Je tiens bien sûr à remercier toute l'unité de génétique constitutionnelle de l'Institut Curie, notamment Marion Gauthier-Villars, Catherine Dehainault et Dorothée Michaux, avec qui nous pouvons parler de « rétinoblastome ». L'origine de la variabilité de l'expression phénotypique du rétinoblastome peut être due à (i) l'existence de mutations mosaïques, (ii) des mutations RB1 et (iii) des facteurs de modification génétique indépendants du locus RB1. Dans ces familles, certains embryons porteurs d'un allèle à risque identifié par une approche indirecte basée sur une étude microsatellite au locus RB1 ne portaient pas la mutation du parent atteint, lui-même atteint d'un rétinoblastome bilatéral.
La conséquence de l’existence de patients présentant une mosaïque dans le cadre d’un conseil génétique a été discutée. Les liens étroits entre MDM2, TP53 et RB1 dans la tumorigenèse du rétinoblastome ont fait de ce SNP fonctionnel un très bon candidat pour contrôler l'expressivité de la maladie. Des études de liaison familiale menées dans toutes les familles de rétinoblastomes identifiées au niveau national ont montré une association significative entre la présence de l'allèle G du SNP309 et la survenue d'un rétinoblastome (p = 0,001).
Le conseil génétique du rétinoblastome étant parfois compliqué par l’existence de familles à pénétrance variable, nous avons souhaité explorer la relation entre génotype et phénotype dans le rétinoblastome. Au cours de ce travail, nous avons abordé trois origines possibles de variabilité phénotypique : l'existence de mosaïques, la nature de la mutation affectant le gène RB1 et la présence de facteurs de modification génétiques indépendants de RB1.
Introduction
Généralité
- Epidémiologie
- Diagnostic et traitement
- a. Diagnostic
- b. Traitement
Le diagnostic différentiel doit être réalisé avec la maladie de Coats (développement anormal des vaisseaux de la rétine), certaines pathologies infectieuses (maladie des griffes du chat ; Bartonella henselae) ou parasitaires (larva migrans, Toxocara canis), affections déformantes de l'œil et bénignes. astrocytomes. En cas d'énucléation primaire, un examen histopathologique est réalisé, qui évalue l'envahissement tumoral intra- et extra-oculaire et définit les facteurs de risque histologiques (notons ici qu'il n'y a pas de facteurs de risque biologiques). Le recours à la radiothérapie en cas d'invasion est aujourd'hui évité en raison des risques de séquelles liées aux radiations (impact sur la croissance osseuse, cataracte et rétinopathie externe, risque de tumeur radio-induite).
De plus, chez les patients atteints de forme familiale de Rb, la radiothérapie externe est associée à un risque accru de cancer radio-induit (voir Chez les patients atteints de Rb bilatérale, un traitement conservateur de l'œil est privilégié et repose sur la chimiothérapie, la radiothérapie, la photocoagulation). , la cryothérapie ou la protonthérapie. Si la tumeur est unilatérale, de petite taille et épargne la macula, un traitement conservateur de l'œil est également envisagé.
Même si les stratégies thérapeutiques ont beaucoup évolué, le pronostic visuel reste inquiétant, d’autant que la lésion est proche de la macula. Il s'agit de programmes multidisciplinaires visant à améliorer la compréhension de la génétique du rétinoblastome (secteur auquel appartient cette thèse), à identifier de nouvelles cibles thérapeutiques en définissant les voies de signalisation nécessaires au développement tumoral, mais également à développer de nouveaux traitements comme de nouvelles méthodes. suivi de l’efficacité thérapeutique (IRM du sodium).

La génétique du rétinoblastome
- Le modèle de Knudson complété par Comings
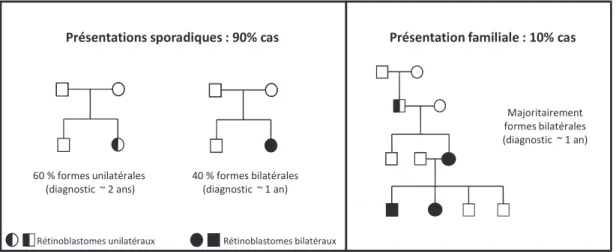
- a. Présentation familiale du rétinoblastome
- b. L’hypothèse de Knudson complétée par Comings
- c. Prédisposition génétique et risque de deuxième tumeur
- Le gène RB1
- a. Identification du gène RB1
- b. Description du gène RB1 et son produit
- Fonction de la protéine pRb
- a. RB1 et contrôle du cycle cellulaire
- b. RB1 et contrôle de l’apoptose
- c. RB1 et contrôle de la différenciation cellulaire
- Pathologie moléculaire
- a. Méthodes du diagnostic moléculaire du gène RB1
- b. Les mutations constitutionnelles du gène RB1
- c. Les mutations somatiques du gène RB1
- d. Les relations génotype/phénotype : La nature de la mutation comme support
- d.i Les mutations de l’exon 1 de RB1
- d.ii Les délétions complètes de RB1 : faible pénétrance et retard du
- d.iii Les mutations affectant l’épissage de RB1 et la régulation de la
- d.iv Les mutations faux-sens de RB1
- d.v Les mutations du promoteur de RB1
- e. Modification de l’empreinte: effet parental de la mutation et risque lié à la
- f. Les néomutations de RB1 : impact des mosaïques sur l’appréciation de la
- Le conseil Génétique dans le rétinoblastome
- a. En l’absence d’étude moléculaire de RB1
- b. Apport des études moléculaires constitutionnelles directes ou indirectes et des
- c. Défaut de pénétrance et mosaïque : impact pour le conseil génétique
La protéine pRb joue également un rôle dans le contrôle de l'apoptose et le contrôle de la différenciation cellulaire. Des modèles murins ont mis en évidence le rôle du pRb dans le développement embryonnaire de la rétine. Modification de l'empreinte : effet parental de mutation et risque associé à la fécondation in vitro fécondation in vitro.
Ainsi, l’apparition d’un phénotype altéré pourrait être associée à une compensation de la perte de RB1 par surexpression de l’allèle sauvage sous le contrôle de l’empreinte parentale. Selon les auteurs, ce pourcentage est probablement sous-estimé, car il est difficile de prouver directement la présence d'une mutation mosaïque. L’impact de la révélation d’une mutation est réel pour la fratrie car elle libère les proches non porteurs de la surveillance ophtalmologique.
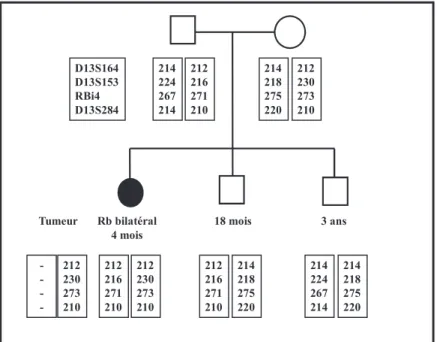
L’analyse des antécédents familiaux est essentielle pour orienter le suivi ophtalmologique des membres de la famille. Le DER est le rapport entre le nombre d’yeux atteints et le nombre de porteurs de mutations dans la famille.
Génétique tumorale du rétinoblastome
- Modèle général de la tumorigénèse du rétinoblastome
- Les événements nécessaires au développement du rétinoblastome
- Rétinoblastome et inhibition de la voie p53
One of the 6 patients with a molecular microdeletion and 5 of the 6 patients with a cytogenetic deletion showed psychomotor delay (Table 1). Supplementary Table 1: Brief description of the 25 Refseq genes found in the zone of interest (see text for definition). Refseq Gene OMIM OMIM Summary EntrezGene Summary Preferred Tissue Expression (GNF Expression Atlas 2 Data) of the RAC1 GTPase.
In the context of genetic predisposition to retinoblastoma, the MDM2 SNP309 G/G genotype could therefore exacerbate any effects of RB1 haploinsufficiency (13,14). Because the magnitude of the genetic effect, in terms of the odds ratio (OR), cannot be estimated using FBAT, a classical transmission disequilibrium test (TDT) was also performed using PLINK software (Purcell, 2007). 34; Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype." Proc Natl Acad Sci U S A.
This point is discussed in more detail in the "Notes" section of the chapter on. Molecular genetic studies of the RB1 gene can now be suggested to all patients with familial or sporadic unilateral or bilateral retinoblastoma. In familial forms comprising two accessible cases, indirect genetic testing rapidly demonstrates the mutant allele of the RB1 gene.
The severity of the risk can be assessed by the disease-to-eye ratio (DER), which is a good indicator of penetrance and expression level (Lohmann et al., 1994). These mutations occur throughout the coding sequence and in the promoter region with the notable exception of the last 2 exons (Figure 3). Direct testing involves looking for a germline change in the RB1 gene that indicates a predisposition to retinoblastoma.
Specific gene analysis techniques must be used to distinguish between 2 copies of the target (wild-type status), one copy (deletion), or 3 copies (duplication). Hypermethylation of the promoter region is a common mutational event found in tumor (Richter et al., 2003). Cytogenetic analysis includes a standard karyotype and analysis of the RB1 gene by FISH or CGH array.
Objectifs de la thèse et presentation des travaux de thèse
Population d’étude
Jusqu'à plusieurs cas consécutifs de patients atteints de rétinoblastome ont bénéficié d'une analyse moléculaire du gène RB1 dans le cadre de leur prise en charge thérapeutique. Les patients porteurs d'une délétion de l'intégralité du locus RB1 ont été étudiés par analyse CGH matricielle afin d'élucider la relation entre les retards de développement psychomoteur et la nature des délétions génomiques (taille et localisation). En revanche, l'âge au moment du dernier examen a été collecté pour chaque patient, permettant de déterminer clairement le phénotype (plus l'âge est élevé, plus le phénotype est fiable).
Les analyses d'association familiale utilisées dans le troisième article de cette thèse portent sur des patients dont le diagnostic moléculaire a été réalisé à l'Institut Curie jusqu'en avril 2008. Durant cette période, 650 patients ont bénéficié d'une analyse moléculaire du gène RB1 au cours de leur traitement thérapeutique. Dans ces 70 familles, 212 porteurs de la mutation RB1 ont été détectés (113 patients avec Rb bilatéral, 40 avec Rb unilatéral, 53 étaient porteurs sains et 6 présentaient un rétinome terminal).
De plus, l'ADN de 114 parents de ces familles, non porteurs de la mutation familiale, a été utilisé pour effectuer des analyses d'association familiale. Nous avons également eu 209 patients atteints de rétinoblastome bilatéral sans antécédents familiaux de mutation causale de RB1. L'âge au diagnostic entre les patients atteints de rétinoblastome familial par rapport à l'âge au diagnostic des patients bilatéraux diagnostiqués dans un contexte sporadique ne montre pas de différence significative (test de Student, p = 0,78).
Étonnamment, l'âge au moment du diagnostic des patients atteints d'un rétinoblastome familial bilatéral est plus précoce que celui des patients atteints d'un rétinoblastome familial unilatéral (test t de Student, p = 0,017). Cependant, il n’y a pas de différence significative si l’on limite la comparaison aux patients atteints de rétinoblastome unilatéral familial qui ne sont pas porteurs de première génération de la mutation familiale (test de Mann-Whitney, p = 0,15). En revanche, la différence reste significative si la comparaison est limitée aux patients atteints de rétinoblastome unilatéral familial de première génération.
De ce fait, l’âge au diagnostic des patients de la première génération de porteurs est plus élevé que celui des patients ayant reçu la mutation de leurs parents (test de Mann-Whitney, p=0,03). Ces observations confortent l'hypothèse selon laquelle l'expressivité variable de la pathologie et les défauts de pénétrance observés chez les patients porteurs de première génération sont au moins partiellement liés à la présence d'un mosaïcisme somatique. Par conséquent, et pour éliminer les biais pouvant être introduits par la présence d'un mosaïcisme cellulaire, les patients unilatéraux et les porteurs sains de première génération ont été exclus des analyses de liens familiaux.
Articles
- Première publication
- a. Introduction au premier travail
- b. Résumé
- c. Tiré à part du premier travail
- d. Discussion
- Deuxième publication
- a. Introduction au deuxième travail
- b. Résumé
- c. Tiré à part du deuxième travail
- d. Supplementary Information (données publiées)
- e. Discussion
- Troisième publication
- a. Introduction du troisième travail
- a.i Polymorphismes communs et risques associés
- a.ii Polymorphismes fonctionnels et modulation des risques majeurs de
- a.iii Polymorphismes fonctionnels et rétinoblastome
- a.iv Hypothèses de l’étude
- b. Résumé
- c. Tiré à part du troisième travail
- d. Données complémentaires
- d.i Supplementary Information (données publiées)
- d.ii Données complémentaires (données non publiées)
- e. Discussion du troisième travail
Deletion thresholds in patients with normal development showed that DLEU2, CDACC1, EPST1 and. Effect of MDM2 SNP309 and p53 Arg72Pro polymorphisms on age of tumor onset in Li-Fraumeni syndrome. Approximately 650 consecutive retinoblastoma patients underwent germline molecular analysis of the RB1 gene at the Institut Curie from September 2000 to April 2008.
If one of the two parents carries the mutation, prenatal diagnosis can be suggested for a subsequent pregnancy. Somatic mosaics may also be observed, as a change of the RB1 gene may occur in the patient during embryonic development and may not be present in leukocyte DNA. In non-familial forms, reassortment of the affected child's alleles reveals the two alleles thought to be related to a change in the RB1 gene.
Splicing abnormalities represent 20% of the mutational spectrum of RB1 and are therefore important to characterize. It is therefore essential to ensure the integrity of the system every day by using control samples.

Discussion générale et perspectives