Synthèse de motifs diols-1,3 syn α -
Introduction
- L’addition conjuguée d’Evans-Prunet
- Préparation de 1,diols syn α -éthyléniques : précédents au
- Présentation du travail : nouvelle stratégie
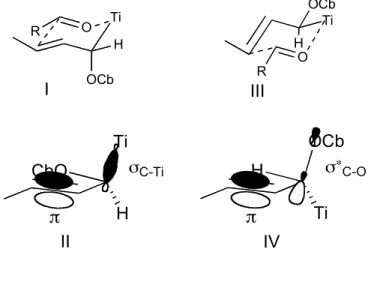
L'hémiacétal II intermédiaire est formé par addition d'alcoolate secondaire I au benzaldéhyde, puis l'hémiacétal II est ajouté en position 1,4 de la vinylsulfone. En effet, Lebel a montré qu'en présence de KHMDS et d'hydrocinnamaldéhyde dans le toluène à −78 °C, le produit de β-élimination I-59 β-.
Préparation des précurseurs pour l’addition conjuguée .34
- Addition radicalaire de thiols
- Autre méthode basée sur une réaction SN 2
- Par couplage croisé
- En utilisant un intermédiaire dianionique
- Une dernière méthode basée sur une réaction d’Hosomi-Sakurai
- Synthèse des précurseurs pyridinyl vinyl sulfones par allylation 48
Les rapports diastéréoisomères au niveau de la double liaison sont de 9 : 1 en faveur de l'isomère E pour les produits I-79 et I-82. La géométrie de la double liaison Z ne peut être obtenue que si le substituant carbamate est en position pseudo-axiale dans l'état de transition.
Addition conjuguée et aménagements fonctionnels
- L’addition conjuguée sur accepteurs de type pyridinyl vinyl sulfone
- Aménagements fonctionnels
Nous avons finalement obtenu un rendement de 66% et un ratio syn/anti-diastéréosélectif >95:5 (Graphique 56). Ensuite, nous avons utilisé de l'acide camphorsulfonique (CSA) dans un mélange méthanol/THF 4:1 pendant 12 h à 20 °C : une conversion de 73 % a été observée, 27 % de diol I-130 et 45 % d'alcool homoallylique ont été obtenus.
Oléfination de Julia modifiée
- Oléfination de Julia modifiée : Introduction
- Oléfination de Julia : Résultats obtenus
- En utilisant les BT sulfones
- En utilisant les PYR sulfones
En effet, le caractère électrophile de la sulfone BT permet la formation de l'oléfine en une seule étape. Comme mentionné précédemment, il est encore difficile de prédire la géométrie de la double liaison formée en réalisant une oléfination Julia modifiée. Il est possible de détecter la formation de benzaldéhyde par chromatographie sur couche mince, ce qui montre la réversibilité de la réaction.
Pensant qu'il s'agissait d'intermédiaires réactionnels décomposés sur de la silice, nous avons tenté d'identifier ces intermédiaires mais sans succès. Malheureusement, aucun produit d'oléfination I-151 n'a été observé après plusieurs tests ; Cependant, le produit de la réaction rétro-Michael a été isolé. Nous avons donc utilisé ces conditions pour notre substrat et seule une dégradation a été observée (schéma 76).
De plus, l'anion sulfone du I-156 bénéficie d'une stabilisation car il est en position allylique.
Conclusion et perspectives
On constate que, d'après ce qui est décrit dans la littérature, seuls des cas particuliers ont été rapportés pour l'oléfination de Julia modifiée sur cétones. Ces deux réactions ont été réalisées dans des conditions de type Barbier, mais il apparaît que la formation d'énolate se produit plus rapidement que la déprotonation de la sulfone et on observe la formation de benzaldéhyde et d'alcool homoallylique de départ. Jouant sur un équilibre thermodynamique de la réaction rétro-Michael, il semble difficile de réaliser l'addition nucléophile à une cétone.
Les accepteurs de type pyridinylvinylsulfone ont donné de meilleurs résultats en termes de rendement et de diastéréosélectivité que les benzothiazolylvinylsulfones ; A l’inverse, pour l’oléfination du Julia modifié, de meilleurs résultats ont été constatés pour les benzothiazolylsulfones. Un meilleur contrôle de la géométrie des doubles liaisons pourrait peut-être être envisagé en utilisant par exemple les sulfones PT ou les sulfones TPT41 qui ont été largement étudiées par le groupe de Kocienski. Ces auteurs ont obtenu un meilleur contrôle de la diastéréosélectivité avec ces sulfones par rapport aux sulfones PYR et BT.
Les meilleurs résultats ont été obtenus en utilisant du 1,2-diméthoxyéthane (DME) ; ce solvant devrait également être testé.41a.
Vers la synthèse du tricycle ABC de l’acide
L’acide hexacyclinique
Zeeck,50,51 L'acide hexacyclique a une structure originale comprenant un système tricyclique ABC 5:6:5 attaché à un tricycle, cette fois ponté DEF composé d'une δ-lactone et d'un hémiacétal cyclique (Figure 35). Zeeck a proposé une hypothétique biosynthèse basée sur une réaction intramoléculaire de Diels-Alder (IMDA) qui donne accès au cycle AB, suivie d'une réaction Vinyl-Prins qui permet l'établissement du cycle C. Il a ensuite été démontré qu'une molécule d'eau interviendrait pour former l'hémiacétal cyclique (Figure 36).52.
L'acide hexacyclique présente des similitudes structurelles frappantes avec le FR182877 ; Ce dernier composé a également été isolé en 1998 à partir d'une souche de Streptomyces (Streptomyces sp. No9885) par l'industrie pharmaceutique Fujisawa Pharmaceutical Company.53 Les seules différences structurelles notables résident dans l'inversion de la configuration à deux centres, entourée en rouge et jaune dans Figure 37, les différents degrés d'oxydation au niveau du carbone C25, l'alcool C14 protégé sous forme d'acétate, et enfin l'hydratation de la liaison C5-C20, entourée en vert dans l'acide hexacyclique. Bien que des recherches approfondies aient été réalisées, seul le diastéréomère conduisant au FR182877.54 a été obtenu. Zeeck et ses collègues ont montré que l'acide hexacyclique avait une activité cytotoxique modeste avec une CI50 de 14 µmol.L-1 sur trois lignées cellulaires (HM02, HEP G2 et MCF7).2.
Il a été démontré que l'activité cytotoxique du FR-182877 peut être liée à la présence de la double liaison pontée qui, en raison de l'environnement structurel qui l'entoure, semble être un excellent accepteur de Michael.
Quelques approches de synthèse de l’acide
- Le tricycle 4-desméthyl DEF par P. A. Clarke et ses
- Le tricycle ABC par P. A. Clarke et ses collaborateurs
- L’approche de Y. Landais concernant le tricycle ABC
- L’approche de Kalesse et ses collaborateurs pour le tricycle ABC
Concernant ce dernier, la lactonisation en conditions acides conduit au tricycle 4-desméthyl-DEF II-8 de l'acide hexacyclique avec un rendement quantitatif. Clarke et son groupe de recherche souhaitaient également développer une méthodologie permettant d'accéder au tricycle ABC de l'acide hexacyclique dans une série racémique.61. Un découplage net au niveau de l'espaceur nous amène aux précurseurs des réactions de Diels-Alder VII et VIII (Figure 39).62.
La formation du produit II-29 résulte de l'une des deux possibilités représentées sur la figure 41. En traitant le 1,2-éthanedithiol en présence de tétrachlorure de titane, le produit II-32 est isolé avec un rendement de 79% puis subit une réaction d'oxydation de l'alcool primaire pour donner l'aldéhyde II-33. L'idée serait d'obtenir le tricycle V par une addition intramoléculaire de Michael sur la roue IV équipée de tous les centres asymétriques des anneaux A et B de l'acide hexacycline.
L'oléfine finale II-71 est ensuite impliquée dans une métathèse croisée avec l'acrylate de méthyle, et l'alcool secondaire est protégé sous forme d'éther silylé TBDPS.
Précédents effectués au laboratoire
Enfin, une réaction d'addition conjuguée intermoléculaire entre le double accepteur Michael VI et l'énolate V permettrait d'accéder au précurseur IV. Il était prévu que l'addition de l'énolate devait se faire dans l'anti-isopropényle. L'étape suivante de cyclisation radicalaire utilisant de l'acétate de manganèse et de l'acétate de cuivre a été réalisée avec un rendement de 37 % du produit II-78 souhaité dans des conditions non optimisées.
Le tricycle ABC serait obtenu par une réaction de décarboxylation de l'ester, puis une réaction d'oléfination de Corey-Winter permettrait d'obtenir une cétone α,β-insaturée sur ce cycle C. Le composé II pourrait être obtenu par une réaction d'addition conjuguée entre le double L'accepteur Michael II-75 et l'énolate énantiopur, formés in situ par l'ajout de n-BuLi à l'éther énol silylé III (Figure 48). Cette cyclisation radicalaire initiée par l'acétate de manganèse(III) a été réalisée dans un mélange très spécifique 2,2,2-trifluoroéthanol/acide acétique.
La stéréochimie α de l'ester II-81 a ensuite été déterminée après réduction de l'ester en alcool primaire II-83 et il n'a pas été prouvé que ce substituant est dirigé vers le plan β (figure 49).72.
Présentation du travail
Synthèse des précurseurs de la réaction de Michael
- Synthèse du donneur de Michael : éther d’énol silylé
Il était donc préférable de n'ajouter qu'un équivalent d'aldéhyde pour perdre un minimum de matière. Avec le produit aldol de Crimmins en main, il a procédé directement à la protection de l'alcool secondaire sous forme d'éther tert-butyldiméthylsilyle suivi d'une réduction en hydrure de diisobutylaluminium et ainsi l'aldéhyde chiral II -94 a été obtenu avec un rendement de 81 % en deux étapes. . (schéma 111). Peut-être que la réaction s'est déroulée plus rapidement en raison de la β-élimination préliminaire au niveau de l'aldéhyde, qui a activé l'aldéhyde conjugué, puis la réaction de Baylis-Hillman s'est produite sur cet aldéhyde insaturé, ce qui pourrait expliquer le temps de réaction plus court observé.
En effet, après recristallisation de l'oxyde de triphénylphosphine dans l'éther de pétrole, une bonne partie de l'oléfine reste piégée avec lui. Également lors de la chromatographie sur silice, une partie de l'oléfine est perdue avec cet oxyde. En répétant ces conditions opératoires, nous nous sommes rendu compte qu'il était possible d'améliorer le rendement en éther d'énol silylé II-108 lors de l'addition du conjugué.
Même lorsque la silice est neutralisée par prétraitement à la triéthylamine, ou lorsqu'on utilise de la silice neutralisée au bicarbonate de sodium, un mélange de produit silylé et de cétone est récupéré alors que le brut réactionnel contient presque exclusivement l'éther d'énol silylé.
Réaction de Michael entre les deux partenaires
- Réaction radicalaire initiée par Mn(III) et Cu (II)
- Application à la formation du cycle B de l’acide hexacyclinique . 147
Parallèlement, une addition conjuguée à la cyclopenténone a également été réalisée avec ajout de cuprate de Lipshutz, piégeant également l'anion qui s'est formé sous forme d'éther silylé racémique II-109.82. Nous avons donc tenté de minimiser cette hypothétique complexation en substituant le DMF à des solvants plus complexes tels que le HMPA, le TMEDA, etc., augmentant ainsi le rapport en faveur du diastéréomère résultant de l'anti-addition du groupe OTBS. Les réactions radicalaires initiées par l'oxydation d'espèces énolisantes à l'aide de manganèse(III)83 sont d'un grand intérêt car aucun dérivé halogéné ou étain n'est nécessaire et, de plus, les précurseurs 1,3-dicarbonyle sont généralement facilement disponibles.
Sans acétate de cuivre(II) dans le milieu réactionnel, l'oxydation du radical primaire par Mn(III) étant lente, l'abstraction des protons de la molécule de solvant sera plus rapide (généralement acide acétique), mais inefficace, avec un rendement de 24 % . obtenu dans le produit VII. En revanche, si de l'acétate de cuivre(II) est présent dans le milieu, le radical primaire instable IV sera piégé dans l'intermédiaire en cuivre(III). L'élimination rapide des radicaux et l'oxydation finale du produit d'addition radicalaire par les espèces de cuivre (II) permettent un contrôle cinétique de la réaction et aboutissent généralement à des réactions plus propres.85.
Malheureusement, cette isomérisation s'est accompagnée de l'élimination de l'OTBS pour donner une cétone α,β-insaturée, ainsi que de l'hydrolyse de l'acétonide protégeant le 1,2-diol.72.
Conclusion et Perspectives
The aqueous layer was extracted three times with CH 2 Cl 2 , the combined organic layers were washed with saturated aqueous NaCl and dried over MgSO 4 . The aqueous layer was extracted three times with Et2O, the combined organic layers were washed with a saturated aqueous NaCl solution. The aqueous layer was extracted three times with diethyl ether, the combined organic layers were washed with saturated aqueous NaCl solution.
The aqueous layer was extracted three times with Et 2 O, then the combined organic layers were washed with saturated aqueous NaCl and dried over MgSO 4 . The aqueous phase was extracted three times with 15 mL of Et 2 O and the combined organic phases were washed with saturated aqueous NaCl and dried. The aqueous phase was extracted three times with 15 mL of Et 2 O and the combined organic phases were washed with saturated aqueous NaCl, dried over anhydrous MgSO 4 , filtered and concentrated.
The aqueous phase was extracted three times with 5 mL of Et 2 O and the combined organic phases were washed with saturated aqueous NaCl, dried over anhydrous MgSO 4 , filtered and concentrated in vacuo. The aqueous layer was extracted three times with Et 2 O and the combined organic layers were washed with saturated aqueous NaCl, dried over MgSO 4 and concentrated in vacuo. The combined organic layers were washed with 15 mL of saturated aqueous NaCl, dried over anhydrous MgSO 4 , filtered, and concentrated in vacuo.
