Az elmúlt évek során - amíg a PhD munkámmal voltam elfoglalva - sok embertől kaptam segítséget, támogatásuk nélkül ez a dolgozat nem jöhetett volna létre. Itt ragadom meg az alkalmat, hogy kifejezzem köszönetemet mindazoknak, akik ösztönző és értékes tanácsaikkal segítették doktori munkámat, nem sajnálva a belefektetett időt és energiát. Segítette Halász Juditot és az Egis Műszerspektroszkópiai Laboratórium valamennyi dolgozóját az NMR és IR spektrumok elkészítésében, hozzárendelésében és a röntgendiffrakciós mérésekben.
Sam Erikának, Abonya Attilának és Kertész Ferencnek a hasznos elméleti és gyakorlati tanácsokért és az Egis Gyógyszergyár Zrt.-ért.
Bevezetés
Célkitűzés
Klasszikus izokinolin, dihidroizokinolin és tetrahidroizokinolin szintézisek
- Bischler-Napieralski szintézis
- Pictet-Gams szintézis
- Pictet-Spengler szintézis
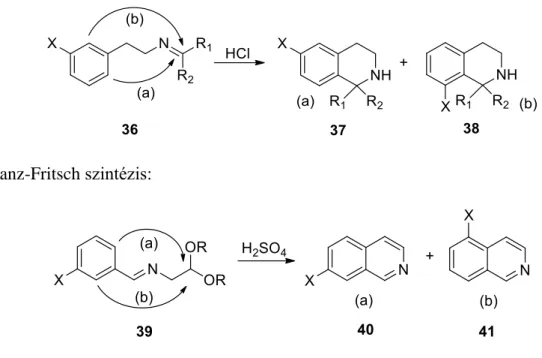
- Pomeranz-Fritsch szintézis
Az izokinolin vegyületek előállításának klasszikus módszerei a Bischler-Napieralski (B-N), Pictet-Gams (P-G), Pictet-Spengler (P-S) és Pomeranz-Fritsch (P-F) szintézisei.9 Az első három eljárásban a heterogyűrű fenil-etilamin származékból, benzilamino-letamin származékából és phenzilamino-letamin származékából jön létre. benzil-aminban és egy elektrofil származékban. ic szubsztitúciós reakció. A P-F módszer kiindulási anyaga az előző háromtól eltérő szerkezettel lehetővé teszi más szubsztitúciós mintázatú izokinolinok szintézisét.
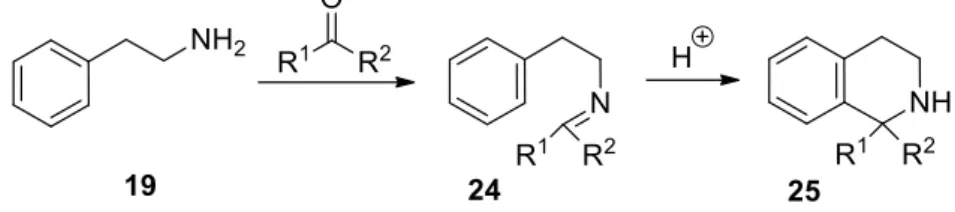
Klasszikus izokinolin szintézisek alkalmazása az aminoalkil oldallánchoz képest
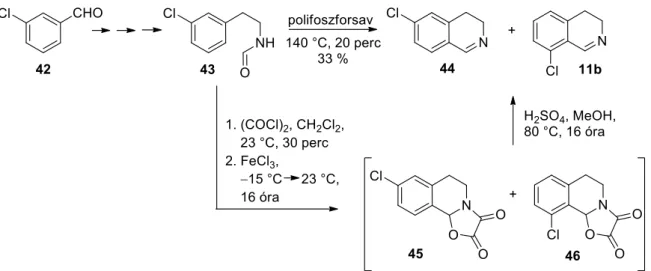
A Bischler-Napieralski módszert alkalmazva Böhme és munkatársai a 6- és 8-helyzetű klór-szubsztituált 3,4-dihidroizokinolinok keverékét kapták. A reakcióban képződő 6-klór- és 8-klór-3,4-dihidroizokinolinok (44, 11b) arányát nem határoztuk meg.11 A Lalonde és munkatársai által publikált eljárás termékei a 44-es és 11b-es vegyületek is. A termékek aránya körülbelül 13 volt. Bondinell és kutatócsoportja ezzel a módszerrel szintetizálta a 8-as helyzetben (50) klóratommal szubsztituált izokinolint is, 6%-os hozammal. 14.
A reakció során a kívánt (54) izokinolin-származékot mindössze 3%-os hozammal kaptuk.15 Ennek oka, hogy az aromás gyűrűhöz kapcsolódó fluorszubsztituens meta-helyzetében lejátszódó aromás elektrofil szubsztitúciós reakció kedvezőtlen. Az izokinolin-származékok előállítására szolgáló korábbi szintézismódszerekhez képest a közleményben említett reakció során magas, 60%-os hozammal kapjuk a kívánt vegyületet.16 Ezért munkánk során a szintézis reprodukálására törekedtünk, a hatékonyságról a dolgozat megfelelő fejezetében lesz szó (3.6.3. fejezet). Ezt követően triklórizocianursav (TCCA) reagenssel egy klór szubsztituenst vittünk be a 8-as pozícióba, majd a védőcsoportként használt brómatomot reduktív módszerrel eltávolítottuk, így 44%-os kitermelést kaptunk.
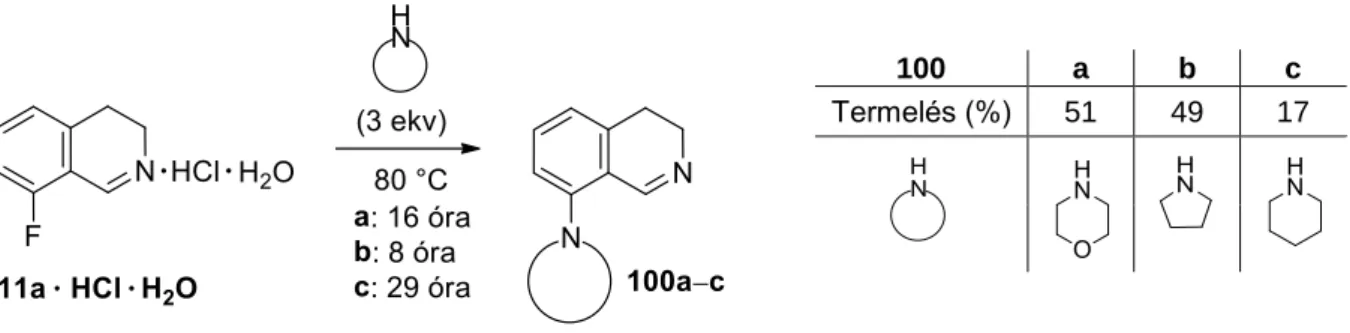
Benzolszármazékok irányított lítiálása
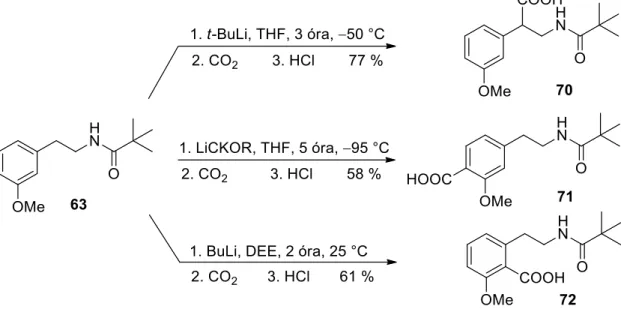
A regioszelektivitást az irányító csoportok minőségén kívül befolyásolhatja az alkalmazott lítiumképző szer, oldószer, komplexképző szer, hőmérséklet, reakcióidő, valamint a szubsztrát és elektrofil térbeli és elektronikus tulajdonságai is. Az alkalmazott reagensektől, oldószertől, hőmérséklettől és reakcióidőtől függően a kiindulási anyag három különböző pozíciójában hidrogén-fém cserét tudtak elérni. Alacsonyabb hőmérsékleten, -95 °C-on LiCKOR szuperbázissal (BuLi és kálium-terc-butoxid keveréke) a (71) vegyületet a metoxi szubsztituens orto pozíciójában, az alkil oldallánctól távolabbi helyzetben lítiumiztuk 58%-os hozammal.
Szobahőmérsékleten, DEE oldószerben és BuLi felhasználásával 61%-os hozammal sikerült előállítani a (72) terméket a két irányítócsoport közös orto-helyzetében lítiumozottan.19.
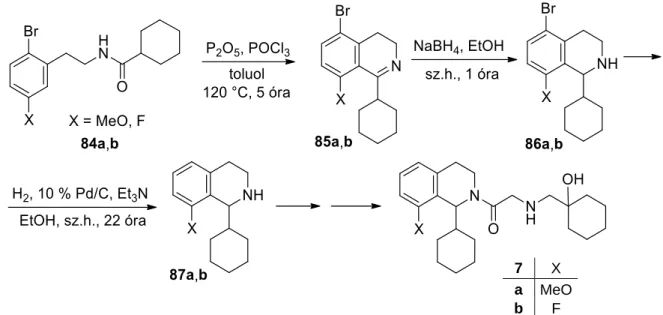
Saját munka: 8-fluor- és 8-klór-szubsztituált 3,4-dihidroizokinolinok szintézise és
- A 8-fluor-3,4-dihidroizokinolin átalakításai
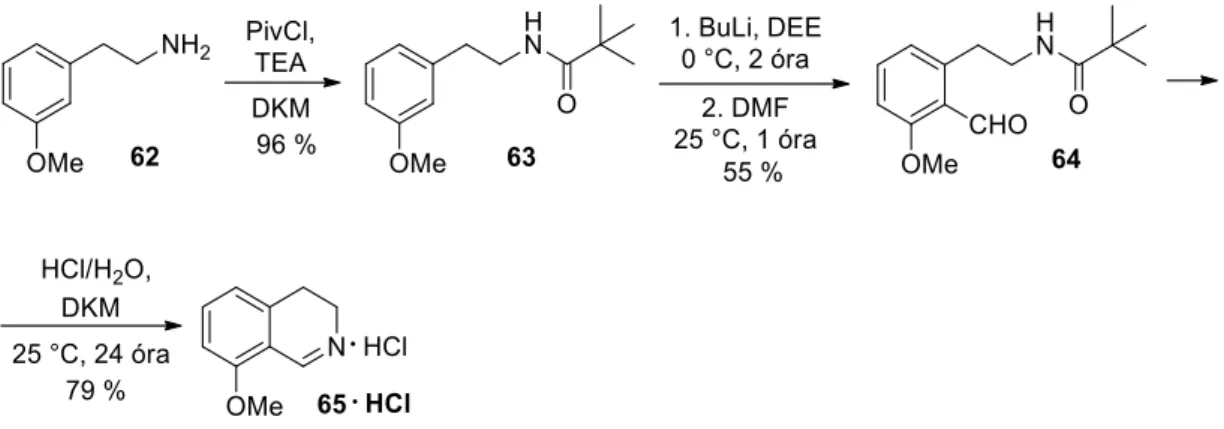
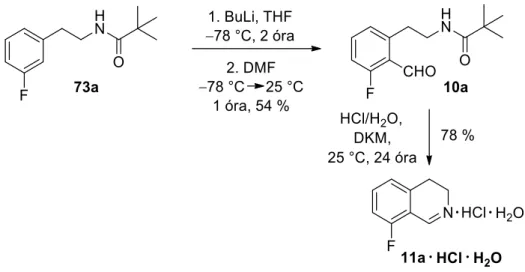
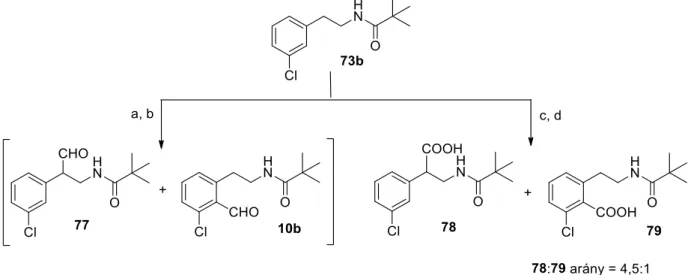
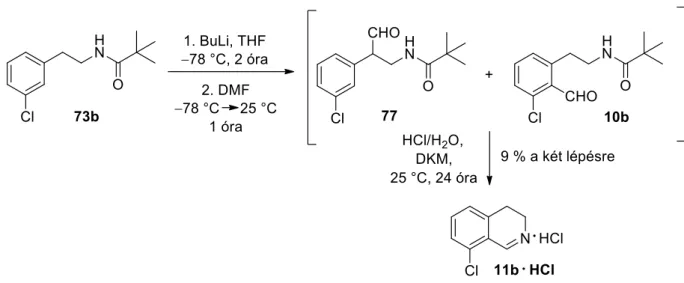
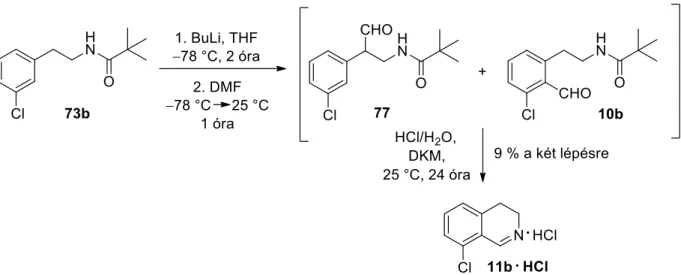
A reakció feldolgozása és tisztítása után a várt (10a) vegyületet 54%-os hozammal kaptuk, így a lítiumozás a szubsztituensek közös orto-helyzetében ment végbe. A reakcióelegy feldolgozása után a vizes fázisból 78%-os hozammal nyertük ki célvegyületünket, a 8-fluor-3,4-dihidroizokinolin-hidroklorid sót monohidrát formában (11a ∙ HCl ∙ H2O). Mivel további terveink megvalósításához 8-klór-3,4-dihidroizokinolint (11b) szerettünk volna nagyobb mennyiségben előállítani, ezért a vegyület szintézisének hatékonyabb útját kerestük.
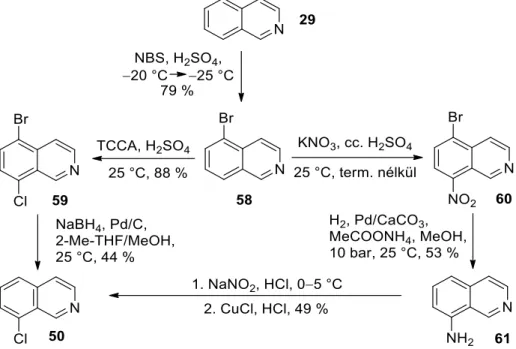
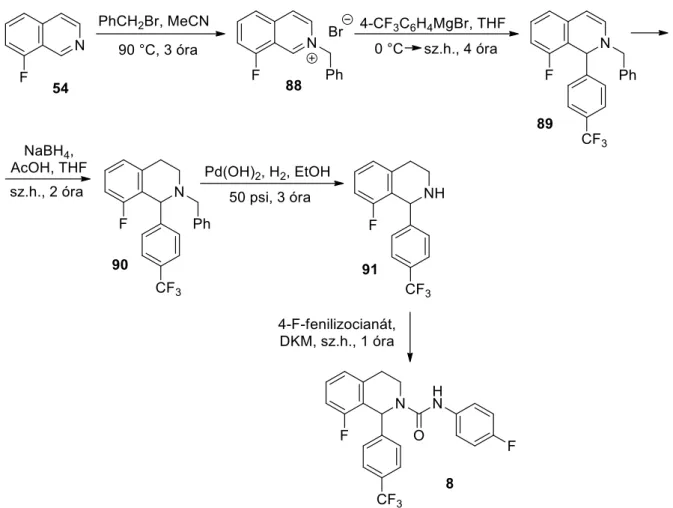
Henrich és munkatársai Bischler-Napieralski szintézissel állítottak elő 6- és 8-fluor-3,4-dihidroizokinolinok keverékét, amelyek az 1-es helyzetben metil-szubsztituenst tartalmaztak. Meg kell említeni, hogy a Grignard-vegyületekhez hasonlóan a szerves lítiumvegyületek is alkalmasak lehetnek C(1)=N kettős kötést tartalmazó izokinolin-származékok 1-es helyzetű szubsztituenseinek bevitelére.25 A szerves lítiumvegyületek még enyhébb körülmények között is sokkal gyorsabban reagálnak, mint a megfelelő Grignard-reagensek. Az eljárások során a (92) 8-brómo-kinolint különféle aminokkal reagálnak Ullmann26,27,28 vagy Buchwald-Hartwig állapotban. -
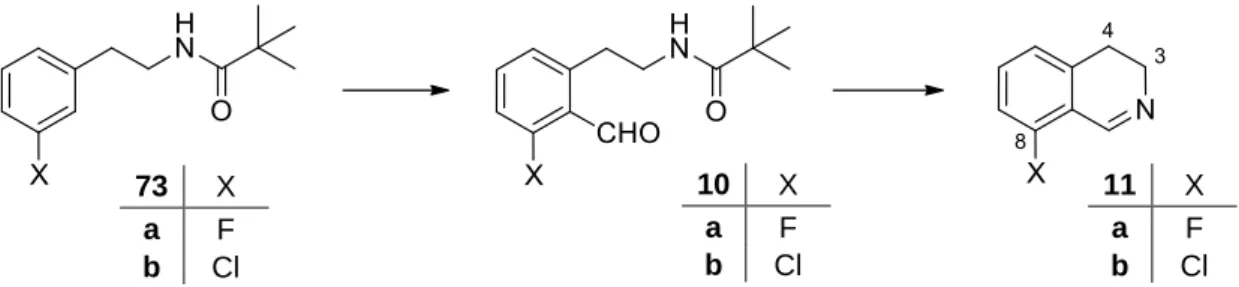
Saját munka: 1,8-diszubsztituált 1,2,3,4-tetrahidroizokinolinok előállítása
Előzetes várakozásaink szerint a fluoratom kellően reagál a gyűrűs aminok nukleofil támadásaira, így megfelelő prekurzora lehet 8-amino-szubsztituált 3,4-dihidroizokinolinok (100) előállításának, majd célvegyületeink (101) könnyen előállíthatók a kettős C szerv vegyület hozzáadásával. Célvegyületeink, az 1,8-szubsztituált tetrahidroizokinolinok (103) az így kapott 8-aril-3,4-dihidroizokinolinok (102) C=N kettős kötésének hozzáadásával állíthatók elő szerves lítiumvegyületek felhasználásával. A meta-helyzetben diizopropil-karbamoil-csoportot tartalmazó N-pivaloil-fenil-etil-amin lítiációs reakciójának vizsgálata és a megfelelő 8-diizopropil-karbamoil-3,4-dihidroizokinolin előállítása.
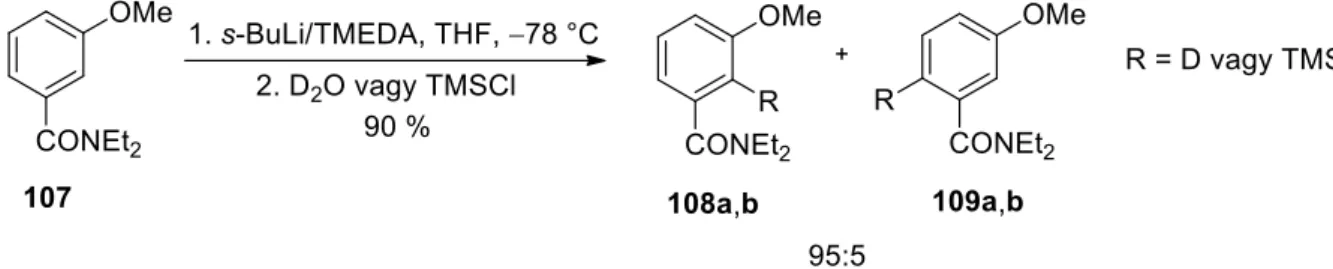
Célmolekulaként a 8-as pozícióban karboxilcsoportot tartalmazó vegyületet, vagy azzal azonos oxidációs állapotú rokon funkciós csoportot kívántunk kiválasztani, mivel ezek a csoportok segíthetik a szintézis kulcslépését jelentő közös orto pozícióba történő fémezést, valamint sokoldalú funkciós csoportok lehetnek a szintetizálandó izokinolin-származék esetleges további átalakulása szempontjából. A meta-helyzetben lévő karboxilcsoport orto-irányító tulajdonsága miatt az aromás gyűrűhöz kapcsolódó pivaloil-amino-etil-csoporthoz képest kezdetben ígéretesnek tűnt, esetünkben azonban kedvezőtlen választásnak bizonyult a dianion rossz oldhatósága miatt, amely valószínűleg a kiindulási anyag lítiációs lépésben történő kettős deprotonálódása miatt alakult ki.
Aromás gyűrűn karboxil- vagy karbamoilcsoporttal szubsztituált vegyületek lítiálási
A tercier amidok is erős irányító csoportok, ezért a dimetil- és dietil-karbamoilcsoportok választását is mérlegeltük, hátrányuk azonban, hogy irodalmi adatok alapján a ketonképződés elkerülése érdekében a fémezés során sec-BuLi-t kellett volna használnunk. Dimetil- és dietil-karbamoilcsoportokkal szubsztituált vegyületek esetén a BuLi általában nem használható fémezőszerként, mert mellékreakcióként aril-butil-keton képződik.47.
Saját munka: meta-helyzetben diizopropilkarbamoil csoportot tartalmazó N-pivaloil-
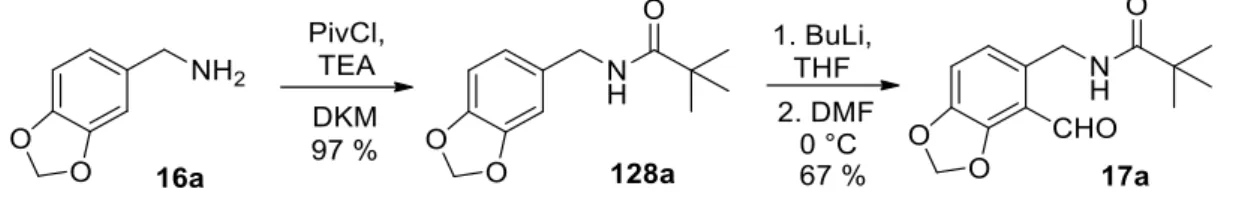
A 13. vegyület előállítása után következő feladatunk a Schlosser-Simig szintézis kulcslépésének, a hagyományos ortopozíciós litiációnak a megvalósítása volt. A 3-fluor- és 3-klór-N-pivaloil-fenil-etil-amin vegyületekkel (73a,b) alkalmazott körülmények között (BuLi, THF, –78 °C) sajnos nem sikerült regioszelektív lítiációt elérni a diizopropil-karbamoil-szubsztituált vegyület közös orto-helyzetére. A nyers keverék kezelését és gyorskromatográfiás tisztítását követően sikeresen kinyertük a reakcióban fő termékként képződő (124) vegyületet 15%-os hozammal, amely formilezett származéknak bizonyult a diizopropil-karbamoilcsoport kevésbé zsúfolt orto-helyzetében.
Általánosságban elmondható, hogy a hőmérséklet emelése és a reakcióidő változtatása nem befolyásolta jelentősen a regioszelektivitást, 14 vegyület mennyisége nem nőtt, viszont az egyéb melléktermékek képződése fokozódott. Még a BuLi felesleg 3 ekvivalensről 2 ekvivalensre csökkentésének sem volt jelentős hatása a szelektivitásra, az egyéb melléktermékek képződése kismértékben csökkent. A szokásos orto-helyzetben lévő formilezett vegyületet (14) ezért nem tudtuk tisztán kivonni a nyers keverékből, de savas kezelés és tisztítás után 3%-os hozammal izolálhattuk célvegyületünket, a 8-diizopropil-karbamoil-szubsztituált 3,4-dihidroizokinolint (15).
Az orto-(pivaloilaminometil)benzaldehidek átrendeződési reakcióinak vizsgálata
- Az orto-(pivaloilaminometil)benzaldehidek átrendeződési reakciójának irodalmi
- Saját munka: metiléndioxi-csoporttal szubsztituált orto-(pivaloilaminometil)-
- Saját munka: különböző szubsztitúciós mintázatú orto-(pivaloilaminometil)-
- Saját munka: orto-(pivaloilaminometil)benzaldehid savkatalizált átalakulása
- Saját munka: különböző szubsztitúciós mintázatú orto-(pivaloilaminometil)-
Saját munka: metiléndioxicsoporttal helyettesített orto-(pivaloilaminometil)-benzaldehid átrendeződési reakciójának vizsgálata benzaldehid átrendeződési reakciójának vizsgálata. HPLC-MS mérésekkel kimutattuk, hogy kisebb mennyiségben más dimerszerű aldehidek is jelen vannak a reakcióelegyben, de ezeket nem izoláltuk. A metilén-dioxi-szubsztituált származéknál tapasztalt átrendeződések és a dimerszerű aldehidek képződésének alaposabb megértése érdekében az aromás gyűrűn különböző szubsztituenseket tartalmazó orto-(pivaloilaminometil)benzaldehidek átrendeződési reakcióinak további vizsgálatát tűztük ki célul.
A trimetoxi-szubsztituált orto-(pivaloilaminometil)benzaldehiddel (17d) végzett reakciók során egyetlen oldószerben sem figyeltük meg az átrendezett aldehid (18d) képződését, a szóban forgó dimer aldehidek (129a,b és 137b) kb. MR-spektruma a hidrogénatomhoz kötődő konstans hidrogénatom oldószerben mérve, a szénatomok konstansa a CDCl3-ban. körülbelül 0,5%.
Az általunk eddig vizsgált, differenciálisan szubsztituált orto-(pivaloilaminometil)benzaldehidek (17a-d) savkatalizált reakcióiban elméletileg négy dimerszerű regioizomer képződhet, amelyek mindegyike két diasztereomer racemátja. A modell egyszerűsítése érdekében az aromás gyűrűn szubsztituenssel nem rendelkező orto-(pivaloilaminometil)benzaldehid savkatalizált reakcióját is vizsgálni kívántuk. Ebben az esetben az aromás gyűrűn lévő két funkciós csoport kicserélődése nem figyelhető meg, és elméletileg csak két diasztereomer racemát dimer-szerű termék képződik.
A 142a,b dimer-szerű aldehidek CDCl3 oldatban mért 1H-NMR spektrumában a kémiai eltolódás értékei meglehetősen hasonlóak, de a jelek gondos hozzárendelése után érdekes eltérések figyelhetők meg. Köztudott, hogy az aromás gyűrű anizotróp árnyékolókúpjában található magok jelentős diamágneses eltolódást mutatnak. A 129a,b dimerszerű aldehidek esetében a 132a,b kationok stabilabbak, mint a 131a,b kationok, és az aromás intermedier (126a,b → 132a,b) protonálódásával összefüggő szabadentalpia-változás exoterm.
Ennek megfelelően a 17a,b vegyületek savkatalizált reakciójában elsősorban a 132a,b kationok indítanak elektrofil támadást a 126a,b izoindolok ellen, ami a legnagyobb mennyiségű dimerszerű 129a,b aldehidet eredményezi.
Összefoglalás
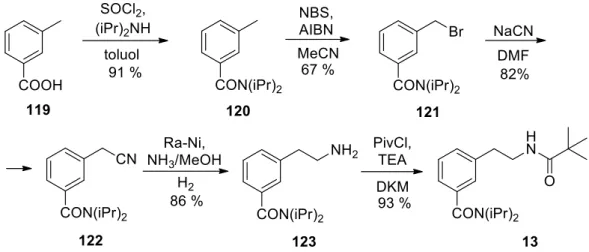
A 13-as vegyület litiálása és formilezése után sikerült izolálni a formilezett származékot (124) a diizopropil-karbamoilcsoport, mint fő termék kevésbé zsúfolt orto-helyzetében. A 17. számú vegyületeket a megfelelő (16) benzil-aminokból állítottuk elő pivaloil-kloriddal végzett acilezéssel, majd formilezéssel, majd irányított orto-litációval. Az aromás gyűrűn metilén-dioxit (17a) és a pivaloilaminometil oldallánc (17b) para-helyzetében metoxiszubsztituenst tartalmazó vegyület esetében az átrendeződött aldehidek (18a,b) mellett a 129a,b dimerszerű vegyületeket is kaptuk.
Az aromás gyűrűn szubsztituált orto-(pivaloilaminometil)benzaldehidek ( 17a-d ) esetében elméletileg négy dimerszerű regioizomer képződhet, amelyek mindegyike két diasztereomer racemátja. Szubsztituensek hiányában ennél a modellvegyületnél az átrendeződés nem vizsgálható, de a dimerszerű aldehidek képződése ebben az esetben leegyszerűsödik, mivel a reakcióban csak két diasztereomer racemát képződhet. A feltételezett átrendeződési mechanizmust és a dimerszerű vegyületek képződését DFT számításokkal vizsgáltuk, az így kapott eredmények megegyeztek a kísérleti tapasztalatokkal.
Az értekezés tézisei
Ez lehetővé tette az átrendeződés és a "dimerképződés" mechanizmusának pontosabb kimutatását, amit a DFT számítások is alátámasztottak.
Saját publikációk és előadások
Az értekezés alapjául szolgáló publikációk
Egyéb publikáció
Konferencia előadások
Kísérleti rész
Alkalmazott berendezések
A disszertáció alapját képező publikációkban ismertetett vegyületek
A disszertáció alapját képező publikációkban nem ismertetett vegyületek előállítása
A reakcióelegyet szobahőmérsékleten 24 órán át keverjük, majd 5%-os vizes nátrium-karbonát-oldatot (7 ml) és DKM-et (4 ml) adunk hozzá.
Irodalomjegyzék
Reduction of the C=N double bond of intermediate 6 with sodium borohydride leads to the corresponding tetrahydroisoquinoline 7. Reduction of the C=N double bond of intermediate 6 with sodium borohydride leads to the corresponding tetrahydroisoquinoline 7. Acid-catalyzed cyclization of the latter was accompanied by the loss of the pivaloyl moiety, resulting in 8-methoxy-3,4-dihydroisoquinoline hydrochloride (26 ∙ HCl) in 79% overall yield [21].
Based on this, we succeeded in extending the aforementioned method for the synthesis of 8-fluoro-3,4-dihydroisoquinoline (23) by a significant modification of the reaction conditions in the metalation step. Cyclization of aldehyde 29 in acidic medium occurred with concomitant loss of the pivaloyl unit to give 8-fluoro-3,4-dihydroisoquinoline (23), which was prepared as the hydrochloride hydrate (23·HCl·H2O). Cyclization of aldehyde 29 in acidic medium occurred with concomitant loss of the pivaloyl unit to give 8-fluoro-3,4-dihydroisoquinoline (23), which was prepared as the hydrochloride hydrate (23 ∙ HCl ∙ H2O).
The 8-chloroisoquinoline (11) thus obtained was reduced to tetrahydroisoquinoline19 (Scheme 4) [5], a potential precursor of the corresponding 3,4-dihydro6 derivative. Two synthetic variants have been disclosed for the preparation of the difficult-to-access 8-chloro-3,4-dihydroisoquinoline. The corresponding chloride salt was prepared by adding a solution of hydrochloric acid gas in isopropyl acetate to a solution of the base (27) in toluene, followed by filtration.
The corresponding hydrochloride salt was prepared by adding a solution of hydrochloric acid gas in isopropyl acetate to the solution of the base in toluene, followed by filtration. Soos, Regio-exhaustive functionalization of the carbocyclic core of isoquinoline: concise synthesis of oxoaporphine core and ellipticine, Synthesis e2190. As a continuation of our previous studies on the lithiation-based synthesis of 8-methoxy-, 8-fluoro- and 8-chloro-3,4-dihydroisoquinoline, a similar approach was explored for the preparation of the 8-diisopropylcarbamoyl congener.
The combined organic layer was washed with brine (4 mL) and dried over MgSO 4 . The solvents were evaporated, the residue was purified by flash chromatography (0-1% MeOH in DCM). To a vigorously stirred mixture of the residue in DCM (5 mL) and water (2 mL), aqueous sodium carbonate solution (10 w/w %, 1 mL) was added. The putative mechanism of the 1 → 5 rearrangement and the existence of equilibrium in single reaction steps are demonstrated by the acid-catalyzed reaction of aldehyde 5 with N -phenylmaleimide to form the Diels–Alder cycloadduct 4 , i.e., the same compound as the isolated one.