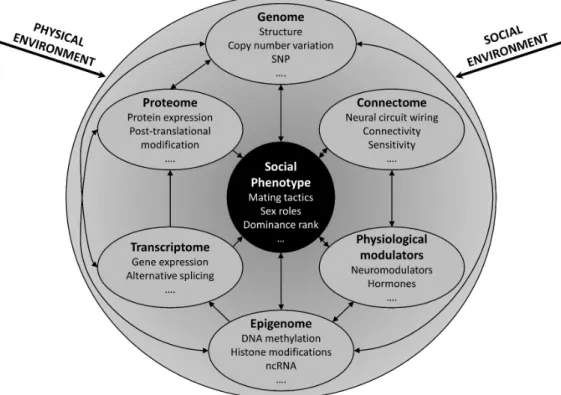
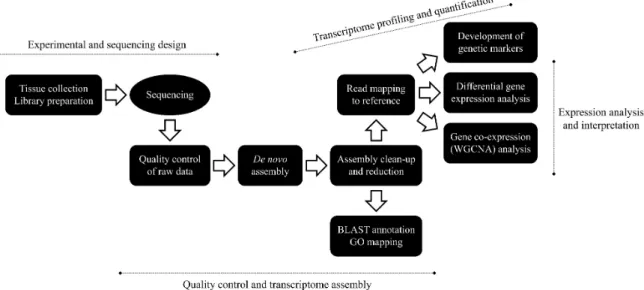
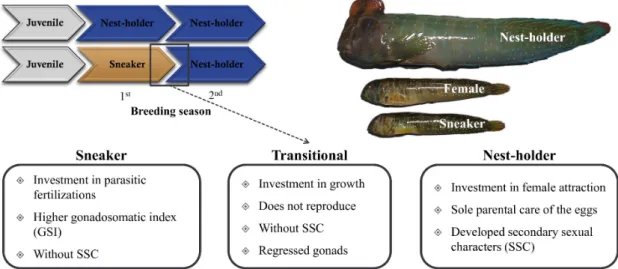
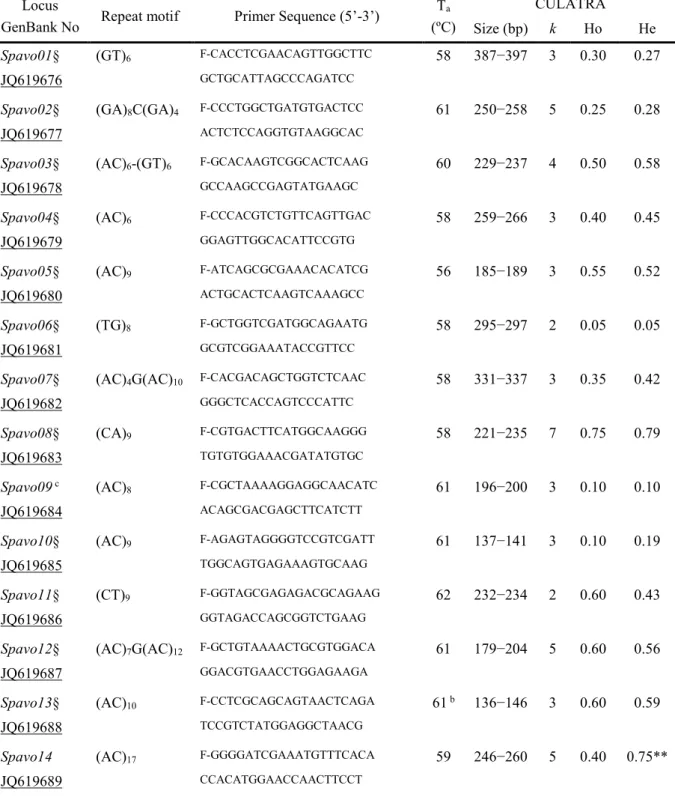
RNA-SEQ applied to the peacock blenny Salaria pavo: unveiling the gene networks and signalling pathways behind phenotypic plasticity in a littoral fish
308
0
0
Texto
Imagem
+7

Documentos relacionados