I would also like to thank him for his valuable advice during my research for this dissertation. The second special person is my dear daughter Emilia, born during the preparation of the manuscript for this thesis.
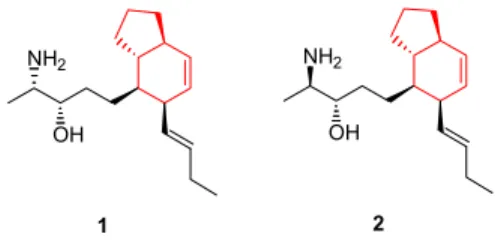
Aliphatic amino alcohol relatives of amaminol A and B
Amaminol A 1 and B 2 are closely related to aliphatic amino alcohols isolated from marine sponges, Xestospongia sp. These are also biogenetic products of (S)-alanine 3. R)-alanine 4 derived aminoalcohols were isolated from tunicates.
Cyclic relatives of amaminol A and B
Pulo’upone and isopulo’upones
- Burke
- Oppolzer
- Takano
- Evans
- Hase
Pyrroloketoindanes
- Indanomycin 72
- Stawamycin 166
Silylation of 4-pentynol 65 was followed by lithiation of the triple bond and iodination to give iodoalkyne. The coupling of the pyran derivative 142 and bicyclo[4,3,0]nonane fragment 141 was achieved using the Lythgoe–Kocienski modification of the Julia reaction (29).
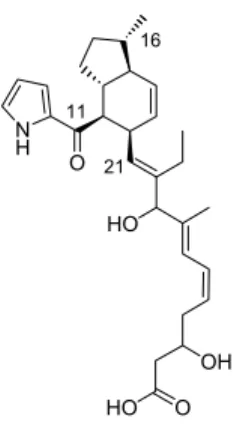
Cochleamycin A 175
Reduction of the alkyne to E,Z,E-triene 188 and IBX oxidation provided the substrate for the IMDA thermal reaction. The IMDA reaction was immediately followed by reduction of the bicyclic aldehyde to yield the cycloadduct 190 via the endo transition state.
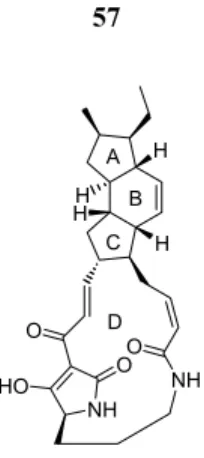
Ikarugamycin 191
- Boeckman
- Kurth
- Jones
- Roush
- Whitesell
- Paquette
Boeckman completed the synthesis of icarugamycin 191 using a transannular Dieckmann cyclization and deprotection of the N-aryl protection. Finally, the tetraene ketone precursor for IMDA addition was obtained by the HWE reaction of triene aldehyde 227 with β-ketophosphonate 228 .
Lepicidin A 272
- Evans
Lewis acid-promoted aldol reaction of 275 with silylenolate 276 gave Felkin adduct 277 with high diastereoselectivity (>20:1). Lactonization was used to protect the hydroxyl and carboxyl groups, and the lactone ring 279 was expected to provide better stereocontrol in the conjugate addition of vinylstannane 280. The diene fragment 289 of the IMDA precursor was prepared from the TBS-protected 3-hydroxy 1,5-dicarboxylate anhydride 285 (Scheme 41) (53).
Aldehyde 286 was reacted with iodoform and chromium chloride to afford vinyl iodide as a stereoisomeric mixture (E:Z, 9:1). 2,3,4-tri-O-methyl-D-rhamnose was coupled to (+)-lepicidin A aglycon 294 after chemoselective desilylation of the TIPS group.(53) This was followed by deprotection of the TBS group and glycosidation with L-forosamine. (Scheme 43).
Summary
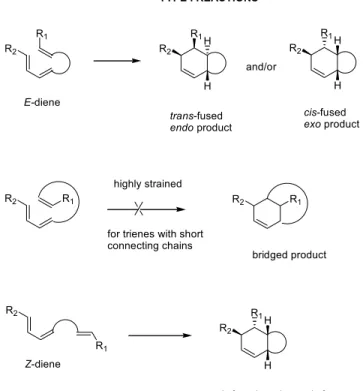
This model assumes that the selectivity of the reaction is controlled by the timing of bond formation in the transition state. IMDA reactions can be divided into two categories based on the point of connection between the diene and the dienophile. In the type I reactions, the connecting chain is at the terminus of the diene (Figure 17).
The LUMO coefficient at the α-carbon of the carbonyl group is greater than at the β-carbon. The formation of the peripheral bond is likely to be more advanced in the transition state.
Thermal cycloadditions
A larger group (i-propyl) at the terminal end of the E,E,E-triene 300 slightly changed the selectivity of the thermal IMDA. If the α-carbon of the diene in the connecting chain is substituted as in substrates 301 and 310, the thermal cycloaddition produces a mixture of endo- and exo-isomers by favoring the endo-cycloadducts with E,E,E-trienes and the exo-adducts with Z, E,E triene. Furthermore, the steric demand of the substituent at this position does not significantly alter the endo/exo selectivity (68).
A mixture of E,Z isomers of the tribenzyloxytriene 342 derived from D-xylose can be cyclized to a single cis-fused adduct 343 (Scheme 52) (75). The sterically demanding tert-butyldimethylsiloxy group effectively controls the transition state of the reaction to give the endo-adduct.
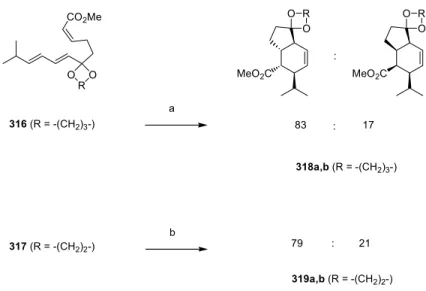
Lewis acid promoted cycloadditions
Stereoselectivity is not greatly affected by the dienophilic substituent in Lewis acid-catalyzed IMDA reactions of nona-trienes (69b).
Asymmetric IMDA cycloadditions
Chiral auxiliary induced IMDA
Saig's chiral triene 359 (82), obtained as an auxiliary oxazolidinone, gave the best enantioselectivity (96%ee) compared to other investigated chiral trienes 356-360. In some cases, the reactions were not complete even after 4 days of stirring at -25 °C. 83) studied asymmetric thermal IMDA reactions of sulfoximine-substituted trienes (Scheme 58). The triflyl sulfoximine derivatives of triene 366 were found to give slightly better endo/exo selectivities and diastereoselectivities compared to 4-tolylsulfonyl and 2,4,6-triisopropylphenylsulfonyl sulfoximines when diene terminated.
Without the terminal methyl group in the diene, the selectivities were similar with all the above-mentioned sulfoxymintriene derivatives.
Catalytic asymmetric IMDA cycloadditions
The enantiomeric excess in this reaction was only 46 %ee. 86) developed Brønsted acid-supported chiral boron catalysts and the binaphthyl derivative 376 catalyzes the IMDA reaction of trienaldehyde 373 in high yield (95. The cyclization gave only the endo adduct with 80 %ee (Scheme 60).. 87). developed titanium-based catalysts for asymmetric IMDA cycloadditions.
Other methods
This was suggested to be due to rapid isomerization of the E,E,Z-triene to the corresponding E,E,E-triene. Although the reaction of triene 386 took 72 h to complete at room temperature, the reaction afforded the cycloadduct in good yield (78%) and the endo:exo ratio was high ≤98:2. The cycloadduct was reduced to the corresponding alcohol 387 with lithium aluminum hydride for analytical purposes.. 93) formed hydrindene 392 in the synthesis of conocephalenol 390 by dehydration of the bicyclic alcohol 391 (Scheme 66).
Cycloadduct 407 was obtained by palladium(II)-catalyzed Heck reaction of the triflate derivative 403 in high yield (85. Palladium acetate-catalyzed cycloalkenylation of 406 afforded 410 in good yield. Low-valent rhodium complexes were reported to catalyze the IMDA cycloaddition between unactivated dienophile and diene (Scheme 72).
Summary of preparation of bicyclo[4.3.0]nonane derivatives
Introduction
Retrosynthetic analysis of amaminol A (1)
Synthesis of E,E,E-trienes for IMDA cycloadditions
Chiral auxiliary promoted asymmetric IMDA
- Removal of the chiral auxiliary
- Reactions of the five membered lactol
However, only minor racemization of the product was observed during the reaction, which was carried out according to the procedure developed by Greene et al. Product 455 was crystallized from methanol and the crystal structure of cycloadduct 455 is presented in Figure 24. Treatment of cycloadduct 455 with DDQ in a mixture of dichloromethane and water (20:1) resulted in cleavage of the benzyl group and immediate formation of lactone 458.
However, a concentrated solution of hydrochloric acid (6 M HCl) in dioxane led to lactonization of amide 456. The slow reaction was also accompanied by racemization of the amino group, which was not observed in the reaction in ethanol.
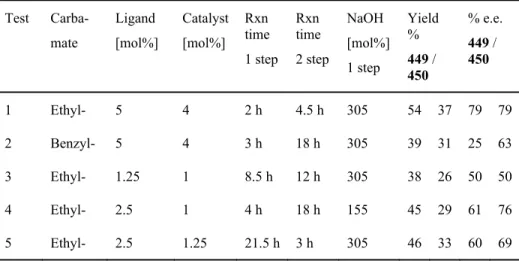
Organocatalytic asymmetric IMDA
I decided to replace the N-methyl group of organocatalyst 475 with an N-benzyl group because I felt that this would make the catalyst more rigid. The linear triene aldehyde 470 was more prone to polymerization, so some experiments were performed with starting materials containing more branched triene aldehyde 471. Conditions and IMDA results of the reaction of triene aldehyde 470 catalyzed by organocatalysts 474a, 475, 476 and 479a .
The yields were correlated with the amount of linear aldehyde 470 at the beginning of the reaction. The iminium ion 483 is active enough to allow asymmetric IMDA cycloaddition with the diene portion of the molecule.

Preparation of amino alcohol side chain
Chirality derived from L-alanine
- HWE based approach
Significant racemization (>96% ee) of the reduction product was not observed by chiral GLC or HPLC analyses. In this model, the metal in the Lewis acid (M) is chelated to two carbonyl oxygen atoms. One of the chelating oxygens belongs to the target carbonyl and the other to the N-BOC carbonyl oxygen.
The two dd patterns were adopted as a result of two different conformations of the acyclic double bond (Figure 26). The extra signals in the 13C NMR spectrum were assumed to originate from a conformer of the product 489a.
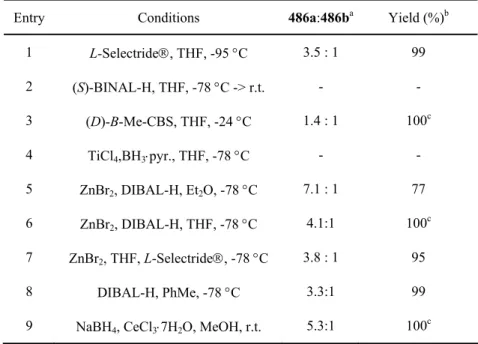
Wittig approach
- Crotonate oxyamination based approach
Enantiopure amino alcohol 498 was converted to oxazolidine 499 with 2,2-dimethoxypropane using p-toluenesulfonic acid as catalyst (Scheme 100). (101f) The ester group was selectively reduced with lithium aluminum hydride to the oxazolidine alcohol 500. The phenylsulfoxide derivative 504 was prepared for the Julia-type coupling of the amino alcohol fragment to the cyclic aldehyde 480. However, the coupling of Julia 480 and 504 was not attempted because it was assumed that that the hydroxyl group of coupling product 505 would be too hindered to be reduced to the corresponding E-olefin 506 by sodium amalgam (Scheme 102).
The hydroxyl group of 505 was assumed to be stabilized by the lone pairs of oxygens in the oxazolidine ring (SE(1)) and the benzyloxy chain (SE(2)). Next, I tried to prepare the mesylate from the alcohol 481 to activate the alcohol 481 for iodo substitution, but unfortunately no mesylation of the alcohol 481 was observed at room temperature.
Preparation of the olefinic side chain
The results of the reactions were very different because the products 511a,b were very volatile and were easily lost during concentration of the product.
Elaboration of amaminol A (1) analog
The diastereoselectivity of the reduction was high and the (S,R)-diastereomer was not observed by 1H NMR measurement from the crude product 513. Attempt to protect the hydroxyl group of 513 with tert.butyldiphenylsilyl chloride and imidazole in DMF solution (153). at 0 → 50 °C proved unsuccessful. It seems difficult to activate lactone 455 to introduce side chains.
The alcohol 481 was surprisingly unreactive due to hydrogen bonding of the hydroxyl group to other oxygen atoms in the side chain. Unfortunately, the diastereomer 519 of amaminol A 1 was obtained as the final product, because the wrong enantiomer of the organocatalyst was used for the preparation of the aldehyde 480.
Preparation of triene derivatives
Acetic acid 6-(tert-butyl-dimethyl-silanyloxy)-hexa-2E,4E-dienyl ester
The colors of the reaction mixture changed during the addition in the following order: orange-brown-green-yellow/green-brown-purple red. The reaction mixture was diluted with ether (25 mL) and washed with a saturated solution of NH 4 Cl (2 x 10 mL). The layers were separated and the aqueous layer was extracted three times with ether (ml).
The organic layer was filtered and the solvents were evaporated to yield 1.09 g of yellow liquid. The organic layer was dried with MgSO 4 for 30 minutes and the solvents were evaporated to yield 0.125 g of a clear oil.
Methyl 6-bromohexa-2E,4E-dienoate (429)
Methyl 6-hydroxyhexa-2E,4E-dienoate (435)
Stirring was continued for 40 min at -78 °C and then the reaction mixture was immersed in a -25 °C cooling bath (ethylene glycol/CO 2 ). The reaction mixture was poured into a beaker containing 1 M solution of HCl (300 mL). The mixture was stirred for 15 minutes. Stirring was continued for 1.5 hours, after which the reaction mixture was allowed to warm to room temperature.
The aqueous layer was extracted once with EtOAc (5 mL) and the combined organics were dried with Na2SO4, filtered and the solvents evaporated to yield 161 mg of yellow oil. 5 ml) was added and the mixture was stirred vigorously for 30 minutes. Stirring was continued for 5 hours and the reaction mixture was filtered through a thin pad of Celite.
Chiral auxiliary induced preparation of bicyclo[4.3.0]nonene derivatives and
Stirring was continued for 6 hours between – 45 – 20 oC and then the reaction mixture was allowed to warm slowly to room temperature. Stirring was continued for 2 days and the reaction mixture was cannulated into an ice-cold solution of 1M HCl (10 ml). After the addition, the reaction mixture was immersed in a cooling bath to – 25 °C (ethylene glycol/CO2).
After that, the reaction mixture was heated to 0 °C for 30 minutes and finally the reaction mixture was allowed to warm to room temperature. Stirring was continued for 20 minutes and the reaction mixture was immersed in an ice bath for 1.5 hours.
Organocatalytic preparation of bicyclo[4.3.0]nonane derivatives
The reaction mixture was stirred for 20 hours and the reaction mixture was diluted with ether (200 mL) and washed with H2O (50 mL) and brine (50 mL) mixture. The reaction mixture was stirred for 5 hours and the reaction mixture was diluted with ether and the combined organics were washed with brine (10 mL). Stirring was continued for 2 h and the reaction mixture was neutralized by adding a saturated solution of NaHCO 3 (12 mL).
Stirring was continued for 2 hours and the reaction mixture was neutralized by adding saturated NaHCO3 solution (15 ml). The reaction mixture was diluted with ether (5 ml) and the combined organics were washed with brine (3 ml).