemas orgânicos. 16 Figura 3 – Orbitais híbridos do carbono que formam ligações σ e π [12]. . . 17
Figura 4 – Diagrama da diferença entre as bandas de energia de alguns materiais em temperaturas acima de 0K. . . 18 Figura 5 – Diagrama de energia dos orbitais, representando a sua evolução em
relação ao aumento da cadeia e os respectivos “gaps” de energia (Eg). . 18
Figura 6 – Representação estrutural do poli(acetileno). . . 19 Figura 7 – Esquema da estrutura básica de um OLED, constituído por cinco
camadas: substrato, ánodo, camada condutiva, camada emissiva e cátodo [18]. . . 20 Figura 8 – Esquema de copolimerização: duas ou mais moléculas são agregadas
(copolimerizadas) e podem emitir em diferentes faixas do espectro ele-tromagnético. Na parte supeior, estão três substâncias que combinadas formam o copolímero CF108 e o CF136, este último é formado quando o fluoreno é associado apenas ao MEH-PPV. . . 22 Figura 9 – Representação estrutural do polifluoreno, onde “n” é o número de
unidades básica da molécula (unidades monoméricas).. . . 22 Figura 10 – Representação estrutural dos copolímeros CF108 (a) e CF136 (b), onde
“n” é o número de unidades básica da molécula (unidades monoméricas). 23 Figura 11 – Representação esquemática do primeiro teorema de Hohenberg-Kohn,
o qual ilustra a influência de um elétron sobre a densidade eletrônica total de um sistema. . . 28 Figura 12 – Esquema que representa interdependência das variáveis básicas no
Teorema de Hohenberg-Kohn: densidade eletrônica (ρ), potencial externo
(ν), hamiltoniano (H), número de elétrons (N) e a energia do estado
fundamentel (E). . . 28
Figura 13 – Representação das etapas realizadas por um algoritmo genético, onde uma população evolui após sofrer operações genéticas (cruzamento e mutação) por gerações produzindo indivíduos mais aptos.. . . 31 Figura 14 – Representação esquemática da metodologia utilizada neste trabalho,
Figura 15 – Representações estruturais planares e não-planares para os dímeros dos copolímeros estudados: (a) 2CF108: planar, (b) 2CF136: planar, (c) 2CF108: não-planar e (d) 2CF136: não-planar. Os átomos em azul(formam um ângulo diedro) e cinza representam carbonos, os ver-melhos correspondem aos átomos de oxigênio e os brancos hidrogênios. 36 Figura 16 – Representações estruturais do oligômero 11CF108: (a) vista frontal e
(b) vista de cima. . . 38 Figura 17 – Representações estruturais do oligômero 11CF136: (a) vista frontal e
(b) vista de cima. . . 39 Figura 18 – Energia total calculada para oligômeros do nCF108 e nCF136, sendo
n=1,2,3,5,7,9,11, otimizados em nível semi-empírico. . . . 40
Figura 19 – Energia total calculada para oligômeros do nCF108 e nCF136, sendo
n=1,2,3,5,7 em nível PBE0/6-31G(d,p). . . . 42
Figura 20 – Gap de energia dos orbitais de fronteira HOMO-LUMO calculado para oligômeros do nCF108 e nCF136, sendo n=1,2,3,5,7 em nível PBE0/6-31G(d,p). . . 43 Figura 21 – Momento de dipolo total calculado para oligômeros do nCF108 e nCF136,
sendo n=1,2,3,5,7 em nível PBE0/6-31G(d,p). . . . 44 Figura 22 – Relação entre as energias dos orbitais de fronteira (HOMO e LUMO) e o
número de unidades monoméricas (n) do oligômero 11CF108, calculadas com o método PBE0/6-31G(d,p). . . 45 Figura 23 – Relação entre as energias dos orbitais de fronteira (HOMO e LUMO) e o
número de unidades monoméricas (n) do oligômero 11CF136, calculadas com o método PBE0/6-31G(d,p). . . 46 Figura 24 – Representações dos orbitais de fronteira, HOMO (a) e LUMO (b), do
oligômero 11CF108. As intensidades dos orbitais moleculares estão representadas numericamente como positiva (azul) e negativa (vermelho). 48 Figura 25 – Representações dos orbitais de fronteira, HOMO (a) e LUMO (b), do
oligômero 11CF136. As intensidades dos orbitais moleculares estão representadas numericamente como positiva (azul) e negativa (vermelho). 50 Figura 26 – Espectros de absorção estimados de diferentes tamanhos de cadeia
oligomérica do nCF108 (a) e nCF136 (b), utilizando a metodologia ZINDO/S-CI. Sendo n= 1,2,3,5,7,9,11. . . 52 Figura 27 – Espectros de absorção teóricos e transições normalizadas para os
Lista de tabelas
Lista de abreviaturas e siglas
CI Interação de Configurações (Configuration Interaction)
CIS Interação de configurações de excitações simples (Configuration Inte-raction Singles)
DFT Teoria do funcional da densidade (Density Functional Theory)
Eg Diferença de energia entre os orbitais LUMO e HOMO (“gap” ou lacuna
de energia)
EF Nível de Fermi
HF Hartree-Fock
HOMO Orbital molecular de maior energia completamente preenchido (Highest Occupied Molecular Orbital)
EO Eletrônica Orgânica
GGA Aproximação de Gradientes Generalizados (Generalized Gradient Ap-proximation)
LDA Aproximação de Densidade Local (Local Density Approximation)
LUMO Primeiro Orbital Molecular Desocupado (Lowest Unoccupied Molecular Orbital)
OLED Diodo emissor de luz orgânico (Organic Light Emitting Diode)
MEH-PPV Poli(2-metóxi-5-(2-etil-hexilóxi)-p-fenilenovinileno)
NDDO Negligência da Sobreposição Diferencial Diatômica (Neglect of Diatomic Differential Overlap)
PDI Pesquisa, Desenvolvimento e Inovação
PF Polifluoreno
PM6 Método Paramétrico 6 (Parametric Method 6)
PM6-DH+ Método Paramétrico 6, com correções de terceira geração
TV Televisor
Sumário
Introdução . . . 13
1 Eletrônica Orgânica . . . 15
1.1 Semicondutores Orgânicos . . . 17
1.1.1 Aspectos Fundamentais . . . 17
1.1.2 Propriedades Eletrônicas de Sistemas Conjugados . . . 17
1.1.3 Propriedades . . . 20
1.2 Sistemas Baseados em Fluoreno . . . 21
2 Objetivos . . . 24
3 Métodos de Mecânica Quântica Molecular e Algoritmo Genético. . 25
3.1 Métodos Semi-empíricos. . . 26
3.1.1 PM6-DH+ . . . 26
3.1.2 Interação de Configurações de Excitações Simples (CIS) . . . 27
3.1.3 Método ZINDO/S-CI . . . 27
3.2 Teoria do Funcional da Densidade (DFT) . . . 28
3.2.1 Funcional de Correlação e Troca . . . 29
3.3 Algoritmo Genético . . . 31
3.4 Considerações Computacionais . . . 31
4 Metodologia . . . 33
5 Resultados e Discussões . . . 35
5.1 Propriedades Estruturais . . . 35
5.2 Propriedades Eletrônicas . . . 40
5.3 Orbital Molecular . . . 47
5.4 Espectroscopia de Absorção Eletrônica . . . 51
Conclusão. . . 56
Trabalho em andamento . . . 58
Produção Científica . . . 59
15
Introdução
A busca por materiais adequados para serem utilizados em dispositivos optoeletrô-nicos e fotôoptoeletrô-nicos tem ganhado caráter e interesse interdisciplinares. Materiais que exibem propriedades eletrônicas e ópticas ajustáveis se fazem indispensáveis para a indústria fabri-cante de tais dispositivos. Por mais de duas décadas, os polímeros e oligômeros conjugados têm sido intensivamente investigados como novos materiais funcionais para aplicações em áreas como eletrônica, fotônica, em aplicações como transistores, diodos, transdutores, entre outras [1–4]. Estes materiais combinam as propriedades elétricas dos semicondutores com propriedades típicas de polímeros convencionais (plásticos) como versatilidade de síntese, relativa facilidade de processamento, transparência óptica e flexibilidade mecânica. Além destas vantagens, estes materiais podem ainda ser quimicamente funcionalizados, o que lhes conferem grande versatilidade, pois assim é possível realizar ajustes de suas propri-edades optoeletrônicas. No ano de 2007, a Sony anunciou ao mercado a primeira TV-OLED (Organic Light Emitting Diode) [5]. As espectativas atuais, referentes às aplicações de
materiais orgânicos emissores de luz em dispositivos, são bastante otimistas. Especialistas do mercado de eletrônicos estimam para o próximo ano (2015) uma demanda de mais de 2 milhões de unidades de painéis, cuja matriz ativa são polímeros semicondutores, como apresentado na Figura 1.
Previsão de Demanda Global de Painéis OLED
(Em Milhões de Unidades)
16
1 Eletrônica Orgânica
A eletrônica orgânica (EO) é uma área emergente e muito ativa devido, tanto à ampla variedade de aplicações de sistemas orgânicos semicondutores, como também por suas interessantes características físico-químicas que podem determinar seu funcionamento. O uso de materiais orgânicos em dispositivos eletrônicos vem sendo objeto de pesquisa há mais de três décadas e começou a ser visto como ponto estratégico na política de Pesquisa desenvolvimento e inovação (PDI) [7] de países desenvolvidos como Reino Unido e Alemanha, bem como de países emergentes como o Brasil [8]. Essas medidas visam substituir, com grandes vantagens, os usuais sólidos inorgânicos em vários segmentos da indústria, dentre outras ações que impulsionem o desenvolvimento de novas tecnologias à base de materiais poliméricos, em especial para a indústria de eletrônicos, que tem sido uma das maiores investidoras de PDI para o desenvolvimento de semicondutores orgânicos [9]. Conforme pode ser observado na Figura 2, esses materiais orgânicos podem ser utilizados para diversas aplicações e finalidades, como informática, comunicação, saúde e sensoriamento, entre eles estão televisores, painéis de alta resolução e outros acessórios de informática, sensores de pressão flexíveis de alta sensibilidade, dispositivos biocompatíveis, entre outros (ver Figura 2).
Saúde
Sensores
Informática
Comunicações
Capítulo 1. Eletrônica Orgânica 17
Dentre os exemplos apresentados, naFigura 2há uma representação de um sensor de pressão de alta sensibilidade capaz de detectar a presença de uma mosca, cuja massa implica uma pressão equivalente 3 P a (Pascal) sobre a área do dispositivo [10].
1.1 Semicondutores Orgânicos
1.1.1 Aspectos Fundamentais
O silício é, sem dúvida, o material semicondutor mais usado em dispositivos eletrônicos [11]. Entretanto, há atualmente demandas reais que não são atendidas em sua plenitude por materiais convencionais como, por exemplo, a flexibilidade exibida nos plásticos. Esta impossibilidade foi um prelúdio de uma nova revolução tecnológica, que vem ocorrendo em função da utilização de sistemas orgânicos π-conjugados para fabricação
de dispositivos eletrônicos.
As características semicondutoras de sistemas orgânicosπ-conjugados são
prove-nientes da alternância entre ligações σ e π, e apenas σ que lhes confere a propriedade
de conduzir carga. A Figura 3 representa a ligação dupla entre dois átomos de carbono adjacentes, em que a sobreposição de dois orbitais 2sp2 forma uma ligação σ e os dois
orbitais 2pz, a ligação π.
Figura 3 – Orbitais híbridos do carbono que formam ligações σ e π [12].
A diferença de simetria destes orbitais leva à separação dos estados eletrônicos moleculares e como as ligações π são mais fracas que as σ, os elétrons daquelas ligações
em sistemas conjugados costumam ser deslocalizados ao longo de toda a cadeia molecular principal.
1.1.2 Propriedades Eletrônicas de Sistemas Conjugados
Capítulo 1. Eletrônica Orgânica 18
de energia se tornam tão próximos que são indistinguíveis. Um sólido qualquer possui grande quantidade de bandas de energia, entretanto nem todas estão preenchidas por elétrons. A probabilidade de uma banda particular ser preenchida é dada pela estatística de Fermi-Dirac, Equação 1.1, que estima, a temperatura ambiente, que as bandas são ocupadas até o nível de Fermi (EF) [13].
f(E) = 1 eE−EF
KT + 1
(1.1)
, onde E é energia em um dado estado,k é a constante de Boltzmann eT é a temperatura.
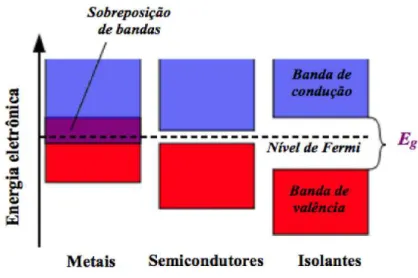
Entre duas bandas energéticas acessíveis (banda de valência - a mais alta ocupada, e de condução - mais baixa desocupada) há uma região proibida aos elétrons, conhecida como banda ou lacuna proibida, ou ainda pelo termo em inglês “gap”. O valor desta grandeza, à temperatura ambiente, determina se o sólido comporta-se como condutor (quando a banda de condução está parcialmente preenchida), isolante (quando a banda de valência está completamente ocupada e a de condução completamente vazia) ou semicondutor (quando o gap entre as bandas de valência e condução é pequeno o bastante para que
elétrons da primeira banda consigam acessar a segunda). Na Figura 4, metais apresentam alta condutividade elétrica em decorrência da sobreposição das bandas de valência e de condução. Entretanto, em materiais isolantes existe uma distância considerável entre as bandas que dificulta a passagem dos portadores de carga. Enquanto que os semicondutores apresentam estrutura de bandas e propriedades elétricas intermediárias entre os dois últimos materiais.
Figura 4 – Diagrama da diferença entre as bandas de energia de alguns materiais em temperaturas acima de 0K.
Capítulo 1. Eletrônica Orgânica 19
Molecular Orbital), também conhecida “gap” ou “band gap” (Eg), é normalmente uma
região de energia proibida, e portanto esta deve ser a energia mínima necessária para promover um elétron do orbital molecular HOMO para o LUMO, além de ser responsável pelas intrínsecas propriedades optoeletrônicas de sistema π-conjugados. A capacidade de
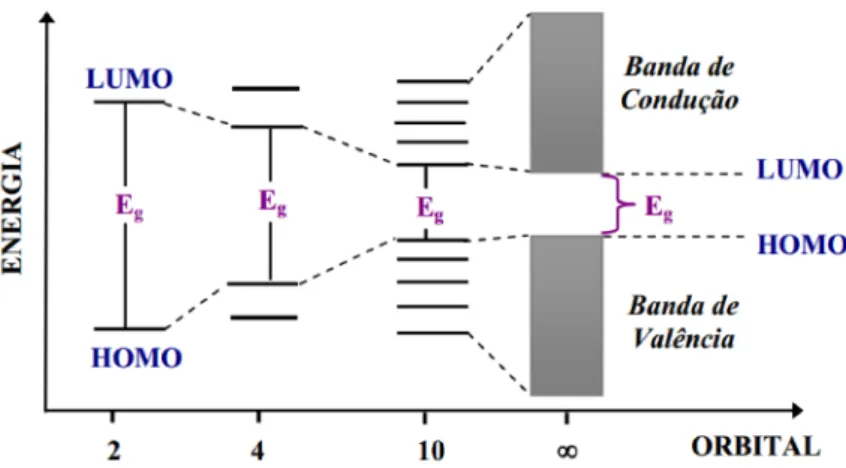
controlar tal energia é desafio fundamental na área dos semicondutores orgânicos por sua inerente conexão com a dinâmica e propriedades destes sistemas. A Figura 5 representa, esquematicamente, as bandas de energia para sistemas orgânicos conjugados, ou seja bandas de energia dos orbitais moleculares.
Figura 5 – Diagrama de energia dos orbitais, representando a sua evolução em relação ao aumento da cadeia e os respectivos “gaps” de energia (Eg).
O poliacetileno (Figura 6) foi o primeiro polímero conjugado a apresentar con-dutividade elétrica, e entre outras utilidades, foi usado no desenvolvimento de células fotovoltaicas [14]. Mesmo representando o sistema conjugado mais simples existente, um grupo de pesquisadores liderados por A. J Heeger, A. G. MacDiarmid e H. Shirakawa demonstraram que a magnitude da condutividade elétrica do poliacetileno poderia ser aumentada por fator de ordem 13 pela dopagem com sistemas aceitadores e doadores de elétrons [15].
Capítulo 1. Eletrônica Orgânica 20
1.1.3 Propriedades
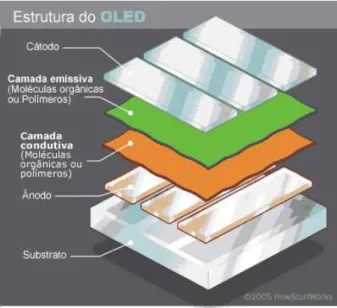
Os semicondutores orgânicos combinam propriedades semicondutoras e mecânicas com a facilidade de deposição em grandes áreas e nas mais variadas superfícies [1]. Esses materiais têm maior facilidade para funcionalização e fabricação, bem como menor custo em relação aos materiais inorgânicos. Estes compostos possibilitam a manufatura de filmes finos flexíveis, da ordem de nanômetros, o que é impraticável com qualquer material inorgânico, uma vez que estes são constituídos por estruturas cristalinas rígidas [16]. Além disso, formam uma classe muito importante de materiais eletro e fotoativos e o uso destes sistemas em dispositivos optoeletrônicos tem atravessado as fronteiras dos laboratórios e institutos de pesquisa para fazerem parte do cotidiano da população, seja em forma de sensores, televisores, celulares entre outros produtos de alta tecnologia, como discutido na seção 1 [17]. A Figura 7 apresenta a estrutura e os componentes de um diodo orgânico emissor de luz.
Figura 7 – Esquema da estrutura básica de um OLED, constituído por cinco camadas: substrato, ánodo, camada condutiva, camada emissiva e cátodo [18].
A base da estrutura do OLED é o substrato, que pode ser vidro, óxido de índio e estanho, ou um plástico (painéis OLED flexíveis). Um eletrodo (ânodo) remove elétrons quando uma corrente circula pelo dispositivo. Então, as camadas orgânicas começam a operar. A primeira é condutora, que transporta os portadores de carga do ânodo. A segunda camada é emissora, constituída por moléculas diferentes da camada condutora e que transporta os elétrons para outro eletrodo, o catodo.
1.2 Sistemas Baseados em Fluoreno
Capítulo 1. Eletrônica Orgânica 21
aplicações em dispositivos eletrônicos. Os éxcitons formados nas cadeias deste tipo de sistema, pela absorção de energia eletromagnética, são capazes de migrar ao longo da cadeia antes de serem desativados [19,20]. Este deslocamento ocorre por efeitos radiativos e/ou não radiativos até o retorno ao estado fundamental, ou pela dissociação em portado-res de carga. Materiais usados para o desenvolvimento de diodos orgânicos emissoportado-res de luz (OLED) devem ser eficientemente fluorescentes, logo deverão apresentar moderada mobilidade excitônica.
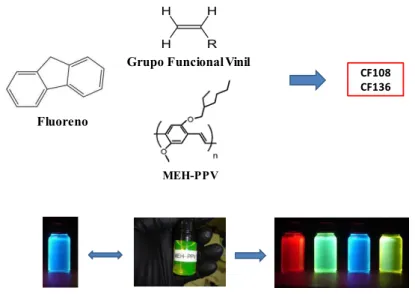
O comprimento de onda emitido por um polímero semicondutor pode ser modulado através da copolimerização, ou seja, pela polimerização simultânea de uma quantidade igual ou maior que duas espécies químicas. A Figura 8 apresenta um esquema que mostra três moléculas distintas, cada uma com suas características específicas, por exemplo o poli(2-metóxi-5-(2-etil-hexilóxi)-p-fenilenovinileno), conhecido como MEH-PPV, que emite por volta de 600nm; contudo quando agregado ao fluoreno, passa a emitir em
outro comprimento de onda como ilustrado. Portanto, é possível funcionalizar um sistema orgânico para lhe conferir emissão de radiação em outro comprimento de onda através da copolimerização.
Grupo Funcional Vinil
CF108 CF136
Fluoreno
MEH-PPV
Figura 8 – Esquema de copolimerização: duas ou mais moléculas são agregadas (copoli-merizadas) e podem emitir em diferentes faixas do espectro eletromagnético. Na parte supeior, estão três substâncias que combinadas formam o copolímero CF108 e o CF136, este último é formado quando o fluoreno é associado apenas ao MEH-PPV.
Dentre a vasta gama de compostosπ-conjugados com potencialidades para serem
empregados na eletrônica orgânica, particularmente, os polifluorenos (PFs), como apre-sentado na Figura 9, os quais são compostos policíclicos aromáticos formados por uma região π-conjugada e uma apolar(insaturada) têm se destacado como uma importante
Capítulo 1. Eletrônica Orgânica 22
Além disso, são capazes de exibir uma eficiente eletroluminescência, alta mobilidade de carga e boa estabilidade térmica [21].
Figura 9 – Representação estrutural do polifluoreno, onde “n” é o número de unidades básica da molécula (unidades monoméricas).
A química do fluoreno teve inicio em 1867 quando Marcellin Berthlot isolou uma nova substância a partir do fracionamento do óleo de antraceno durante suas pesquisas com hidrocarbonetos. Ele ficou impressionado pela emissão fluorescente do material, mais profunda que a do próprio antraceno, que lhe deu o nome fluoreno. Mais tarde, em 1883, Hodgkinson e Matthews demonstraram que esta intensa emissão era proveniente de impurezas que poderiam ser removidas. No entanto, o nome inicialmente atribuído ao novo composto por Berthlot permaneceu [22].
O fluoreno, na forma de homopolímero emite intensa cor azul e, através de dopagem ou copolimerização, é possível manipulá-lo quimicamente para que possa emitir em ampla faixa do espectro visível [23]. O fluoreno contendo pirenil como substituinte foi reportado e caracterizado como injetor de buracos e emissor de luz azul simultaneamente [24,25].
Sistemas aceitadores e doadores baseados no fluoreno tem sido estudados com relação à absorção de dois e três fótons em função do tamanho da cadeia e da eficiência da fluorescência. Foi observado que o aumento da conjugação nem sempre melhora a eficiência quântica de fluorescência e a absorção de dois fótons [26,27].
Um estudo comparativo entre as propriedades óticas de polímeros emissores em função da conformação da cadeia principal quando aplicada uma pressão nesta também foi reportado, onde os autores informam ser possível verificar os mecanismos de transferência energética através dos efeitos da pressão sobre a cadeia principal do sistema composto por polifluorenos com cadeias laterais substituídas por grupos alquila. Observou-se que sobre pressão, o sistema que emitia no comprimento de onda referente a cor azul passa a emitir na cor verde [28].
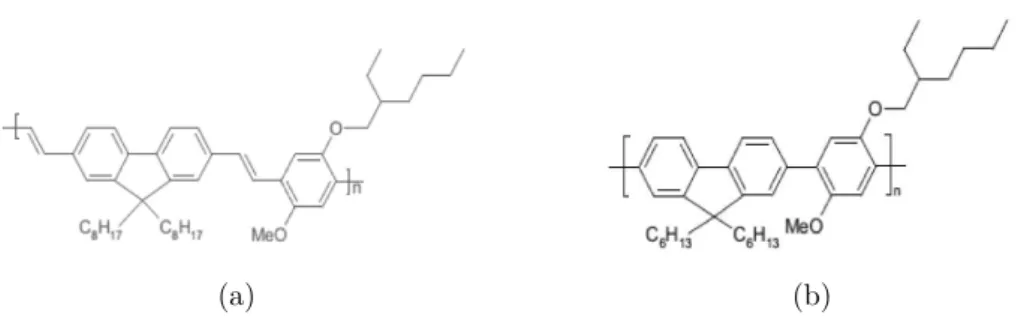
poli[(9,9-dioctil-2,7-divinilenofluorenileno)-Capítulo 1. Eletrônica Orgânica 23
alt-co-(2-metoxi-5-(2-etilexiloxi)-1,4-fenileno)] e poli[(9,9-diexilfluorenil-2,7-diil)-alt-co-(2-metoxi-5-2-etilexiloxi-1,4-fenileno)], que neste trabalho são denominados de CF108 e CF136, respectivamente.
(a) (b)
Figura 10 – Representação estrutural dos copolímeros CF108 (a) e CF136 (b), onde “n” é o número de unidades básica da molécula (unidades monoméricas).
O CF108 apresenta coloração amarelada, temperatura de decomposição acima de 100oC e máximo comprimento de absorção igual a 481nm [29]. O composto CF136 possui
coloração amarela mais clara e máximo comprimento de absorção igual a 374nm [29], o
que representa uma diferença de mais de 100nm em relação ao do sistema anterior. Como
24
2 Objetivos
25
3 Métodos de Mecânica Quântica Molecular
e Algoritmo Genético
Métodos teóricos de mecânica quântica (MQ) implementados em sistemas computa-cionais têm contribuído de forma importante para a investigação de sistemas moleculares. A teoria aliada ao avanço da capacidade de processamento dos computadores tem possi-bilitado o tratamento de moléculas cada vez mais complexas. Desta forma, nos últimos anos tem sido possível compreender e estimar propriedades de sistemas moleculares de maneira a complementar as caracterizações experimentais [20,32] e fazer inferências mais abrangentes acerca do seu comportamento físico-químico.
Para obtenção das propriedades eletrônicas e estruturais de moléculas e sólidos é preciso conhecer a função de onda do sistema. Tal função é obtida através da solução da equação de Schr¨odinger, ou Dirac, que descreve o sistema quântico [33], cuja forma é dada
pela seguinte expressão (para o primeiro caso):
ˆ
H(~r, ~R)Φ(~r, ~R) = EΦ(~r, ~R) (3.1)
que depende das coordenadas espaciais dos elétrons (~r) e núcleos (R~) do sistema, o
que leva a existência de termos de interação coulombiana elétron-núcleo, elétron-elétron, núcleo-núcleo e do operador Hamiltoniano ( ˆH(~r, ~R)), e é utilizada a aproximação de Born-Oppenheimer.
Esta leva em consideração que os núcleos possuem massa muito maior que a dos elétrons (me
mp ≈ 10
−4) e sua energia cinética será muito pequena em relação à energia
cinética dos mesmos. E, portanto, a aproximação de Born-Oppenheimer consiste em considerar que o movimento dos elétrons não sofre influência do movimento dos núcleos, o que equivale a dizer que estes estão em repouso (ou movendo-se muito lentamente em relação aos elétrons) [34]. Desta maneira é possível separar a função de onda eletrônica da nuclear e uma integral do hamiltoniano deixa de ser calculada. Essas considerações são utilizadas na maioria dos métodos de MQ aplicados a sistemas complexos.
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 26
quântica e algoritmo genético utilizados neste trabalho.
3.1 Métodos Semi-empíricos
A modelagem de sistemas orgânicos, utilizando métodos semi-empíricos, é uma importante maneira de avaliar as propriedades eletrônicas e estruturais de sistemas com muitos elétrons, inclusive de sistemas biológicos (como proteínas) ou cadeias oligoméricas, dentre outros. Os métodos semi-empíricos de Mecânica Quântica são baseados no forma-lismo de Hartree-Fock, porém possuem algumas aproximações e determinados parâmetros são obtidos a partir de dados experimentais. Eles são muito importantes para a física e química computacional para o tratamento de sistemas contendo muitos átomos, para os quais o método de Hartree-Fock completo, sem as aproximações, é bastante “caro” computacionalmente. A utilização de parâmetros empíricos possibilita a inclusão de efeitos de correlação eletrônica na metodologia autoconsistente. Existem várias aproximações e variações de métodos semi-empíricos [35–38], entretanto neste trabalho serão tratados apenas os que foram utilizados.
3.1.1 PM6-DH+
O método semi-empírico PM6 (Método Paramétrico 6) é um método semi-empírico
oriundo do aprimoramento do formalismo NDDO, do termo em inglês“Neglect of Diatomic Differential Overlap”, ou Negligência da Sobreposição Diferencial Diatômica, aproximado
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 27
3.1.2 Interação de Configurações de Excitações Simples (CIS)
Cálculos de interações de configurações podem ser formulados a partir do estado fundamental de Hartree-Fock, Φ0(r), que corresponde ao melhor determinante de Slater [39]
que descreve o estado de menor energia do sistema, que pode ser escrito como:
Φ0(r) =|φ1(r)φ2(r)φ3(r)...φn(r)| (3.2)
Para determinar os estados excitados é necessário impor que a função de onda do estado i deve ser ortogonal às funções de onda dos estados i-1 anteriores. Entretanto,
é mais conveniente utilizar o método de interação de configurações (CI) para estimar a energia dos estados excitados. Então, com base no princípio de Franck-Condon [40], é possível calcular o espectro de absorção eletrônico na região do UV-Vis. A função de onda eletrônica Ψ é representada por uma combinação de determinantes de Slater e os elétrons dos orbitais ocupados da função de onda de Hartree-Fock são promovidos para orbitais virtuais. Contudo, tem-se que
Ψ =a0ΦHF +
X
S
asΦS+...=
X
i=0
aiΦi (3.3)
A série representada pela equação 3.3 deve ser truncada no índiceS, uma vez que
este refere-se às excitações simples. Portanto, os termos de ordens superiores não serão incluídos. Dado este fato, o método de CI que considera apenas excitações simples (CIS) não considera efeitos de correlação eletrônica.
3.1.3 Método ZINDO/S-CI
Este método supera a incapacidade de desdobrar estados provenientes de uma mesma configuração eletrônica inerente de aproximações, que desconsideram todas as integrais de repulsão eletrônica em que hajam produtos envolvendo orbitais distintos. O método ZINDO/S-CI (Zerner’s Intermediate Neglect of Differential Overlap/Spectroscopic-Configuration Interaction) foi desenvolvido por Michael C. Zerner e colaboradores, abrange
grande gama de átomos da tabela periódica, bem como sistemas orgânicos, além de ser parametrizado para estados excitados e introduzir o efeito de correlação eletrônica [41]. Portanto, este modelo teórico tem apresentado desempenho satisfatório para o tratamento de transições eletrônicas de compostos aromáticos [38,42] por levar em consideração integrais de troca necessárias para separar diferentes termos em uma configuração e aumentar a interação entre estados oriundos de transições π→π∗ eσ →σ∗ melhorando
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 28
3.2 Teoria do Funcional da Densidade (DFT)
A Teoria do Funcional da Densidade (DFT -Density Functional Theory) é uma
alternativa à teoria de estrutura eletrônica de Hartree-Fock para sistemas no estado fundamental, onde a distribuição da densidade eletrônica (ρ(r)) tem o papel principal ao
invés da função de onda multi-eletrônica. Esse método foi desenvolvido por Hohemberg e Kohn (HK) [43] e destacou-se por sua maneira diferente de descrever propriedades químicas e físicas de sistemas em estado sólido, moléculas e átomos em estado fundamental em relação a metodologia utilizada pelos métodos ab initio e semi-empíricos. O foco da DFT é a densidade eletrônica e a de correlação e troca, e descreve como a presença de um elétron em um dado ponto do espaço afeta a densidade total, conforme ilustra a Figura
11. Ou seja, a densidade eletrônica determina o potenial externo (ν(r)). O Hamiltoniano
(H) é definido a partir da aproximação de Born-Oppenheimer e ρ(r) determina a energia
do sistema e todas as propriedades eletrônicas no estado fundamental. Na Figura 12, a energia E é um funcional de ρ(r).
Figura 11 – Representação esquemática do primeiro teorema de Hohenberg-Kohn, o qual ilustra a influência de um elétron sobre a densidade eletrônica total de um sistema.
Estas quantidades são físicas e independentes de representação. Assim, a equação de Schr¨odinger polieletrônica, com sua função de onda de muitas variáveis, é reescrita
como uma função da densidade eletrônica ρ(r) com apenas três variáveis.
Figura 12 – Esquema que representa interdependência das variáveis básicas no Teorema de Hohenberg-Kohn: densidade eletrônica (ρ), potencial externo (ν), hamiltoniano
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 29
Supondo a existência de tal função, esta corresponde à densidade do estado funda-mental de um átomo ou molécula. De acordo com o primeiro teorema de HK, esta função corresponde a um número N de elétrons e um agregado de núcleos contendo quantidade,
carga e posição [44].
O segundo teorema de HK foi elaborado como uma maneira para encontrar a den-sidade (ρ) que minimiza a energiaE baseada no calculo de variações, onde o multiplicador
de Lagrange contém o denominado funcional de Hohenberg-Kohn (FHK) que por sua vez
possui o funcional de correlação e troca EXC[ρ], e corresponde ao termo que deve ser
aproximado na teoria [45]. Assim, a qualidade dos resultados do funcional da densidade depende principalmente da escolha da aproximação utilizada no funcional de correlação e troca (EXC) que podem ser do tipo LDA, GGA, híbridos e outras. Entre funcionais
disponíveis, os mais usados são B3LYP, B3PW91 e PBE.
A DFT tornou o problema de muitos corpos mais simples de ser tratado (“menor custo computacional”) e com satisfatória aproximação dos resultados, mesmo havendo um termo aproximado.
3.2.1 Funcional de Correlação e Troca
A efetiva implementação do formalismo de Kohn-Sham depende de uma aproxima-ção para o termo de correlaaproxima-ção e troca. Tal termo é devido ao fato de que elétrons tendem a se afastar por conta de suas cargas elétricas, e a energia associada a esta repulsão e movimento é dada quanticamente levando-se em consideração o spin eletrônico, uma vez
que o princípio de Pauli estabelece que dois férmions (partículas comspin semi-inteiro)
não podem ocupar o mesmo estado quântico, ou de forma equivalente, que a função de onda deve ser antisimétrica com relação a troca de duas partículas quaisquer.
A primeira aproximação sugerida foi a Aproximação da Densidade Local (Local Density Approximation - LDA), que ignora os aspectos não locais do potencial de correlação
Vxc (Eq. 3.5) que é uma derivada funcional da energia de correlação e troca em relação a
densidade, e assume que Exc pode ser escrita como:
Exc=
Z
εxcn(~r)d~r (3.4)
Vxc=δExc/δn (3.5)
Esta expressão depende apenas da densidade local, onde o termoεxcn(~r) é a energia
de correlação e troca por partícula de um sistema homogêneo de densidade n. Na LDA, é
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 30
proposta a substituição do termo εxcn(~r) por um que depende também do gradiente de
n(~r). Assim tem-se a chamada Aproximação do Gradiente Generalizado (Generalized Gradient Approximation - GGA) [46]:
EGGA
xc [n;∇~n] =
Z
d3~rf(n;∇~n). (3.6)
A escolha de f(n, ~∇n) depende do comportamento de εxcn(~r) em situações particulares.
Lembrando que se um funcional F[n] é dado por F[n] =R f(n(~r))d~r, então sua derivada
funcional é:
δF δn |~r0 =
df
dn |~r0 . (3.7)
Neste trabalho foi utilizado um funcional GGA híbrido que mistura a energia de troca exata(EHF
x ) com aproximações do funcional da densidade para o termo de correlação
e troca, cuja forma é:
ExcP BE0[ρ] =E P BE
xc [ρ] +α(E HF
x [Φ]−E
P BE
x [ρ]), (3.8)
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 31
3.3 Algoritmo Genético
Algoritmo genético é uma técnica de busca e otimização bioinspirada cuja idéia principal é que a partir de uma “população” inicial de estruturas, compostas por “cro-mossomos” e “genes”, dois indivíduos podem transmitir suas cargas genéticas para seus “filhos”. Neste processo é permitido ainda “mutações genéticas” nos indivíduos.
A Figura 13representa a idéia geral de um algoritmo genético que inclui operadores de cruzamento e mutação.
Figura 13 – Representação das etapas realizadas por um algoritmo genético, onde uma população evolui após sofrer operações genéticas (cruzamento e mutação) por gerações produzindo indivíduos mais aptos.
Pode se dizer que neste trabalho foi utilizado um modelo elitista, uma vez que indivíduos pior avaliados, baseado em uma função de qualidade, foram retirados da população e os melhores foram selecionados para próxima “geração” em proximidade com o princípio de sobrevivência do mais forte.
Considerando uma molécula contendo n ângulos diedros que podem constituir
10n diferentes conformações. Com uma população de digamos 150 estruturas distintas
caracterizadas por uma função de qualidade, por exemplo pelo momento de dipolo total, energia total ou alguma informação experimental. No caso da energia total, indivíduos com menor energia são melhores. Todos os indivíduos podem se repoduzir com probabilidade dependente de sua função de qualidade. Duas conformações filhas podem ser geradas tomando-se n-p ângulos diedros de uma das estruturas pai e o complementar da segunda.
Capítulo 3. Métodos de Mecânica Quântica Molecular e Algoritmo Genético 32
3.4 Considerações Computacionais
Em cálculos de mecânica quântica o preço “pago” por utilizar métodos com maior rigor teórico para tratar sistemas complexos, em termo de número de átomos, é a redução do espaço de Hilbert ou o uso de aproximações. Hartree-Fock (HF) é um dos métodos quânticos mais importantes, uma vez que considera até 99% da energia eletrônica, porém não considera os efeitos de correlação entre elétrons. Ainda assim é base para os métodos semi-empíricos. As equações de Hartree-Fock dependem de suas próprias soluções, portanto devem ser resolvidas iterativamente. Quando os orbitais moleculares são expandidos por um conjunto de funções de base, as equações resultantes são escritas como problemas de autovalores. Os elementos da matriz de Fock correspondem às integrais envolvendo um e dois elétrons (estas duas integrais apresentam termos fatorias de N, o número de elétrons
do sistema) sobre funções de base, multiplicadas pelos elementos da matriz densidade. Então, executando repetidas diagonalizações da matriz de Fock é possível obter a solução das equações de HF expandida em funções de base no espaço de Hilbert.
Dadas as operações mencionadas acima e o fato de que o problema deve ser resolvido de forma autoconsistente, o modelo de HF é classificado quanto à sua complexidade computacional como NP-Completo (Não Polinomial Completo), ou seja, não pode ser resolvido em tempo polinomial ou requer uma busca exaustiva no espação de configurações [48]. A metodologia empregada na teoria do funcional da densidade se compara, em termos da complexidade computacional, ao modelo de Hartree-Fock. Entretanto, o primeiro necessita menor capacidade de memória de acesso aleatório, uma vez que o problema deixa de depender da função de onda do sistema (3N variáveis mais spins) e passar a ser tratado
33
4 Metodologia
Neste trabalho foi utilizada a metodologia “stepping-stone”, aumentando gradu-almente o rigor teórico dos métodos para diminuir o tempo gasto durante o processo de otimização estrutural. Foram seguidas as metodologias PM6-DH+ [36] e DFT [43,44,50], como implementados nos aplicativos ORCA 2.9 [51] e MOPAC2012 [52]. Neste último, a energia de correlação e troca foi tratada de acordo com a aproximação do gradiente gene-ralizado (GGA), com o funcional híbrido de Perdew, Burke, e Ernzerhof (PBE0) [46,53], ambos associados à função de base de Pople 6-31G(d,p) [54]. A Figura 14 mostra esque-maticamente as etapas seguidas neste trabalho.
Capítulo 4. Metodologia 34
Em suma, os principais focos de cada etapa foram:
1. Relacionar o aumento do número de meros com as propriedades eletrônicas e ópti-cas utilizando Mecânica Quântica Molecular. Esta metodologia foi seguida até ser observado que para oligômeros com número de meros, n, maior que quatro não há
diferença significativa nas propriedades estruturais estimadas com PM6 e DFT. As análises foram realizadas desde monômeros até estruturas contendo onze unidades monoméricas;
35
5 Resultados e Discussões
5.1 Propriedades Estruturais
As propriedades eletrônicas e ópticas em sistemas π-conjugados estão intimamente
ligadas ao arranjo estrutural molecular. Para caracterizar as estruturas com ângulos diedros entre anéis aromáticos vizinhos menores e maiores que 300 ou complementares,
utilizou-se a nomenclatura “planar” e “não-planar”, respectivamente. AFigura15contém representações dos sistemas 2CF108 e 2CF136.
(a) (b)
(c) (d)
Capítulo 5. Resultados e Discussões 36
O aumento do número de unidades monoméricas afeta a planaridade dos oligômeros favorecendo torções entre meros e “intrameros” (torção entre o fluoreno e MEH-PPV em um mesmo monômero) da cadeia principal. Tal fato leva à formação de um zig-zag na estrutura que pode ser visto nas Figuras 16e 17.
(a)
(b)
Figura 16 – Representações estruturais do oligômero 11CF108: (a) vista frontal e (b) vista de cima.
(a)
(b)
Figura 17 – Representações estruturais do oligômero 11CF136: (a) vista frontal e (b) vista de cima.
Capítulo 5. Resultados e Discussões 37
5.2 Propriedades Eletrônicas
A energia total contém contribuições das energias de repulsão eletrônica e nucleares. Na Figura 18 é apresentado o gráfico de energia total em função do número de unidades monoméricas calculada para oligômeros, n= 1, 2, 3, ..., 11, para ambos os sistemas em análise, empregando-se o método semi-empírico.
Figura 18 – Energia total calculada para oligômeros do nCF108 e nCF136, sendo
n=1,2,3,5,7,9,11, otimizados em nível semi-empírico.
Foi observado para os dois sistemas estudados, que a energia decresce com o aumento da cadeia, ou seja, a inclusão gradual de unidades monoméricas. Conforme mostra a Figura 18há duas tendências de evolução energética. A primeira referente a oligômeros com até três unidades monoméricas em que as estruturas não possuem quantidade de meros necessários para formar estruturas torcidas e a segunda tendência foi observada para as estruturas que têm tamanho suficiente para tal formação. A partir de cinco unidades monoméricas são observadas estruturas torcidas.
Os coeficientes dos ajustes lineares efetuados para a primeira tendência das curvas de energia são apresentados na Tabela 1.
Capítulo 5. Resultados e Discussões 38
Tabela 1 – Resultado do ajuste linear calculado a partir das energias totais em função do número de unidades monoméricas dos dois oligômeros
Oligômeros a b
CF108 -0.674 -0.054 CF136 -0.684 -0.013
Estas informações ajudam a estimar a tendência energética em função do cres-cimento da cadeia para sistemas (mais) planares. Vale observar que as estruturas com torções na cadeia tendem a ser energeticamente mais estáveis.
Sabe-se que qualquer material exposto a um campo eletromagnético externo sofrerá perturbação em algumas propriedades e isso pode ser avaliado através do momento de dipolo (Equação 5.2) e da energia total (Equação 5.3).
µi =µ(0)i +
X
j
αijEj +
X
jk
βijEjEk+
X
jkl
γijklEjEkEl+..., (5.2)
sendo µi a i-ésima componente do momento de dipolo,Ei é o campo eletromagnético, µ(0)i
é o momento de dipolo estático, α é a polarizabilidade, eβ, γ,... são a primeira, segunda,...
hiperpolarizabilidades. Para a energia total, temos [56]
Etot =Etot(0)−
X
i
µiEi, (5.3)
onde Etot(0) é a energia total sem a presença de um campo externo.
Agora são mostrados resultados obtidos para algumas propriedades eletrônicas avaliadas como: a energia total, gap de energia HOMO-LUMO e momento de dipolo total dos oligômeros nCF108 e nCF136, sendo n = 1, 2, 3, 5 e 7. A magnitude das grandezas
Capítulo 5. Resultados e Discussões 39
Assim como observado para a energia total proveniente de cálculos semi-empíricos (Figura 18), os resultados calculados com a DFT mostraram a mesma tendência de diminuição. Verifica-se que a energia total tende a diminuir com relação ao aumento gradual das cadeias oligoméricas (Figura 19).
1 2 3 5 7
Meros(n) 0.00 10.00 20.00 30.00 40.00 50.00 60.00 70.00 En er gi a To ta l ( − 10
4 e
V)
CF108 CF136
Figura 19 – Energia total calculada para oligômeros do nCF108 e nCF136, sendo
n=1,2,3,5,7 em nível PBE0/6-31G(d,p).
A Figura 20 mostra que para o gap de energia HOMO-LUMO ocorre uma tentedência parecida com a observada para a energia total.
1 2 3 5 7
Meros(n) 0.00 1.00 2.00 3.00 4.00 5.00 6.00 7.00 8.00 9.00
Energia HOMO-LUMO (eV)
CF108 CF136
Capítulo 5. Resultados e Discussões 40
Durante o processo de análise conformacional foi observado que torções na cadeia principal tendem a aumentar a magnitude do momento de dipolo total. A Figura 21
apresenta a magnitude desta grandeza para alguns oligômeros, onde é possível ver que, em geral, esta tende a ser maior para o sistema CF108 em relação ao CF136.
1
2
Meros(n)
3
5
7
0.00 1.00 2.00 3.00 4.00 5.00 6.00 7.00 8.00
Momento de Dipolo Total (Debye)
CF108 CF136
Figura 21 – Momento de dipolo total calculado para oligômeros do nCF108 e nCF136, sendo n=1,2,3,5,7 em nível PBE0/6-31G(d,p).
Capítulo 5. Resultados e Discussões 41
Relacionando as energias dos orbitais moleculares de fronteira (HOMO e LUMO) com o número de unidades monoméricas dos oligômeros analisados, foi observada uma evolução primordialmente linear, como apresentam as Figuras 22 e 23.
Gap (Eg)
Figura 22 – Relação entre as energias dos orbitais de fronteira (HOMO e LUMO) e o número de unidades monoméricas (n) do oligômero 11CF108, calculadas com
o método PBE0/6-31G(d,p).
Gap (Eg)
Figura 23 – Relação entre as energias dos orbitais de fronteira (HOMO e LUMO) e o número de unidades monoméricas (n) do oligômero 11CF136, calculadas com o método PBE0/6-31G(d,p).
Capítulo 5. Resultados e Discussões 42
5.3 Orbital Molecular
Os orbitais moleculares podem prover informações acerca de algumas propriedades estruturais e físicas de sistemas moleculares. Os orbitais moleculares de fronteira estão relacionados à região em que um sistema conjugado poderá absorver e emitir radiação eletromagnética com maior probabilidade.
Nas Figuras 24 e 25, estão representados os orbitais moleculares estimados para estruturas constituídas por onze unidades monoméricas do CF108 e CF136. Deve-se notar que os valores das intensidades dos orbitais estão representados numericamente como positivo (azul) e negativo (vermelho).
(a)
(b)
Figura 24 – Representações dos orbitais de fronteira, HOMO (a) e LUMO (b), do oligô-mero 11CF108. As intensidades dos orbitais moleculares estão representadas numericamente como positiva (azul) e negativa (vermelho).
Capítulo 5. Resultados e Discussões 43
O orbital LUMO (Figura 25(b)) é atribuído à mesma região, mas a distribuição no oitavo fluoreno restinge-se ao primeiro anel aromático deste. Portanto, a distribuição é ainda mais localizada e, novamente, ocorre a inversão entre orbitais atômicos ligantes e antiligantes, bem como uma inversão de fase da distribuição no sétimo fluoreno comparado ao HOMO.
(a)
(b)
Figura 25 – Representações dos orbitais de fronteira, HOMO (a) e LUMO (b), do oligô-mero 11CF136. As intensidades dos orbitais moleculares estão representadas numericamente como positiva (azul) e negativa (vermelho).
Capítulo 5. Resultados e Discussões 44
5.4 Espectroscopia de Absorção Eletrônica
A fim de correlacionar o comportamento das bandas de UV-Vis com as conformações analisadas foram calculados espectros de absorção eletrônico empregando a metodologia ZINDO/S-CI. Os resultados espectroscópicos estimados foram comparados com os dados experimentais dos sistemas CF108 e CF136, encontrados na literatura [29].
Os espectros de UV-Vis teóricos calculados para os oligômeros (no vácuo) estão representados simultaneamente na Figura 26, onde pode ser observada a evolução da banda principal com o aumento da cadeia.
(a)
(b)
Capítulo 5. Resultados e Discussões 45
O deslocamento do espectro para regiões de menores energias, em geral, reflete um aumento no grau de conjugação do sistema π-conjugado [57]. De acordo com os espectros
de UV-Vis ocorre um deslocamento batocrômico das bandas de absorção no espectro dos oligômeros, a medida em que o tamanho do sistema aumenta, como pode ser visto na Figura 26. Contudo, o grau de torção do sistema também é um fator importante para localização da banda principal.
Este trabalho segue com uma análise mais detalhada dos espectros teóricos dos oligômeros 11CF108 e 11CF136 (ver Figura 27).
(a) 11CF108
(b) 11CF136
Figura 27 – Espectros de absorção teóricos e transições normalizadas para os oligômeros 11CF108 (a) e 11CF136 (b), utilizando a metodologia ZINDO/S-CI.
Capítulo 5. Resultados e Discussões 46
(Figura 27(a)) e as transições são do tipo HOMO-1→LUMO+2 e HOMO→LUMO (π → π∗) para o primeiro estado, ambas contribuindo com aproximadamente 11%. É
possível observar um segundo estado excitado associado à uma banda por volta de 400nm,
devido principalemte à uma transição HOMO-4→LUMO+6 (≈24%).
No espectro do 11CF136 (Figura27(b)), verifica-se uma banda (361nm) principal
devido a três estados, o primeiro é caracterizado misto composto por duas transições principais do tipo HOMO-3→LUMO+4 e HOMO→LUMO (π →π∗), esta última parece
dominar o estado (≈ 52%). O segundo estado também misto, envolve duas transções principais HOMO-3→LUMO+4 (≈ 19%) e HOMO-1→LUMO+1 (≈ 35%). O terceiro estado característico desta banda tem menor intensidade em relação aos dois últimos (mais de 50% em relação ao primeiro estado), envolve sete confugurações onde a configuração HOMO-2→LUMO+3 possui maior contribuição, mais de 61%. Os modelos moleculares apresentados podem ser validados observando-se os espectros experimentais dos polímeros em estudo.
AsFiguras 28e29apresentam os espectros de absorção eletrônicos experimentais dos sistemas CF108 e CF136 medidos pela American Dye Source [29].
Figura 28 – Espectros experimental de absorção eletrônica dos sistemas CF108 [29].
Capítulo 5. Resultados e Discussões 47
No caso do CF136, observa-se uma banda localizada por volta de 370nm.
Figura 29 – Espectros experimental de absorção eletrônica dos sistemas CF136 [29].
48
Conclusão
Neste estudo teórico, dois copolímeros do fluoreno, CF108 e CF136, foram analisados empregando métodos de estrutura eletrônica (semi-empíricos e DFT). As moléculas foram expandidas de 1 até 11 unidades monoméricas. O interesse nestes compostos se deve ao seu grande potencial para aplicações como matrizes ativas em dispositivos optoeletrônicos. As simulações computacionais efetuadas neste trabalho permitiram a análise mais aprofundada da correlação entre os aspectos estruturais das cadeias oligoméricas e algumas propriedades, como gap de energia, forma dos orbitais moleculares de fronteira, comportamento dos espectros de UV-Vis, dentre outras.
De forma geral, os resultados indicam que a enegia total e ogap HOMO-LUMO
tendem a diminuir com o aumento da cadeia oligomérica. O comportamento da energia total apresenta duas tendências de evolução e a partir de cinco meros a energia tende a diminuir mais rapidamente.
Traçando um paralelo entre a evolução do tamanho das cadeias oligoméricas com as propriedades estruturais estimadas para os dois sistemas, foi evidenciado que estruturas não-planares são mais favoráveis.
De acordo com o comportamento dos orbitais moleculares, o sistema CF108 apre-sentou uma maior facilidade para a ocorrência de mobilidade eletrônica ao longo da cadeia principal do que o CF136, o qual apresentou uma menor densidade eletrônica localizada na região central. Foi observado ainda que a adição do grupo funcional vinil parece favorecer a deslocalização da função de onda molecular, entretanto torções entre monômeros da cadeia oligomérica tendem a diminuir esta deslocalização, afetando diretamente o comprimento de conjugação.
As análises comparativas das propriedades de ambos os sistemas analisados pos-sibilitaram uma melhor compreensão sobre as características peculiares de cada um, separadamente. O CF108 possui duas bandas características (≈436 e 400nm), enquanto
que o CF136 possui apenas uma em torno de 361nm. Contudo, a banda de maior
in-tensidade tem contribuição mais significativa proveniente da transição HOMO-LUMO. Em comparação com as medidas experimentais, aproximadamente 481 e 450nm para o
CF108 e 374nm para o CF136, foi observado um comportamento similar para as formas das bandas. Além de uma diferença relativa entre os máximos comprimentos de onda de aproximadamente 3% para o 11CF136 e 9% para o 11CF108. Portanto, os modelos moleculares propostos se mostram bastante satisfatórios.
Conclusão 49
de sistemas π-conjugados, empregando-se métodos de mecânica quântica apropriados, pode
50
Trabalho em andamento
A realização de análises conformacionais e busca no espaço de configurações é im-portante para o estudo de novos materiais, uma vez que algumas propriedades imim-portantes são dependentes dos arranjos conformacionais moleculares. O esforço de investigações desta natureza pode ser minimizado com a utilização de técnicas de otimização baseadas em inteligência artificial, como os algoritmos genéticos (como introduzido no capítulo 3).
Tendo em vista a relavância do problema citado, um algoritmo genético foi imple-mentado e está em fase de teste, para ser utilizado na etapa de análise conformacional para seleção da melhor estrutura. A Figura 30 mostra o fluxograma do algoritmo proposto.
Figura 30 – Fluxograma do algoritmo proposto. Este fluxo contempla o processo de aqui-sição de estruturas, o algoritmo genético e o procedimento de persistência de dados.
Trabalho em andamento 51
adicionais que desempenham os seguintes papéis:
1. ler e escrever geometrias moleculares;
52
Produção Científica
O presente trabalho originou participações em eventos científicos nacionais e internacionais conforme listado abaixo:
1. MAGALHAES, C. E. T. ; G.R. Ferreira ; MARQUES, Márcio ; Bianchi, Rodrigo F. ; SIQUEIRA, M. F. . Photodecomposition process and structural functionalization effects in fluorene copolymers. In: 9th International Conference on Organic Electronics, 2013, Grenoble. 9th International Conference on Organic Electronics. Grenoble-France, 2013.
2. SIQUEIRA, M. F. ; MARQUES, Márcio . OPTOELETRONIC PROPERTIES OF FLUORENE-MEH-PPV COPOLYMERS. In: XXXVI Encontro Nacional da Física da Matéria Condensada, 2013, Águas de Lindóia. XXXVI ENFMC, 2013. v. 26.
3. MAGALHAES, C. E. T. ; Marques, M. ; SIQUEIRA, M. F. . MOLECULAR QUANTUM MECHANICS MODELLING ON OPTOELECTRONIC PROPERTIES OF FLUORENE-PPV COPOLYMERS. In: XXXVI Encontro Nacional da Física da Matéria Condensada, 2013, Águas de Lindóia. XXXVI ENFMC, 2013.
4. MARQUES, Márcio ; SIQUEIRA, M. F. . PROPRIEDADES OPTOELETRÔNICAS DE COPOLÍMEROS DO FLUORENO. In: II Semana de Física de Materiais, Universidade Federal de Ouro Preto. Ouro preto - MG. 2013.
5. MAGALHAES, C. E. T. ; MARQUES, Márcio ; SIQUEIRA, M. F. . EFEITOS ESTRUTURAIS E DE SUBSTITUINTES RELACIONADOS ÀS PROPRIEDADES OPTOELETRÔNICAS DE COPOLÍMEROS DO FLUORENO. In: SEIC 2013 Seminário de Iniciação Científica da UFOP, 2013, Ouro Preto. Anais do SEIC 2013 -Seminário de Iniciação Científica da UFOP - 2013, 2013.
53
Referências
[1] BURNS, SE et al. A scalable manufacturing process for flexible active-matrix e-paper displays.F. Soc. Inf. Display, v. 13, p. 583–86, 2005.
[2] KELLEY, T. W. et al. Recent progress in organic electronics: Materials, devices, and processes. Chemistry of Materials, v. 16, n. 23, p. 4413–4422, 2004. Disponível em:
<http://pubs.acs.org/doi/abs/10.1021/cm049614j>.
[3] PARASHKOV, R. et al. Large area electronics using printing methods. Proceedings of the IEEE, v. 93, n. 7, p. 1321–1329, July 2005. ISSN 0018-9219.
[4] SOMEYA, T. et al. Conformable, flexible, large-area networks of pressure and thermal sensors with organic transistor active matrixes.Proceedings of the National Academy of Sciences of the United States of America, v. 102, n. 35, p. 12321–12325, 2005.
[5] Sony Corp. Sony Launches World’s First OLED TV. 2007. Disponível em:
<www.sony.net/SonyInfo/News/Press/200710/07-1001E/index.html>.
[6] IHS iSuppli. 2013. Disponível em: <www.isuppli.com>.
[7] ELECTRONICS, Photonics and Electrical Systems - Key Technology Area. [S.l.], 2008.
[8] SEMICONDUTORES Orgânicos - Proposta para uma estratégia brasileira. [S.l.], 2007. Disponível em: <http://www.cgee.org.br/publicacoes/SemicondutoresOrganicos.php>.
[9] ARAI, T. et al. R&D prospects of organic electronic devices. Hitach Review, v. 57,
n. 3, 2008.
[10] MANNSFELD, S. C. B. et al. Highly sensitive flexible pressure sensors with micros-tructured rubber dielectric layers.Nature Materials, v. 9, p. 859–864, Set 2010.
[11] OUELLETTE, J. SiliconGermanium Gives Semiconductors the Edge. 2002.
Http://www.aip.org/tip/INPHFA/vol-8/iss-3/p22.pdf. Acessado em Novembro de 2013.
[12] DE PAULA, J; ATKINS, P. Chemical Physics. 2. ed. London UK: Oxford University
Press, 2004.
[13] ASHCROFT, N. W.; MERMIN, N. D. Solid state physics. Holt, New York: [s.n.], 1976.
[14] WEINBERGER, B.; AKHTAR, M.; GAU, S. Polyacetylene photovoltaic devices.
Synthetic Metals, v. 4, n. 3, p. 187 – 197, 1982. ISSN 0379-6779. Disponível em:
Referências 54
[15] CHIANG, C. K. et al. Electrical conductivity in doped polyacetylene. Phys. Rev. Lett., v. 39, n. 17, p. 1098–1101, 1977.
[16] KATZ, H. E.; HUANG, J. Thin-film organic electronic devices. Annu. Rev. Mater. Res., v. 39, p. 71–92, 2009.
[17] AKCELRUD, L. Electroluminescent polymers. Prog. Polym. Sci., v. 28, p. 875, 2003.
[18] HowStuffWorks. How OLEDs Work. 2013. Disponível em:
<http://www.howstuffworks.com/oled.htm>.
[19] ZHU, X. Y.; YANG, Q.; MUNTWILER, M. Charge-transfer excitons at organic semiconductor surfaces and interfaces.Acc. Chem. Res., 2009.
[20] FARIA, G. C. et al. A multitechnique study of structure and dynamics of polyfluorene cast films and the influence on their photoluminescence. The Jour-nal of Physical Chemistry B, v. 113, n. 33, p. 11403–11413, 2009. Disponível em:
<http://pubs.acs.org/doi/abs/10.1021/jp9043368>.
[21] KLAERNER G.; MILLER, R. D. Polyfluorene derivatives: Effective conjugation lengths from well-defined oligomers. Macromolecules, v. 31, p. 2007–2009, 1998.
[22] RIEVESCHL, G.; RAY, F. The chemistry of fluorene and its derivatives. Chemical Reviews, p. 288–374, 1937.
[23] RONDEAU, D.; LECLERC, M.; RANGER, M. New well-defined poly(2,7-fluorene) derivatives: Photoluminescence and base doping. Macromolecules, v. 30, p. 7686–7691,
1997.
[24] TANG, C. et al. Fluorene-substituted pyrenes-novel pyrene derivatives as emitters in nondoped blue oleds.Org. Electron., v. 7, p. 155–162, 2006.
[25] TANG, C. et al. Efficient 9-alkylphenyl-9-pyrenylfluorene substituted pyrene derivati-ves with improved hole injection for blue light-emitting diodes.J. Mater Chem., v. 16,
p. 4074–4080, 2006.
[26] MONGIN, O. et al. Systhesis, fluorescence, and two-photon absorption of a series of elongated rod-like and banana-shaped quadrupolar fluorophores: A comprehensive study of structure-property relationship. Chem. Eur. J., p. 1481–1498, 2007.
[27] ZHANG, G. et al. Two-photon absorption and optical limiting properties of a new 9-branched fluorene derivative. Acta Chim. Slov, v. 55, p. 315319, 2008.
Referências 55
[29] PRODUCT BULLETIN. [S.l.], 2013. Disponível em: <http://www.adsdyes.com>.
[30] MORGON, N. H.; COUTINHO, K. Métodos de Química Teórica e Modelagem Mole-cular. [S.l.]: São Paulo: Livraria da Fisica, 2007.
[31] BARFORD, W. Electronic and Optical Properties of Conjugated Polymers. 2. ed.
USA: Oxford University Press, 2013.
[32] BERNARDINELLI, O. D. et al. Correlation between molecular conformation, pac-king, and dynamics in oligofluorenes: A theoretical/experimental study. The Jour-nal of Physical Chemistry A, v. 116, n. 17, p. 4285–4295, 2012. Disponível em:
<http://pubs.acs.org/doi/abs/10.1021/jp210953m>.
[33] SCHIFF, L. I. Quantum Mechanics. 3. ed. [S.l.]: McGraw-Hill Education, 1968.
[34] BORN, M.; OPPENHEIMER, J. R. On the quantum theory of molecules.Ann. Phys. (Leipzig), v. 84, p. 457, 1927.
[35] MCWEENY, R. Methods of Molecular Quantum Mechanics. [S.l.]: Academic Press, 1992.
[36] KORTH, M. Third-generation hydrogen-bonding corrections for semiempirical qm methods and force fields. Journal of Chemical Theory and Computation, v. 6, p. 3808–
3816, 2010. Disponível em: <http://pubs.acs.org/doi/abs/10.1021/ct100408b>.
[37] STEWART, J. J. P. Optimization of parameters for semiempirical methods v: Modification of nddo approximations and application to 70 elements. Jour-nal of Molecular Modeling, v. 13, n. 12, p. 1173–1213, 2007. Disponível em:
<http://pubs.acs.org/doi/abs/10.1021/ct100408b>.
[38] ANDERSON, W. P.; CUNDARI, T. R.; ZERNER, M. C., D. Utility of the semiem-pirical INDO/1 method for the calculation of the geometries of second-row transition-metal species.Inorg. Chem., v. 29, n. 1, p. 1–3, 1990. ISSN 0020-1669. Disponível em:
<http://dx.doi.org/10.1021/ic00326a001>.
[39] SLATER, J. C. The theory of complex spectra. Phys. Rev, v. 34, p. 1293, 1929.
[40] CONDON, E. U. The franck-condon principle and related topics. Amer. J. of Physics,
v. 34, n. 5, p. 335, 1947.
[41] ZERNER, M. C. et al. An intermediate neglect of differential overlap technique for spec-troscopy of transition-metal complexes. ferrocene.J. Am. Chem. Soc., v. 102, n. 2, p. 589–
Referências 56
[42] BUNCE, N. J.; RIDLEY, J. E.; ZERNER, M. C. On the excited states ofp-quinones and an interpretation of the photocycloaddition ofp-quinones to alkenes. Theoretica chimica acta, v. 45, p. 283–300, 1977.
[43] KOHN, W.; HOHENBERG, P. Inhomogeneous electron gas. Physical Review, v. 136,
p. B864, 1964.
[44] PARR, R. G.; YANG, W. Density-functional theory of atoms and molecules. New York: Oxford University Press, 1989.
[45] KOCH, W.; HOLTHAUSEN, M. C. A chemist’s guide to density functional theory. 2.
ed. Weinheim: Wiley-VCH, 2002.
[46] BURKE, K.; ERNZERHOF, M.; PERDEW, J. P. Generalized gradient approximation made simple.Physical Review Letters, v. 78, p. 3865, 1996.
[47] GOLDBERG, D. E. Genetic Algorithms in Search, Optimization, and Machine Lear-ning. 1. ed. [S.l.]: Addison-Wesley Professional, 1989.
[48] GREENLAW, R.; HOOVER, H. J.; RUZZO, W. L. Limits to Parallel Computation: P-Completeness Theory. 1. ed. Oxford, England: Oxford University Press, 1995.
[49] JENSEN, F. Introduction to Computational Chemistry. 2. ed. England: John Wiley,
2006.
[50] KOHN, W.; SHAM, L. J. Self-consistent equations including exchange and correlation effects. Physical Review, v. 140, p. A1133, 1965.
[51] Neese, F.ORCA, an Ab Initio, Density Functional and Semiempirical program package.
2005.
[52] Stewart, James J. P. MOPAC2012. 2012.
[53] ADAMO, C.; BARONE, V. Toward reliable density functional methods without adjustable parameters: The pbe0 model. JOURNAL OF CHEMICAL PHYSICS, v. 110, n. 13, p. 6158, 1999.
[54] RASSOLOV, V. A. et al. 6-31g* basis set for atoms k through zn.Journal of Chemical Physics, v. 109, p. 1223, 1998.
[55] BIRKS, J. B. Photophysics of Aromatic Molecules. 1. ed. Londres: Wiley, 1970.
[56] SPRINGBORG, M. et al. Theoretical studies of electronic properties of conjugated polymers. In: FARCHIONI, R.; GROSSO, G. (Ed.). Organic Electronic Materials. [S.l.]:
Referências 57
[57] YANG, S.-C. et al. Geometry-dependent electronic properties of highly fluorescent conjugated molecules. Phys. Rev. Lett., American Physical Society, v. 85, p. 2388–2391,
![Figura 1 – Previsão de crescimento do consumo global de painéis OLED, estimada para o período de 2013 a 2018, segundo pesquisa de mercado realizada pela iSuppli [6].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15752916.638306/15.892.303.643.724.1023/figura-previsão-crescimento-painéis-estimada-período-pesquisa-realizada.webp)

![Figura 3 – Orbitais híbridos do carbono que formam ligações σ e π [12].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15752916.638306/17.892.261.682.660.867/figura-orbitais-híbridos-do-carbono-que-formam-ligações.webp)