UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA ANIMAL
Microglia demonstrate age-dependent performance and
interaction with beta-amyloid peptide
Gonçalo Manuel da Costa Lidónio
Dissertação
Mestrado em Biologia Humana e Ambiente
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA ANIMAL
Microglia demonstrate age-dependent performance and
interaction with beta-amyloid peptide
Gonçalo Manuel da Costa Lidónio
Dissertação
Mestrado em Biologia Humana e Ambiente
Dissertação orientada por:
Professora Doutora Dora Brites (orientação externa)
iMed.UL – Faculdade de Farmácia da Universidade de Lisboa
Professora Doutora Deodália Dias (orientação interna)
Faculdade de Ciências da Universidade de Lisboa
I
N
OTAP
RÉVIAA realização desta dissertação também contou com a co-orientação da Doutora Ana Sofia Falcão, investigadora auxiliar da Faculdade de Farmácia da Universidade de Lisboa.
Parte dos resultados inseridos nesta dissertação foram apresentados nos seguintes encontros:
Lidónio G, Caldeira C, Vaz AR, Santos M, Moreira R, Falcão AS, Brites D. Exploring a therapeutic approach to prevent Ab-induced dysfunctional microglia. 5th Postgraduate iMed.UL Students Meeting, Lisboa, Portugal, 18 Julho, 2013 [Poster]
Caldeira C, Frederico A, Oliveira AF, Lidónio G, Vaz A, Fernandes A, Brites D. Cell
ageing effects on microglia response to Aβ.XIII Reunião da Sociedade Portuguesa de
Neurociências, Luso, Portugal, 31 de Maio - 2 de Junho, 2013 [Poster]
A formatação das referências usada nesta dissertação baseou-se no utilizado pela revista Neurochemical Research da Springer, conceituada na área e na qual o grupo de investigação tem vindo a publicar.
II
[Escre va um
III
AGRADECIMENTOS
Em primeiro lugar, naturalmente, quero agradecer à Prof. Doutora Dora Brites, orientadora externa desta dissertação. Fico muito grato pela oportunidade de realizar a minha dissertação de mestrado no seu grupo de investigação, Neuron Glia Biology in Health and Disease, e por todos os conhecimentos que me transmitiu ao longo deste ano. O seu rigor científico, espírito crítico, empenho e inteligência são admiráveis, e uma fonte de inspiração.
À Doutra Sofia Falcão, co-orientadora deste trabalho, agradeço todo o apoio, dedicação, disponibilidade e preocupação revelados ao longo deste projecto. Sem ti, Sofia, este trabalho não seria possível. Agradeço, profundamente, toda a paciência e compreensão que tiveste comigo e toda a motivação que me deste. Contigo, muito aprendi, tanto a nível profissional como a nível pessoal.
À Prof. Doutora Deodália Dias, orientadora interna desta dissertação e coordenadora do mestrado em Biologia Humana e Ambiente, agradeço por toda a disponibilidade e pelo apoio e conselhos que me deu, não só ao longo deste ano, mas durante toda a minha estadia neste mestrado.
Ao Prof. Doutor Rui Silva e à Prof. Doutora Alexandra Brito agradeço toda a disponibilidade para esclarecer dúvidas que foram surgindo.
Quero também agradecer à Prof. Doutora Adelaide Fernandes e à Doutora Ana Rita Vaz toda a disponibilidade que sempre demonstraram, o conhecimento que partilharam comigo e, principalmente, a ajuda no ultrapassar de muitos dos desafios que encontrei.
Quero também expressar o meu agradecimento para com a Doutora Teresa Pais por gentilmente nos ter cedido a linha microglial N9, e ao Prof. Doutor Rui Moreira por nos ter cedido o composto explorado neste trabalho.
Como não poderia deixar de ser, um grande agradecimento para todos os restantes membros do grupo:
Inês Palmela, ao contrário do que contam, quero salientar a boa disposição que sempre observei e agradecer toda a ajuda que me deste – mesmo pensando que adormeci na tua aula. À Filipa agradeço os bons conselhos, o apoio e, claro, os
IV
conhecimentos para melhorar as minhas imunocitoquímicas. À Andreia Barateiro agradeço toda a disponibilidade e a sua contribuição ao alegrar os nossos dias com os seus bolinhos. Em ti, Cláudia, vejo um exemplo. Acho fascinante a tua força de vontade. Agradeço-te todo o apoio, ajuda e maturidade que partilhaste comigo neste trabalho que tão próximo está do teu. Cátia e Carolina, apesar das dores de cabeça que vocês me deram, tenho muito que vos agradecer. Transmitiram-me conhecimentos preciosos e, passo a passo, contribuíram para que me tornasse um melhor cientista. A ti, Gisela, agradeço o apoio, a disponibilidade e todas as sugestões que me deste em muitos dos problemas que encontrei. Agradeço ainda a simpatia, a motivação que sempre me transmitiste e o ouvido para os meus desabafos nas situações mais complicadas. E não me esqueço de ti, Inês Figueira, que mesmo que não tendo os dois pezinhos no grupo, também contribuíste neste meu percurso. Obrigado.
Um agradecimento também aos mais novos, aos que seguiram ao meu lado nesta viagem e que mais força me deram para chegar ao destino. À Vera, um enorme obrigado. Principalmente por todo o teu positivismo, por partilhares comigo os teus conhecimentos laboratoriais e, acima de tudo, por todos os momentos de convívio que contribuíram para tornar esta viagem menos difícil. A vocês as duas, Marta e Andreia, temo que se deixasse passar tudo o que sinto para o papel, a minha dissertação seria um agradecimento. Desde os dias de faculdade que seguem ao meu lado e que preenchem as minhas boas memórias. Agradeço-vos por toda a amizade, por me puxarem sempre por um sorriso e por todos os momentos de drama, comédia e Sci-Fi com que comigo partilharam. Sem vocês, esta viagem não seria a mesma. Não posso passar também sem agradecer ao telemóvel da Marta, que muitos desabafos partilhou comigo e muito me animou nos dias de chuva.
Agradeço ao grupo, como um todo, pela forma como me receberam. Fizeram-me sentir em casa.
Quero agradecer também aos constituintes do meu grupo de mestrado. Em especial à Marília e ao Gonçalo, pelo seu companheirismo e amizade, que me encheram as horas de risos e desabafos.
Um obrigado muito especial aos meus amigos da terrinha, que estão comigo desde que lembro e me têm acompanhado nos momentos mais importantes da minha vida. Em especial a vocês, Nuno, Miguel e Sandra. De perto ou de longe, tornaram este trabalho não apenas meu, mas também vosso.
V Por último, mas acima de tudo, quero agradecer à minha família. Aos que cá estão para presenciar esta etapa e aos que gostaria que cá estivessem. Sem vocês, não seria o que sou hoje. Agradeço principalmente aos meus pais e à minha irmã, pelo esforço que têm feito para chegar onde cheguei e por nunca desistirem de mim. Não poderiam ser melhores. Vocês estão e estarão sempre no meu coração.
VI
[Escre va um
VII
ABSTRACT
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the elderly and remains without a cure. One of its prominent features is the appearance in the brain of amyloid-beta (Aβ) deposits forming amyloid plaques. Microglia, which are the first line of defense in the brain, are activated by Aβ assuming a phagocytic phenotype followed by the inflammatory state that is thought to play a role in the progression of this disease. However, the important role of microglia in Aβ clearance and neuronal support seems to be decreased with age, through an ensemble of alterations still not clarified that render cells to function abnormally. Indeed, in these circumstances, microglia lose responsiveness to their key regulating factors, thus compromising their neuroprotective or inflammation solving properties. Therefore, in this work we aimed to: a) understand how ageing influences microglia reactivity to Aβ and microglia-neuron cross-talk dynamics, using an in vitro ageing model of primary microglia (isolated or in mixed microglia-neurons cultures) and by evaluating the release of glutamate, adenosine triphosphate (ATP) and matrix metalloproteinases (MMPs); b) explore the therapeutical properties of a vinyl sulfone-based compound (VS) as a modulator of Aβ-induced microglial activation, using the N9 cell line and assessing cell viability, inflammation-related factors, activation receptors and phagocytosis.
Our results demonstrate that microglia behave differently accordingly with age, releasing less glutamate and MMP-9, but more MMP-2. Ageing also seems to render microglia more irresponsive to neuronal-mediated regulation, particularly regarding their ability to modulate the extracellular glutamate content. In addition, in our experimental model of ageing in vitro, Aβ failed to induce any microglia reactivity. In regard to VS testing, no toxicity was observed for this compound, and it successfully diminished the Aβ-induced release of MMP-9 and MMP-2, as well as the expression of high-mobility group box 1 protein, important inflammatory mediators. Moreover, Aβ reduced the level of microglial toll-like receptor 4 and phagocytosis, effects that were counteracted by VS.
Overall, these results deepen our knowledge about the role of ageing in microglial biology and elect VS as a potential new therapeutic approach to modulate Aβ-induced microglial activation and dysfunction in AD.
Keywords: Ageing, Alzheimer disease, Amyloid-beta peptide, Microglia reactivity, Neuroinflammation, Neuron-microglia communication, Vinyl sulfone compound
VIII [Escre
IX
SUMÁRIO
A Doença de Alzheimer (DA) é considerada a doença neurodegenerativa mais comum e a principal causa de demência a nível mundial, afectando cerca de 35 milhões de pessoas. Esta doença é caracterizada por uma perda progressiva das capacidades cognitivas, o que leva a um completo estado de debilitação e, por último, à morte. O principal factor de risco é o envelhecimento e permanece sem tratamento até hoje.
No encéfalo dos pacientes com DA, para além de uma acentuada perda de neurónios e sinapses, são ainda observadas duas características distintas da doença: emaranhados neurofibrilares e placas amilóides. Os primeiros consistem em agregados filamentosos intracelulares de proteína tau hiperfosforilada, enquanto que as últimas se devem a depósitos extracelulares do péptido β-amilóide (βA), um pequeno fragmento de 38-43 aminoácidos que resulta da cisão sequencial da proteína precursora amilóide por duas secretases, a β-secretase e a γ-secretase.
A causa primordial da DA ainda está por desvendar; contudo, o βA, devido aos seus variados efeitos neurotóxicos é apontado como um dos principais factores envolvidos no desenvolvimento da doença. Este, principalmente na forma de 42 aminoácidos, possui uma grande tendência para se auto-agregar, formando estruturas que vão desde oligómeros até fibrilhas, as quais originam as placas amilóides. Apesar de inicialmente se julgarem ser os agregados fibrilares as formas mediadoras de toxicidade, nos últimos anos, as formas oligoméricas têm sido apontadas como as verdadeiramente tóxicas. Porém, além da oligomerização e deposição de βA, outros mecanismos têm vindo a ser estudados pela sua associação com a patofisiologia da DA, nomeadamente a hiperfosforilação da proteína tau, o stress oxidativo, a disfunção mitocondrial e a neuroinflamação.
As células da microglia, consideradas as células imunes do sistema nervoso central, desempenham um papel fundamental na resposta inflamatória. Estas células, conhecidas por constantemente sondarem o tecido cerebral e responderem a vários estímulos patológicos no sentido de reporem a homeostasia, são frequentemente encontradas num estado activado e associadas às placas amilóides. Estudos já realizados evidenciaram que o βA interage com a microglia por meio de vários receptores, induzindo a libertação de diversas moléculas de carácter pro-inflamatório e citotóxico. Neste sentido, a deposição de βA pode induzir uma permanente activação da microglia, conduzindo a uma perpetuação da inflamação e, consequentemente, contribuindo para neurodegeneração. Por outro lado, a microglia também é responsável pela libertação de vários factores neurotróficos e pela degradação e
X
fagocitose de βA, funções fundamentais na restrição da acumulação de βA e suporte dos neurónios, impedindo a progressão da DA.
Vários estudos têm demonstrado que a microglia durante o envelhecimento e em presença de diversos estímulos assume um fenótipo mais do tipo inflamatório, o qual na presença de uma nova indução acaba por ocasionar uma resposta mais exacerbada. Para tal, pode igualmente contribuir qualquer alteração da comunicação entre a microglia e os neurónios, os quais desempenham um papel fundamental no controlo do estado de activação da célula glial. Provavelmente na sequência destas sucessivas activações a microglia pode tornar-se morfologicamente distrófica, sendo assim frequentemente encontrada no cérebro envelhecido e, principalmente, na DA. Tal indica que estas células sofrem senescência e degeneração com o avanço da idade, predispondo à doença, que por sua vez intensifica a disfunção da célula, possivelmente na sequência da exposição ao βA. Assim, com a idade e/ou à medida que a doença progride, a microglia tende a degenerar, deixando de dar suporte aos neurónios, o que contribui para a sua morte. Este ponto de vista também ajuda a explicar a razão pela qual a abordagem terapêutica com fármacos anti-inflamatórios não esteróides só produz efeitos benéficos nos estádios mais iniciais da doença, enfatizando a importância do intervalo de actuação nas abordagens terapêuticas.
Com base no crescente trabalho que tem vindo a ser desenvolvido sobre o papel da microglia na progressão da DA, esta dissertação teve como primeiro objectivo compreender de que modo o envelhecimento microglial influencia a reactividade da microglia ao βA e se a capacidade da microglia responder à sinalização neuronal fica comprometida em resultado do envelhecimento. Para isto, recorreu-se a um modelo de envelhecimento in vitro utilizando células microgliais primárias. Foram escolhidas duas idades celulares diferentes, as de 2 e 15 dias de cultura, de modo a mimetizar um fenótipo microglial jovem e envelhecido, respectivamente. Estas células foram então colocadas na presença, ou não, de neurónios e estimuladas com 50 nM e 1000 nM de βA. Depois da incubação, determinou-se no meio extracelular a concentração de glutamato, de adenosina trifosfato (ATP) e das metaloproteinases de matriz extracelular (MMP), factores que desempenham um papel importante na mediação da resposta inflamatória. Os resultados obtidos demonstraram que a microglia liberta uma menor quantidade de glutamato e de MMP-9 à medida que envelhece. Verificou-se ainda uma ausência de resposta da microglia envelhecida em termos de libertação de glutamato, quando exposta ao βA na presença de neurónios. Em relação à MMP-2, por outro lado, foi demonstrado um aumento na sua libertação pela microglia em resultado do envelhecimento. Quanto à libertação de ATP, em qualquer uma das
XI condições estudadas, não foram observadas alterações. Nesta abordagem, a exposição ao βA também não provocou activação microglial, em qualquer uma das idades avaliadas. Porém, é de salientar que observámos respostas diferentes em termos de libertação de glutamato e MMP-9 e MMP-2 com a idade e, de forma geral, estes resultados fortalecem a ideia de que a biologia microglial se altera como resultado do envelhecimento. Apesar de o envelhecimento poder condicionar a resposta da célula glial ao βA, tal não foi evidenciado pelos parâmetros analisados, pelo que se optará pela determinação de indicadores directos de neuroinflamação no futuro.
O segundo objectivo desta dissertação foi o de avaliar a capacidade de uma vinil sulfona (VS) sintetizada pelo grupo de Química Medicinal como modulador da activação microglial pelo βA. Para isto, a linha microglial N9 foi estimulada com as duas concentrações de βA (50 nM e 1000 nM), na presença ou ausência de duas concentrações de VS (10 µM e 20 µM), sendo posteriormente avaliados os seguintes parâmetros: viabilidade celular, libertação de MMPs, capacidade fagocítica da microglia e a expressão de proteínas como milk fat globule-EGF factor 8 protein (MFG-E8), toll-like receptor 4 (TLR-4) e high-mobility group box 1 (HMGB1).
A nível de viabilidade, não foram observadas alterações significativas por parte deste composto testado, nem pelas concentrações de βA usadas. Tanto quanto à libertação de MMP-2 e MMP-9 como relativamente à expressão de HMGB1, importantes mediadores da resposta inflamatória, observou-se uma notória indução após exposição ao βA, sendo a mesma revertida após tratamento com VS. Quanto à expressão do TLR-4, verificou-se uma redução na presença de βA, sendo este efeito invertido na presença da maior concentração de VS usada. Em relação tanto à capacidade fagocítica microglial como à expressão de MFG-E8, um factor com um papel importante na fagocitose, foi observada uma redução após indução com βA; porém, apenas a capacidade fagocítica retomou os valores do controlo após adição de VS. Assim, de forma geral, o composto por nós estudado demonstrou ser capaz de reverter alguns dos efeitos causados pelo βA, reduzindo de forma significativa a expressão de factores inflamatórios. Porém, mais estudos ainda têm que ser realizados para determinar a verdadeira extensão dos efeitos mediados por este composto, assim como os mecanismos pelos quais actua.
Em suma, estes resultados demonstraram contribuir para o conhecimento de como o envelhecimento da microglia condiciona a funcionalidade da célula e evidenciaram a VS como uma potencial terapêutica a ser explorada na modulação de outros determinantes, quer da activação, quer da latência microglial, observada na DA.
XII
Palavras-chave: Beta-Amilóide, Comunicação neurónio-microglia, Doença de Alzheimer, Envelhecimento celular, Neuroinflamação, Reactividade Microglial, Vinil sulfona
XIII
TABLE OF CONTENTS
NOTA PRÉVIA ... I AGRADECIMENTOS ... III ABSTRACT ... VII SUMÁRIO ... IX INDEX OF FIGURES ... XVIIABBREVIATIONS... XIX
I. INTRODUCTION ... 1
1. Alzheimer’s disease (AD): An overview ... 1
1.1. Disease presentation... 1
1.2. Pathophysiology ... 2
1.2.1. Amyloid beta peptide ... 4
1.2.2. Tau ... 7
1.2.3. Oxidative Stress ... 8
1.2.4. Mitochondrial Dysfunction ... 8
1.2.5. Neuroinflammation... 9
1.3. Diagnosis and treatment ... 10
2. Microglia in AD: A closer look to the central player in neuroinflammation ... 12
2.1. Microglia: origin and functions ... 12
2.2. Activation of microglia in AD ... 14
2.2.1. Interaction of amyloid beta with microglia ... 14
2.2.2. Production of inflammation-related factors by amyloid beta-stimulated microglia ... 16
2.2.3. Amyloid beta-induced microglial phagocytosis ... 17
2.3. Microglial role in the progression of AD: activation vs. dysfunction ... 18
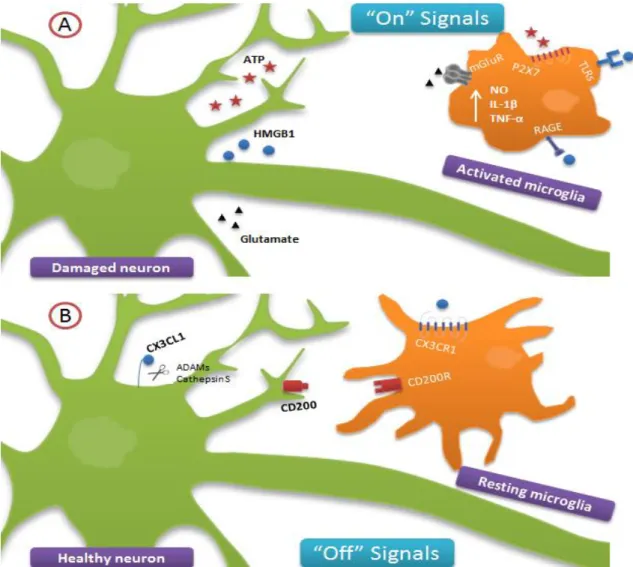
2.4. Microglia-neuron interplay in AD ... 20
2.5. Vinyl sulfones and inflammatory diseases: a possible therapeutic approach for microglial modulation in AD? ... 23
2.6. Microglial in vitro models for studying AD ... 24
3. Aims ... 27
II. MATERIAL AND METHODS ... 29
1. Material ... 29
1.1. Reactives ... 29
1.1.1. Cell culture media ... 29
XIV 1.1.3. Antibodies ... 30 1.2. Equipment ... 30 1.3. Animals ... 31 2. Methods ... 31 2.1. Primary cultures ... 31
2.1.1. Isolation of primary cell cultures ... 31
2.1.2. In vitro treatment of primary cell cultures with amyloid beta ... 32
2.1.3. Quantification of extracellular ATP ... 33
2.1.4. Measurement of extracellular glutamate ... 34
2.1.5. Gelatin zymography ... 34
2.2. Cell line ... 35
2.2.1. N9 microglial cell line culture ... 35
2.2.2. Treatment of N9 microglial cell line ... 35
2.2.3. Determination of Cell viability ... 36
2.2.4. Microglia phagocytic capacity ... 37
2.2.5. Gelatin zymography ... 37
2.2.6. Western blot analysis ... 37
2.3. Statistical analysis ... 38
III. RESULTS ... 39
1. Characterization of young and aged microglia reactivity to Aβ and of modulation by neurons ... 39
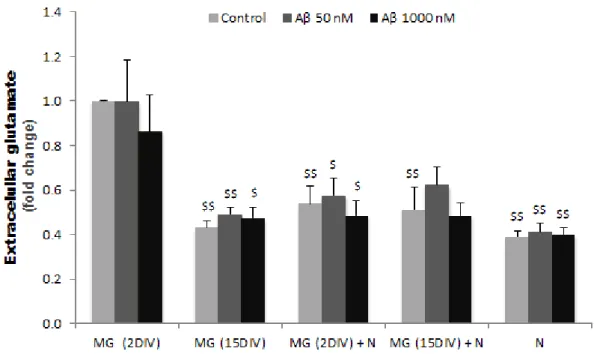
1.1. Young cells release higher levels of glutamate than older ones when in monoculture, but not in mixed culture with neurons, and no alterations are produced by Aβ in each differently aged cell ... 39
1.2. ATP release by microglia in response to Aβ is not affected by ageing or neurons ... 40
1.3. Release of MMP-9 and MMP-2 has opposite profiles in young and aged microglia and is not altered by Aβ or by the presence of neurons ... 41
2. Exploring a new therapeutic approach to prevent Aβ-induced microglial activation ... 43
2.1. Loss of viability in Aβ-stimulated microglia is slightly prevented by VS ... 43
2.2. Release of active MMPs by microglia is enhanced upon Aβ stimulation and decrease by VS ... 45
2.3. Expression of TLR4 is reduced by the highest concentration of Aβ, an effect that is counteracted by VS ... 46 2.4. Aβ triggers a reduction in microglia phagocytosis, which is prevented by VS
XV 2.5. The expression of MFG-E8 is reduced by 50 nM Aβ and VS alone, while increases when the highest concentrations of both compounds are concomitantly used 49
2.6. Aβ induces an increase in the expression of HMGB1 and effect that is
prevented by VS ... 50
IV. DISCUSSION ... 51
Future perspectives ... 59
XVI
[Escre va um
XVII
INDEX OF FIGURES
I. INTRODUCTION
Fig. I.1: Neuropathology of Alzheimer’s Disease ... 3 Fig. I.2: Proteolytic processing of amyloid precursor protein ... 5 Fig. I.3: Amyloid beta assembly states ... 6 Fig. I.4: Microglia activation states ... 14 Fig. I.5: Amyloid beta interaction with microglial receptors ... 16 Fig. I.6: Microglia-neuron communication ... 21
II. MATERIAL AND METHODS
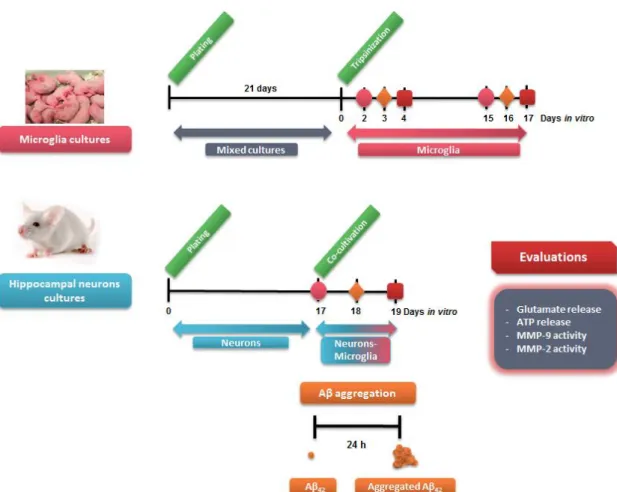
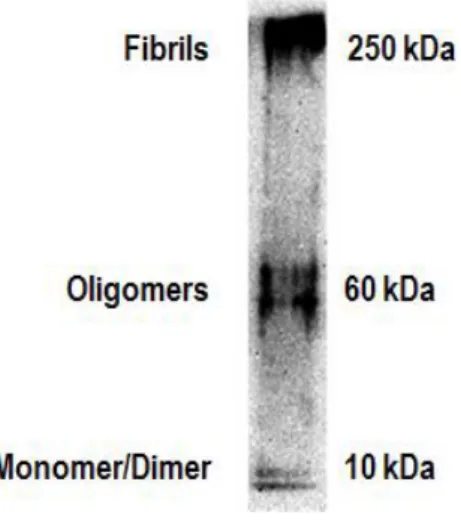
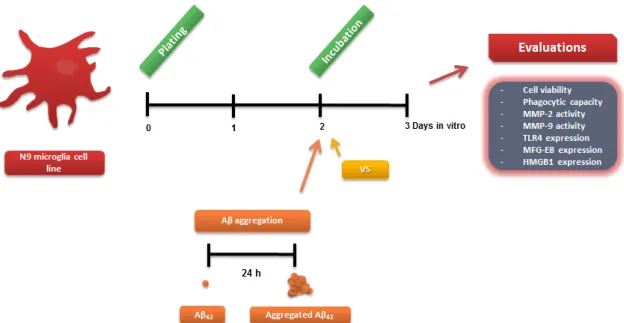
Fig. II.1: Experimental procedure used in the primary cellular cultures and parameters evaluated ... 33 Fig. II.2: Analysis of Aβ aggregation forms present in our experimental conditions ... 34 Fig. II.3: Experimental procedure used in the N9 microglial cell cultures and parameters evaluated. ... 36
III. RESULTS
Fig. III.1: The extracellular levels of glutamate are higher in young microglia than in old microglia, being reduced in the first by the presence of neurons and no effect by the presence of Aβ could be observed in any of the cases. ... 40 Fig. III.2: Microglia release of ATP by Aβ stimulation does not change by age or by incubation with neurons ... 41 Fig. III.3: MMP-2 release from microglia is up-regulated by ageing, while MMP-9 is instead down-regulated and these features are not modulated by Aβ or neuronal signaling ... 42 Fig. III.4: Cell viability shows a trend to be decreased by Aβ and was slightly prevented by VS ... 44 Fig. III.5: Stimulation of microglia with Aβ increases the release of active MMP-2 and MMP-9, which is prevent by VS ... 46 Fig. III.6: Expression of TLR4 is reduced by both Aβ and VS, but revealed to be induced by the highest VS concentration when in the presence of 1000 nM Aβ ... 47 Fig. III.7: Microglia phagocytic ability is decreased upon exposure to Aβ and restored by VS. ... 48
XVIII
Fig. III.8: The expression of MFG- E8 that is reduced by 50 nM and by VS alone, is increased when the highest concentration of both compounds are concomitantly used. ... 49 Fig. III.9: Expression of HMGB1 greatly increases in microglia treated with Aβ, but VS shows ability to prevent such effect from occurring ... 50
XIX
ABBREVIATIONS
ABAD Amyloid-binding alcohol dehydrogenase Ab/Am Antibiotic-antimycotic
AD Alzheimer’s disease
ADAM A desintegrin and metalloproteinase APOE Apoliprotein-E
APP Amyloid precursor protein ATP Adenosine triphosphate
Aβ Amyloid beta
Aβ40 Amyloid beta with 40 amino acids Aβ42 Amyloid beta with 42 amino acids BACE1 β-site APP cleaving enzyme 1 BBB Blood-brain barrier
BSA Bovine serum albumin
CaCl2 Calcium chloride
CD Cluster of differentiation CNS Central nervous system CSF Cerebrospinal fluid
DIV Days in vitro
DMEM Dulbecco’s modified Eagle’s medium EDTA Ethylenediamine tetraacetic acid fAβ Fibrillar amyloid beta
FBS Fetal bovine serum
GLT1 Glutamate transporter 1 HMGB1 High-mobility group box 1 HRP HorseRadish Peroxidase
Iba1 Ionized calcium-binding adaptor molecule 1 IDE Insulin-degrading enzyme
IL Interleukin
KOH Potassium hydroxide
LPS Lipopolysaccharide
MAP Microtubule associated protein MCI Mild cognitive impairment MEM Minimum essential medium MFG-E8 Milk fat globule EGF-like factor 8
XX
MHC Major histocompatibility complex MMP Matrix metalloproteinase
NEAA Non-essential amino acids
NEP Neprilysin
NF-κB Nuclear factor-κB NGF Nerve growth factor NMDA N-methyl-D-aspartate
NO Nitric oxide
NSAID Non-steroidal anti-inflammatory drugs oAβ Oligomeric amyloid beta
PBS Phosphate buffered saline PCR Polymerase Chain Reaction
PDL Poly-D-lysine
PET Positron emission tomography
PI Propidium iodide
PMSF Phenylmethylsulfonyl fluoride PRR Pattern recognition receptors
PS Phosphatidylserine
PSEN1/2 Presinilin 1/2
RAGE Receptor for advanced glycation end-products RNS Reactive nitrogen species
ROS Reactive oxygen species RPMI Roswell Park Memorial Institute SDS Sodium dodecyl sulphate
SDS-PAGE SDS- polyacrilamide gel electrophoresis TLR Toll-like receptor
TNF Tumor necrosis factor VR Vibronectin receptor αvβ3 VS Vinyl sulfone-based inhibitor
1
I. INTRODUCTION
1. Alzheimer
’s disease (AD): An overview
1.1. Disease presentation
In 1901, Alois Alzheimer, a german physician-pathologist, faced the case of August Deter, a 51 years-old woman who suffered from an intriguing set of symptoms, namely, progressive memory loss, confusion, disorientation and delusions [1]. During almost five years, her mental capacities continued to deteriorate rapidly, until she finally died in a completely demented state[2]. After patient’s death, Alzheimer performed an autopsy on her brain, finding significant cerebral atrophy and the presence of unusual plaques and tangles [1]. His characterization and notes on the case led to the discovery of a new neurodegenerative disease, a disorder that now bears his name – Alzheimer’s disease (AD).
One hundred years after its identification, AD has become one of the most serious preoccupations of our society. AD is considered to be the most common neurodegenerative disorder and the leading cause of dementia, accounting for 50-60% of all dementia cases [3]. Presently, the number of people affected by AD worldwide is estimated to be around 35 million [4]; however, this number is expected to increase, reaching more than 115 million by 2050 [5]. Like most dementias, AD’s clinical course begins insidiously, with an unusual gradual decline in memory - classified as mild cognitive impairment (MCI) - and then progresses to involve the deterioration of other cognitive functions, such as thinking and reasoning, and also behavior abnormalities, leaving the individual in a completely debilitated state and totally dependent on others. [6]. Symptoms unroll from several years to a decade and mortality is frequently related to resulting secondary issues, like multi-organ failure or opportunistic infections [7]..There is still no effective treatment for the disease, which in association with the huge need for patients’ care, makes AD an enormous financial burden to families and healthcare systems [8].
The elderly population is the main group at risk of developing AD, since advanced age is the factor most strongly associated with the disease [8] . Based on the age of onset, AD can be classified in two types: late-onset AD and early-onset AD. The late-onset AD is the most common form of the disease, accounting for around 90-95% of cases, and has its onset after age 65.On the other hand, the other 5-10% of AD
2
cases occur between ages 30-65, being defined as onset AD [4]. Some of early-onset AD cases, termed familial AD, are inherited in an autosomal dominant manner and are caused by mutations in three genes: the amyloid precursor protein (APP) gene on chromosome 21, the presinilin 1 (PSEN1) gene on chromosome 14 and the presinilin 2 (PSEN2) gene on chromosome 1 [8]. Nonetheless, most of AD cases are spontaneous - or within familial clusters that present no clear Mendelian inheritance pattern -, representing a complex disorder likely caused by an interplay between multiple susceptibility genes, environmental factors and ageing [5].In the last years, several genome-wide association studies have been done, leading to the identification of different susceptibility genes for AD. Of the genes identified, Apoliprotein-E (APOE) on chromosome 19 presents, by far, the strongest correlation with AD [5]. There are three common variants of this gene: ε4, which is the high risk form, increasing the risk in a dose-dependent manner; ε3, the most common in humans and the neutral allele; and ε2, which appears to be of low risk [9].
1.2. Pathophysiology
On a macroscopic level, AD pathology can be characterized by a progressive atrophy of the brain, affecting some regions more than others [10] (Fig. I.1). At a microscopic level, it is observed that atrophy mainly reflects the loss of neurons and synaptic contacts [11]. This happens in a defined pattern, with neurons in the enterohinal cortex being first affected, followed by hippocampal neurons and, ultimately, surrounding neurons in extra-hippocampal regions, such as neocortical areas [12]. Nonetheless, not all neuronal populations are equally susceptible to AD. For instance, neurons that are capable of projecting over long distance (projection neurons), possessing disproportionately long in relation to cell body size and sparsely myelinated axons seem to be particularly prone to the disease process [13]. It is not yet fully elucidated why this happens; however, these neurons have a large cell surface, a high energy requirement (due to the lack of myelinization) and are very reliant on axonal transport (anterograde and retrograde) for sustained function and trophic support, which might increase their susceptibility to damage [14]. In the case of AD, damage is believed to occur owning to mechanisms outside as well as inside the neuron and it is characterized by the appearance of intracellular filamentous aggregates of tau protein, called neurofibrillary tangles, and by extracellular deposits consisting largely of aggregated amyloid beta (Aβ) peptide, present typically between neurons with dystrophic neurites, called amyloid plaques [15,16]. These changes that where firstly described by Dr. Alzheimer are considered the major histopathological
3 hallmarks of the disease [1]. Besides these, gliosis, the accumulation of reactive astrocytes and microglia near lesions, is also seen in AD [17]. Of the various pathological features of AD, synaptic loss and selective neuronal death (in the limbic system and neocortex) are the ones that correlate best with the progression of cognitive decline, representing the main substrates of dementia [18].
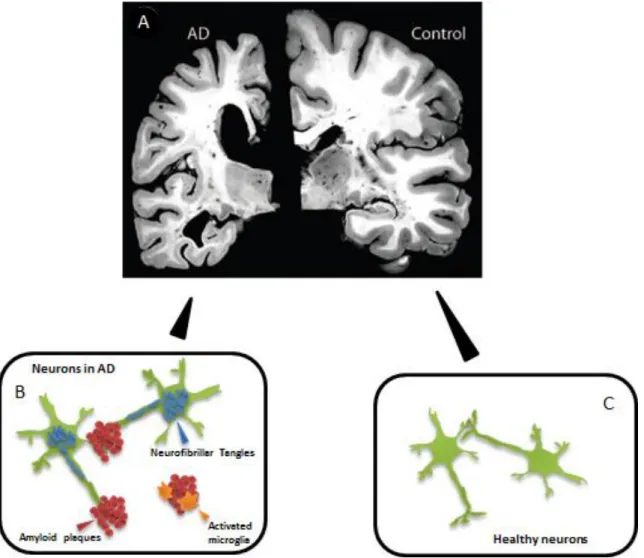
Fig. I.1: Neuropathology of Alzheimer’s Disease. (A) Extensive atrophy is observed in later stages of
Alzheimer’s Disease (AD), which is accompanied by enlarged ventricles, narrowed cortical gyri and widened sulci. Temporal lobes are most severely affected, while occipital lobes and motor cortex are usually spared (adapted from Holtzman [19]). (B) High number of degenerated neurons containing neurofibrillary tangles are found in AD, as well as extracellular deposits of amyloid beta protein (amyloid plaques), which are frequently surrounded by reactive glia cells. (C) In healthy brain, although some characteristics of the disease may be found as a result of the ageing process, they are usually not only present or present in a lower extent.
One of the most important issues for unraveling the AD mystery is finding out the mechanisms underlying the disease process. In this regard, several mechanisms are under study, including Aβ aggregation, deposition and toxicity, tau hyperphosphorylation with tangle formation, as well as oxidative stress, mitochondrial dysfunction and neuroinflammation.
4
1.2.1. Amyloid beta peptide
Aβ, the principal component of amyloid plaques, is considered a central player in AD and therefore it has been a major focus of research in the last 30 years. The main source of Aβ is the processing of APP, a transmembrane protein located on the plasmatic membrane or in intracellular compartments of diverse cells, like neurons and glia cells, whose main function still remains unknown [20,21].
APP can undergo several different endoproteolytic cleavage events, which culminate, or not, in the formation of the Aβ peptide (Fig. I.2). This proteolyic cleavage is sequential, starting with the cleavage by α-secretase or β-secretase, followed in both cases by cleavage by γ-secretase. Under normal conditions, APP is preferentially processed by the so-called non-amyloidogenic pathway, which starts with cleavage by α-secretase [22]. The enzyme is one of the ADAM9, 10 or 17, all a desintegrin and metalloproteinase (ADAM) family members [18], and cleaves within the Aβ domain, resulting in the production of a small peptide presumably non-pathogenic dominated P3, instead of Aβ . Alternatively, APP may be a substrate of β-secretase, a role that has been essentially attributed to the β-site APP cleaving enzyme 1, BACE1 , initiating the amyloidogenic pathway, which results in the formation of Aβ [22]. α-secretase and β-secretase cleave APP at single sites, whereas γ-secretase, an enzyme complex for which PSEN1 and PSEN2 act as catalytic subunits, performs a sequential series of intramembranous cuts, giving rise to products of varying length [23]. Aβ peptide length varies between 38 to 43 amino acids. The most common isoforms within the brain are the ones with 40 (Aβ40) and 42 (Aβ42) residues, representing ~90% and ~10%, respectively of the total [24]. Both Aβ40 and Aβ42 can spontaneously self-aggregate into higher order structures, ranging from low molecular oligomers (oAβ) to insoluble aggregates of fibrils (fAβ) [25] ; however, A42 is more hydrophobic and therefore more prone to aggregation [22] (Fig. I.3).
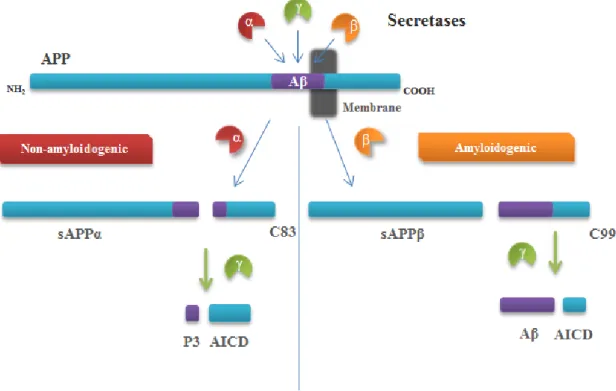
5 Fig. I.2: Proteolytic processing of amyloid precursor protein. There are two different pathways by
which amyldoid precursor protein (APP) can be processed: the non-amyloidogenic pathway and the amyloidogenic pathway. In the non-amyloidogenic pathway, APP is initially cleaved by α-secretase, originating a secreted fragment of APP (sAPPα) and a membrane-bound carboxy-terminal fragment of 83 amino acids (C83). Subsequently, C83 is further processed by γ-secretase, giving rise to P3, precluding amyloid beta (Aβ) formation. Amiloidogenic processing, alternatively, is initiated by β-secretase and results in the release of sAPPβ. The retained membrane-bound fragment of 99 amino acids (C99) is also a γ-secretase substrate, leading to the production of Aβ. APP intracellular domain (AICD) is an end-product in both pathways, being translocated into the nucleous to regulate gene transcription.
Several lines of investigation support the notion that the pathogenesis of AD is directly related to a progressive accumulation of Aβ, resulting from an imbalance between the levels of Aβ production, aggregation and clearance in the brain [26]. The strongest evidence for this is seen in familial AD, where all cases identified carry mutations in APP or presenilin genes, leading to an increase of total Aβ production or a shift in the Aβ40/Aβ42 ratio toward Aβ42. In addition, individuals with Down’s syndrome, who possess an extra APP gene, also develop an AD-like pathology [27]. In sporadic form of AD, the Aβ accumulation is thought to be more related with a dysfunction of its clearance mechanisms [26]. Indeed there is an association between APOE isoforms and Aβ clearance, with the ε4 isoform showing to be the less efficient in clearing Aβ. [11]. Furthermore, the activity of major Aβ-degrading enzymes, like neprilysin (NEP) and insulin-degrading enzyme (IDE), has also been reported to be reduced with age and in brain regions affected by AD [28]. The identification of these AD-related genes has led to the creation of diverse animal models, which although not completely mimicking AD, are a valuable tool to study the mechanisms involved in Aβ accumulation [29].
6
How Aβ contributes to neurodegeneration in AD is still not fully understood, but aggregation seems to be essential for A toxicity [27]. Since extracellular deposition of A is a major hallmark of AD, attention was given to the insoluble fA, the main aggregated forms in the amyloid plaques [22]. Some studies demonstrated that fAβ are neurotoxic in vitro [30] and in vivo [31] and it was then hypothesized that they could represent a primary cause of neurodegeneration in AD. Nevertheless, the number and temporal progression of plaques do not correlate well with the local extent of the neuronal death and synaptic loss, nor with the disease progression [22]. Moreover, some individuals show high levels of plaques without revealing any cognitive impairment [18]. On the other hand, the levels of extracellular soluble oA show a robust correlation with the extent of synapse loss and cognitive impairment [27].A great number of different oA, natural or synthetic, possessing various sizes and shapes, have been reported: dimers and trimers, A-derived diffusible ligands, A56* (56 kDA) and others [32]. These intermediate forms are considerably more toxic than fA, and are appointed as the main culprits for neurodegeneration in AD [33] .
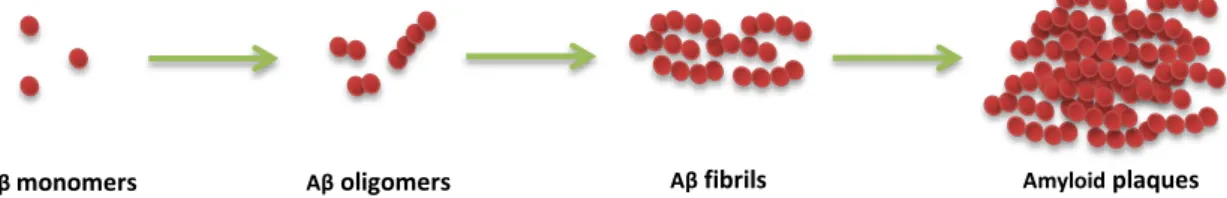
Fig. I.3: Amyloid beta assembly states. Amyloid beta (Aβ) can exist in diverse assembly states, which
include monomers, oligomers or fibrils, with the latter being the state deposited as amyloid plaques. Formation of fibrils is a complex multi-step process that is preceded by the oligomerization and aggregation of monomeric Aβ. The mechanism driving this process is not fully understood, but may be related to protein misfolding
Besides extracellular accumulation, studies indicate that A also deposits inside the cells and that this may be an early event in AD, since it precedes extracellular accumulation and has been reported on brain regions that are more prone to the development of early AD pathology, such as entorhinal cortex and hippocampus. Moreover, it is inside the cells that oligomerization process is thought to begin [34]. The intracellular localization of Aβ is a result of both APP cleavage inside the cell and new uptake of extracellular Aβ through receptors and transporters, such as the receptor for advanced glycation end-products (RAGE) [33].
The mechanisms by which Aβ induce neuron and synaptic degeneration are complex and far from being resolved, but it seems to be influenced by differences in aggregation forms and localization (intracellular or extracellular). Some of the proposed mechanisms are: interaction with diverse cell-surface receptors, conducting to inhibition
7 or aberrant activation of signal transduction pathways; insertion in membrane and formation of ion channels or pores, disrupting ionic homeostasis; disruption of lysosomal membrane; proteasome impairment [35,33].
It is important to have in mind that Aβ is a physiological product of the organism and as so it may have a physiological role. In this regard, some authors suggest a vital function in modulation of synaptic plasticity, neuronal survival and neuronal development, particularly at low concentrations, when in the absence of oligomerization [36].
1.2.2. Tau
Almost concomitantly with the identification of Aβ in amyloid plaques, neurofibrillary tangles were demonstrated to be composed of abnormally hyperphosphorylated tau protein. Tau is a member of the family of microtubule-associated proteins (MAPs) that is particularly abundant in neuronal axons [37]. The best-established functions of tau are the stabilization of microtubules and the regulation of motor-driven axonal transport [38]. The biological activity of tau is dependent on its degree of phosphorylation, which modulates the capacity to bind to microtubules [39]. Several kinases (e.g, glycogen synthase kinase 3 beta) and phosphatases (eg, protein phosphatase 1) are responsible for the fine tuning of tau phosphorylation state [37].
During AD pathogenesis, tau becomes increasingly phosphorylated, which causes its detachment from microtubules, resulting in a loss of its normal function [40]. Unbound hyperphosphorylated tau is able to sequester normal tau and other MAPs, further compromising microtubule structure. Moreover, it possesses an inherent capacity to self-assembly, forming paired helical filaments that will eventually give rise to neurofibrillary tangles [39]. Tau aggregates have been reported to be toxic primarily in the form of oligomers, like in the case of Aβ [41]. Therefore, it is though that the loss of normal functions and the toxic gain of function of tau, resulting from its hyperphosphorylation, might contribute to synaptic dysfunction and neurodegeneration [42]. The causes of abnormal phosphorylation are unclear, but altered function of kinases or phosphatases, or maladaptation by cellular and microenvironmental stress, are thought to be implicated [43] .
The involvement of tau in neurodegenerative process is also supported by the existing correlation between neurofibrillary tangles and the AD onset and progression [39]. Nonetheless, tau mutations have not been connected to AD. Indeed they are used to distinct phenotypic diseases, like frontotemporal dementia, that do not display plaques [42]. This suggests that tau pathology in AD is downstream of Aβ
8
accumulation, and is supported by experimental evidence indicating that Aβ accumulation precedes and drives tau aggregation. Some studies also indicate that the presence of tau might be necessary for Aβ neurotoxicity [37].
1.2.3.
Oxidative Stress
Oxidative stress derives from an imbalance between the production of reactive oxygen/nitrogen species (ROS/RNS) and the cellular anti-oxidant mechanisms [44]. ROS and RNS, resulting from hydrogen peroxide and nitric oxide (NO) accumulation, may damage nucleic acids, carbohydrates, lipids (lipid peroxidation) and proteins (protein oxidation) [45]. Neurons are considered highly susceptible to an increase of ROS/NOS due to their low levels of antioxidants mechanisms when compared with other neural cells [44].
Extensive evidence of increased oxidative damage, including lipid peroxidation, protein oxidation and nucleic acid oxidation, has been found in AD vulnerable neurons and suggested to precede any other feature of the disease[46,47]. In addition, low levels of naturally occurring anti-oxidants, such as α-tocopherol, excessive iron and copper deposits, intense microglial activation and mitochondrial abnormalities, are all known to be involved in the production of ROS/RNS and reported in AD [48,49].
Aβ is able to promote the oxidation of compounds like phospholipids and cholesterol, if an intact methionine in position 35 is present [48]. Furthermore, Aβ also promotes ROS production by directly associating with transition metals such as iron and copper, by stimulating microglial activation or through its effects on mitochondria dynamics [50,51]. Despite promoting oxidative stress, some authors suggest that accumulation of Aβ might also be a protective response against oxidative stress. For instance, Aβ has demonstrated to follow the appearance of oxidative stress markers in AD and to have anti-oxidant properties in cerebrospinal fluid (CSF) and plasma, protecting lipoproteins from oxidation [49].
Oxidative stress further potentiates Aβ oligomerization and aggregation, which in turn produces neuroinflammation, mitochondrial damage, and thus, further ROS generation, leading to a vicious cycle of more oxidative stress and more damage to the cells [49]. Consequently, there is an enhanced cell dysfunction and demise.
1.2.4.
Mitochondrial Dysfunction
Mitochondria are the main producers of both energy and free radicals, through oxidative phosphorylation and endogenous ROS species, playing a key role in neuronal function and survival in the brain, the third most energy-expensive organ in
9 the human body [52,53].
Increasing evidence in patients and disease models indicates that mitochondrial dysfunction accompanies ageing and plays an important role in AD. Indeed, it seems to be present at all stages of AD and worsens as the disease progresses [54]. Abnormalities on mitochondrial function that are observed in AD include a decreased in activity of respiratory chain enzymes (e.g. cytochrome c oxidase) and some Krebs cycle enzymes (eg. α-ketoglutarate dehydrogenase). Moreover, a reduction in the mitochondrial membrane potential and in the levels of adenosine triphosphate (ATP), together with increased oxidative modifications in mitochondrial DNA are observed in the disease [55,56].
The recent intracellular existence of Aβ has led researchers to consider it as a potential cause of mitochondrial dysfunction. Indeed, evidence suggests that Aβ crosses mitochondrial membranes, accumulating in the interior of mitochondria [57], thus influencing the activity of electron transport chain complexes and disturbing calcium storage, leading to apoptotic pathways. Moreover, Aβ interacts with mitochondrial matrix components, causing a decrease in the membrane potential and impairing ATP formation [58]. amyloid-binding alcohol dehydrogenase (ABAD) is one of Aβ targets. Interaction with ABAD leads to an inactivation of the enzyme activity and promotes mitochondrial generation of free radical species [59].It is also important to refer that mitochondria-derived ROS seem to be able to trigger amyloidogenic APP-processing, possibly by inducing BACE1 activation, which further implies an existence of a vicious cycle that contributes to AD pathogenesis [60].
It is now accepted that mitochondria are dynamic organelles and maintain their homeostasis and function by constantly undergoing fission (or splitting) and fusion (or combining) [52]. In AD this dynamic equilibrium is disrupted in favor of fission, which may compromise cellular integrity [61]. Recently, it has also been appointed that APP overexpression, likely through Aβ-mediated effects, could be responsible for abnormal mitochondrial dynamics in AD, including decreased mitochondrial mobility and alterations in fission/fusion processes [54]
1.2.5.
Neuroinflammation
The term neuroinflammation refers to the inflammatory-like process that occurs in the central nervous system (CNS), which is usually a transient, well controlled process, resulting from the response to a toxic insult [62]. In general, an acute neuroinflammatory response is beneficial to the CNS, since it contributes to repair the damaged tissue and to minimize further injury; in contrast, chronic neuroinflammation
10
that persists after an initial toxic insult may produce degenerative changes in neurons and alter brain function [63,64].
Although contribution of chronic neuroinflammation to AD is a controversy issue, namely in late AD stages [65], it may exacerbate the course of the disease, while leading to microglia senescence [66]. This process seems to have a genetic relation with the disease, since AD patients are more represented with pro-inflammatory genotypes than anti-inflammatory genotypes [67]. Signs of inflammation are particularly localized in brain areas exhibiting high levels of AD pathology. Furthermore, high pathology controls (that is, individuals presenting Aβ and tau aggregates at levels similar to AD patients, but that do not develop dementia) show lower signs of inflammation. In addition, the inflammatory mechanisms present in AD patients are comparable to those present in peripheral inflammatory reactions, which are established to be cytotoxic, and therefore are likely to have cytotoxic effects on neurons [68]. AD patients who develop a short-term peripheral infection present a sudden decline in cognitive state, rarely returning to previous level, even after the eradication of the infection [69]. It is worthwhile to mention that although some studies demonstrate that individuals treated (+ 24 months of cumulative use) with non-steroidal anti-inflammatory drugs (NSAIDs) present a 60-80% reduction in the risk of developing AD [70,71], anti-inflammatory drugs are ineffective or even harmful when used in advanced stages of the disease [72,73].
The neuroinflammatory process involves primarily the activation of microglia, the immune cells of the CNS, but also astrocytes, and even neurons. [74]. All these cells are able to produce/release several inflammatory mediators, which include pro-inflammatory cytokines, mainly tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6, as well as ROS/NOS, complement proteins and various proteolytic enzymes [62]. Some of these inflammatory factors by enhancing Aβ deposition and tau phosphorylation, concur to further activation and proliferation of glial cells and to the chronic inflammatory scenario seen in AD, at least in the earlier stages [75,76]. All these factors, alone or in concert, can then contribute to the neuronal dysfunction seen in the disease.
1.3. Diagnosis and treatment
Diagnosis of AD can reach about 95% confidence when realized by highly experienced clinicians, but a full confirmation can only be obtained at autopsy [77,78]. Until recently, the criteria for the diagnosis of AD only considered the late stages of the disease, when dementia is already present [79]. In 2011, however, a new form for
11 diagnosis was proposed [80]. This new diagnostic criteria expanded the definition of AD into 3 phases: a pre-symptomatic phase; a symptomatic, pre-dementia phase (or MCI); and a dementia phase. Moreover, it also incorporated the use of biomarkers to improve diagnosis, which are mainly used for research purposes.
These biomarkers are physiological, biochemical or anatomical parameters that can be measured in vivo and reflect specific features of disease-related pathophysiologic processes [81]. Five biomarkers were incorporated in the new criterion proposed, being divided in two classes. The first set is constituted by biomarkers reflecting Aβ deposition in brain, comprising both CSF evaluation of Aβ42 and positron emission tomography (PET) with the amyloid binding tracer N-methyl-11C-2-(4-methylaminophenyl)-6-hydroxybenzothiazole. The second group reflects neuronal degeneration or injury and includes tau (total and phosphorylated) level in CSF, fluorodeoxyglucose uptake on PET (used to measure brain metabolic energy) and regional brain atrophy (hippocampus, medial, basal and lateral lobes, and the parietal lobe) on structural magnetic resonance imaging [81]. Besides these, other promising biomarkers under investigation are the level of BACE1 [82] and isoprostanes (markers of oxidative stress) in CSF [83]. Plasma or serum biomarkers have also been explored, but none has shown the diagnostic accuracy of CSF biomarkers [84]. Biomarkers might certainly improve diagnosis of AD, principally at early stages. Nonetheless, deficient standardization or their limited access still needs to be solved [80].
After diagnosed, there is still no way to effectively deal with AD. Currently available treatments include cholinesterase inhibitors (donepezil, galanthamine, rivastigmine), which target the impairment of cholinergic function due to loss of basal forebrain cholinergic neurons, and memantine, a N-methyl-D-aspartate (NMDA) receptor antagonist, that aims to reduce excessive glutamate activation [85]. However, although these treatments show some clinical benefit, they are just symptomatic, not stopping disease progression [3]. Therefore, several studies for the development of new treatments are under way. Drugs aiming to prevent Aβ aggregation by decreasing Aβ generation and stimulating Aβ clearance are under investigation, and some of them are even in phases II and III of clinical trials [86]. Treatments to target tauopathy in AD have received less attention than amyloid therapies, but drugs aiming to inhibit tau hyperphosphorylation, tau oligomerization or to promote the degradation of hyperphosphorylated tau are also under study [87]. Additional approaches to treatment intend to improve neuronal function by using trophic factors like nerve growth factor (NGF). Moreover, due to the importance of oxidative stress and inflammation in AD, neuroprotective approaches may also include oxidants (e.g. vitamin E) and
anti-12
inflammatory drugs (e.g. NSAID), although clear positive effects are still missing. In addition, nondrug approaches for cognitive rehabilitation such as cognitive training and cognitive stimulation have shown modest, but promising results [88].
2. Microglia in AD: A closer look to the central player in
neuroinflammation
The inflammatory process is unlikely to be the cause of AD [89]; nevertheless, this does not mean that it is not important. In fact, like it was appointed before, several evidences indicate that it may play a major role in the early disease stage. Of the different cells involved in the inflammatory process, microglia by their immune nature are certainly the most important ones.
2.1. Microglia: origin and functions
Microglia comprise about 12% of all the cells in the brain, occupying both grey and white matter, with a higher density in hippocampus, substantia nigra, basal ganglia and olfactory telencephalon [90]. Although still in debate, the general consensus is that microglia are of hematopoietic origin, being derived from the myeloid precursor cells from yolk sac that enter the developing CNS during embryogenesis [91].
Usually, under normal conditions, microglia are in a surveying state, characterized by a ramified morphology with long and thin processes [92]. These processes are highly mobile, being constantly extending and retracting to monitor microglia’s surroundings [93]. Each microglia covers a defined territory, non-overlapping with neighboring microglial cells [94]. Microglia are considered to be long-lived cells, but might be replaced by low level of local self-renewal[95].
Microglia play numerous important roles through life. In the developing brain, microglia have seen to be involved in the removal of the massive number of cells that undergo apoptosis during developmental programmed cell death [96], in the control of the number of synapses through the process of synaptic pruning [97] and also in CNS vascularization [96]. Nonetheless, within the adult CNS, the primary role of microglia is similar to that of macrophages in other tissues, representing the first line of defense against injury or pathogens[98]. In order to detect and interpret potential insults that disturb CNS homeostasis, a plethora of different surface receptors are expressed by microglia, including neurotransmitter receptors and pattern recognition receptors (PRRs), which represent a vast array of conserved receptors with ability to recognize
13 and bind small molecular motifs present in pathogens (pathogen-associated molecular patterns) or factors associated with tissue damage (alarmins or damage-associated molecular patterns) [99,94]. These signals evoke the activation of microglia, leading to a change of their ramified morphology to an amoeboid shape. Microglia became highly mobile and able to migrate to site of the signal. Furthermore, the activation process is accompanied by the up-regulation of several surface receptors, as well as by the production of a plethora of bioactive molecules. [94]. The type of activation is dependent on factors like the intrinsic properties of the stimulus, duration and exposure to a prior or another existing stimulus [98].
Microglia are highly plastic cells and while displaying similar morphology they may show distinct activated phenotypes. Inspired by studies in peripheral macrophages, the activation of microglia has been generally classified in i) classical activation, ii) alternative activation and iii) acquired deactivation [100]. Classical activation (commonly called M1 phenotype) is induced by pro-inflammatory mediators such as lipopolysaccharide (LPS) or interferon-γ and is characterized by the production of numerous pro-inflammatory cytokines (e.g. TNF-α, IL-1β and IL-6), proteases and ROS/NOS. These molecules play an important role in the defense of the organism to pathogens, but can also damage neurons and glial cells [101]. Alternative activation (M2a phenotype) is induced by IL-4 or IL-13 and is associated with the production of anti-inflammatory cytokines and trophic factors such as insulin-like growth factor, playing a role in tissue repair. [100]. Acquired deactivation (M2c phenotype) is produced by inducing agents like transforming growth factor β and IL-10, and it is associated with a robust suppression of the innate immune system, mainly through an elevated production of the anti-inflammatory cytokine IL-10.[102,100]. Furthermore, the acquired deactivation is also elicited by the exposure to apoptotic cells, permitting the phagocytosis of these cells without triggering an immune response, which is important to tissue maintenance [102]. Both alternative activation and acquired deactivation down-regulate innate immune responses and demonstrate similar, but not identical, gene profiles; therefore, some authors treat them as subgroups of a single category, commonly called M2 or alternative activation [100] . It is also important to refer that these phenotypes are not static. In fact, microglia is able to switch between phenotypes, and may exist in many intermediate states (Fig. I.4).
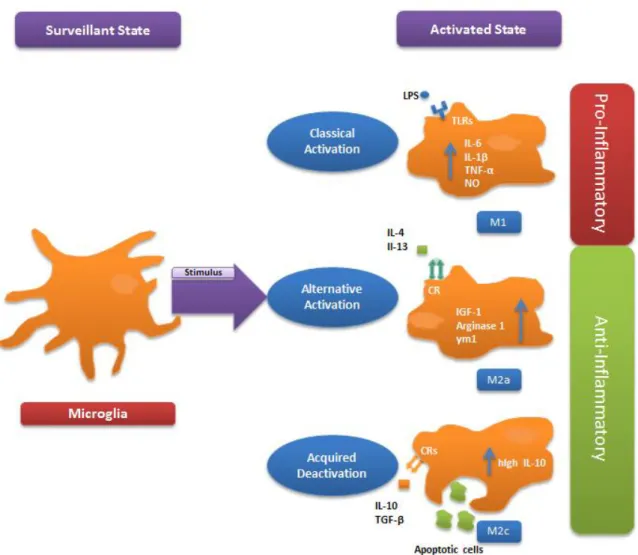
14
Fig. I.4: Microglia activation states. Depending on the stimulus, surveying microglia might respond with different activation profiles. Lipopolysaccharide (LPS) is responsible for inducing a classical activation of microglia, mainly through toll-like-receptor (TLR) 4, resulting in a pro-inflammatory profile characterized by an increase in the production of interleukin (IL) -6, 1β, tumor necrosis factor (TNF)-α and nitric oxide (NO). Although playing an important role in organism defense, the production of these cytokines is commonly related to neurotoxicity. In contrast, alternative activation and acquired deactivation are both associated with an anti-inflammatory, neuroprotective profile, with the former being characterized by the up-regulation of insulin-like growth factor (IGF)-1, arginine 1 and Ym1 and related to tissue repair and extracellular matrix reconstruction, while the later through the production of high levels of IL-10, is more related to a robust suppression of the immune system. Described inducer agents include IL-4 or IL-3 and IL-10 or TGF-β, respectively, which act through different cytokine receptors (CRs).
2.2. Activation of microglia in AD
2.2.1. Interaction of amyloid beta with microglia
Studies evidence the presence of activated microglia clustered near amyloid plaques in AD patients [103], as well as in AD mouse models [104]. This clustering might be explained by chemotactic signaling mediated by A, or by other molecules found to be associated with plaques, such as complement factors or chemokines [105]. Recently, it was visualized by in vivo imaging techniques that microglia migrate to newly formed plaques within 1-2 days [106]. The number and the dimension of
15 microglia around amyloid plaques increase in proportion to the size of plaques, and it was suggested that plaque-associated microglia can regulate plaque dynamics [107]. Accordingly, microglia activation has been considered an early event in the pathogenesis of AD [108].
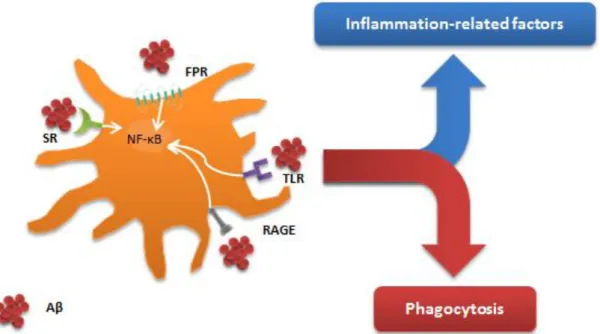
Several triggers for microglial activation are present in AD, which might include either molecules released from damaged neurons or even Aβ [109]. Indeed, Aβ was demonstrated to induce microglial activation and to be one of the main triggers of microglial response in AD [110]. Both oA and fA are able to bind and activate microglia through several receptors, including a number of different PPRs [111] (Fig. I.5). In this context, toll-like receptors (TLR) appear to be particularly important. Among the cell-surface TLRs present in microglia, TLR2 and TLR4 were shown to be involved in Aβ-induced microglial activation [111]. Furthermore, several studies in vitro and in vivo have confirmed that TLR4 mediates the neurotoxicity induced by microglia [112,113]. Besides TLRs, it was demonstrated that receptors like RAGE, scavenger receptors (e.g. cluster of differentiation 36, CD36) or formyl peptide receptors (FPR; eg. FPR-like 1) are also involved in Aβ-induced microglial activation[111]. The binding of Aβ to these receptors is not only responsible for the secretion of inflammatory molecules through the activation of signaling cascades such as nuclear factor-κB (NF-κB), but also for inducing phagocytosis [114,111]. Worth to note that, although they both stimulate a pro-inflammatory activation, oAβ and fAβ present different activation profiles [115], and oAβ revealed to be a stronger M1-inducter than fAβ [116].
16
Fig. I.5: Amyloid beta interaction with microglial receptors. Microglia cells express a panoply of
surface receptors with which they are able to sense their environment and respond to stimuli. Amyloid beta (Aβ) is able to interact with several of these receptors, inducing microglial activation. Among the array of receptors described to be involved in Aβ binding are: toll-like receptors (TLR), like TLR2 and TLR4; scavenger receptors (SRs), like CD36; formyl peptide receptors (FPR); and receptor for advanced glycation endproducts (RAGE). The binding of Aβ to microglial receptors elicits a pro-inflammatory response, with the release of inflammatory cytokines like Interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α, probably through activation of the nuclear factor- κB (NF-κB) signaling pathway, but also seems to induce phagocytosis, which is mediated, at least in some cases, by initiating the Src-Vav-Rac signaling cascade
2.2.2. Production of inflammation-related factors by amyloid
beta-stimulated microglia
As aforementioned, challenging of microglia with A results in a pro-inflammatory response. Inflammatory cytokines like IL-1β, IL-6 and TNF-α have all been reported to be released by A-induced microglia [117]. IL-1β and TNF-α, for instance, can directly injure neurons when at elevated levels [118]. These cytokines may act in an autocrine manner further inducing cytokine production, while also activating astrocytes [17]. Indeed, inflammatory cytokines have been reported to be increased in vulnerable brain regions during AD and where found to be associated with plaques [62]. Interaction of microglia with Aβ also leads to the secretion of chemokines, such as monocyte chemotactic protein-1 (MCP-1, also known as CCL2) and macrophage inflammatory protein-1α (MIP-1α, also known as CCL3) [119,120]. Chemokines play an important role in the recruitment of microglia and astrocytes, and an increased expression of the chemokines receptors CCR3 and CCR5 was found in microglia associated with Aβ deposits [121].
Other inflammatory-related molecules produced by microglia are ROS and NOS. In this regard, stimulation with Aβ, for instance, was demonstrated to lead to the
17 release of NO and superoxide radical [90]. Although these molecules have an important role in the defense against pathogens, high levels are detrimental and might contribute to AD, as it was discussed before. Derivatives of APP, including Aβ, were also reported to induce the release of glutamate from microglia cells [122,123]. Glutamate is an excitatory amino acid that plays an important role in synaptic plasticity, memory and learning, but it can also be highly toxic to neurons, inducing excitotoxicity, which consists on overstimulation of glutamate receptors like NMDA receptors, leading to a massive influx of extracellular Ca2+ that damages cell structures and components [124,125]. However, these reports are questioned by recent findings indicating that Aβ might not affect glutamate release [109]. Also an important molecule that was recently reported to be released by Aβ-stimulated microglia is ATP, which was linked to ROS production by microglia [126].
Several proteases are released by microglia, contributing to their functions. MMPs, for example, which are essential to the maintenance and reconstruction of extracellular matrix, were indicated to be involved in AD [127]. MMP-3, -12, -13 and -9 have all been reported to be released by Aβ-stimulated microglia [128,129]. Moreover, both MMP-9 and MMP-2 seem to be able to degrade Aβ in vitro [130,131], and their relevance to amyloid deposition was further corroborated by knockout mice [132]; however, these proteases also have a role as mediators of tissue degradation and inflammation, damaging the blood-brain barrier (BBB) and processing pro-inflammatory cytokines [127] .
2.2.3.
Amyloid beta-induced microglial phagocytosis
Phagocytosis represents a central housekeeping function played by microglia that is important to the elimination of neurotoxic compounds, cellular debris and pathogens. Accordingly to the receptor that is stimulated, it may be or not coupled with inflammation [133]. Aβ has been documented by several studies to be phagocytized by microglia. For instance, examination of microglia at amyloid plaques using electron microscopy has shown that they are able to engulf Aβ with their processes and indeed Aβ was observed in endosome-like cellular compartments [134]. Moreover, in vitro studies using labeled Aβ [135] in combination with direct injection of fAβ into rat brain [136] further demonstrated the capability of microglia to phagocytize Aβ. The mechanism by which Aβ is phagocytized upon interaction with microglia is dependent on its physical and biochemical properties. Insoluble Aβ aggregates are internalized through receptor mediated phagocytosis (e.g TLR2 and 4), while soluble Aβ has been suggested to not be truly internalized by phagocytosis, but through macropinocytosis