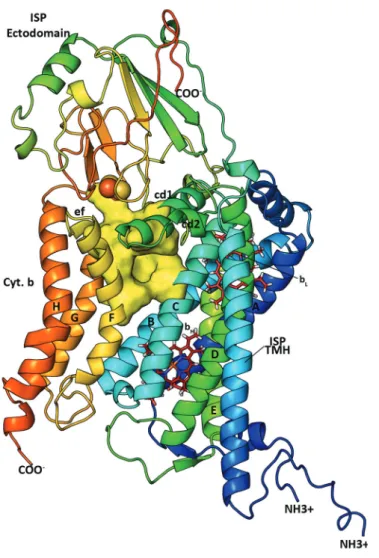
Insights into cytochrome bc
Texto
Imagem



Documentos relacionados
A ligand library of thirty two Knoevenagel condensates of curcumin were designed and docked against PfATP6 protein and six compounds with the best binding scores were synthesized
Results of investigations of influence of small vanadium up to 0.3%, niobium up to 0.16% and nitrogen up to 58 ppm additions and heat treatment of 1080 o C-24h/640 o C and 1080
The structure of the remelting zone of the steel C90 steel be- fore conventional tempering consitute cells, dendritic cells, sur- rounded with the cementite, inside of
Fractures were made in all samples. Figure 1 shows fractures of test bars cast from the examined high-aluminium iron in base condition and with the addition of vanadium
These authors used the RAPD technique to detect mutants of Trichoderma harzianum obtained by gamma radiation, observing an evident DNA polymorphism among the wild strain and
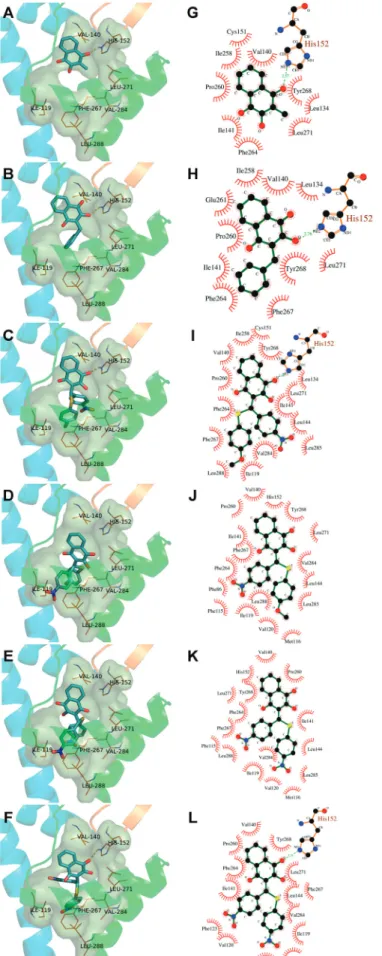
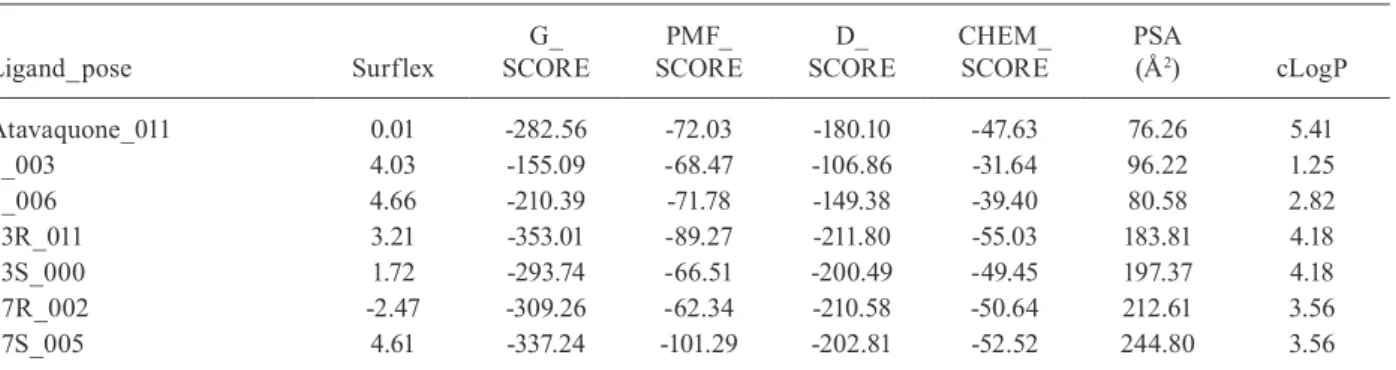
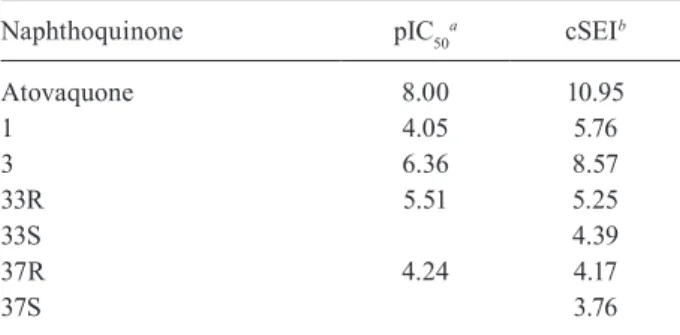
This could be verified through a significant correlation between the inhibitory activity of indene pyrazol ligands and the total score of the complex docking, with a coefficient
The integration of enzyme kinetics, structural analysis and molecular modeling studies provided important insights into the molecular basis underlying ligand binding afinity.
All inhibitors were subsequently docked into the binding site obtained from the receptor and conformation of the inhibitors with the lowest binding free energy was used to
