Transcriptomes
Sven-Eric Schelhorn1*, Matthias Fischer2, Laura Tolosi1, Janine Altmu¨ller3, Peter Nu¨rnberg3, Herbert Pfister4, Thomas Lengauer1, Frank Berthold2
1Max-Planck-Institute for Informatics, Campus E1.4, Saarbru¨cken, Germany,2Department of Pediatric Oncology and Hematology, Children’s Hospital, and Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany,3Cologne Center for Genomics, University of Cologne, Cologne, Germany,4Institute of Virology, University of Cologne, Cologne, Germany
Abstract
In excess of12% of human cancer incidents have a viral cofactor. Epidemiological studies of idiopathic human cancers indicate that additional tumor viruses remain to be discovered. Recent advances in sequencing technology have enabled systematic screenings of human tumor transcriptomes for viral transcripts. However, technical problems such as low abundances of viral transcripts in large volumes of sequencing data, viral sequence divergence, and homology between viral and human factors significantly confound identification of tumor viruses. We have developed a novel computational approach for detecting viral transcripts in human cancers that takes the aforementioned confounding factors into account and is applicable to a wide variety of viruses and tumors. We apply the approach to conducting the first systematic search for viruses in neuroblastoma, the most common cancer in infancy. The diverse clinical progression of this disease as well as related epidemiological and virological findings are highly suggestive of a pathogenic cofactor. However, a viral etiology of neuroblastoma is currently contested. We mapped14 transcriptomes of neuroblastoma as well as positive and negative controls to the human and all known viral genomes in order to detect both known and unknown viruses. Analysis of controls, comparisons with related methods, and statistical estimates demonstrate the high sensitivity of our approach. Detailed investigation of putative viral transcripts within neuroblastoma samples did not provide evidence for the existence of any known human viruses. Likewise,de-novoassembly and analysis of chimeric transcripts did not result in expression signatures associated with novel human pathogens. While confounding factors such as sample dilution or viral clearance in progressed tumors may mask viral cofactors in the data, in principle, this is rendered less likely by the high sensitivity of our approach and the number of biological replicates analyzed. Therefore, our results suggest that frequent viral cofactors of metastatic neuroblastoma are unlikely.
Citation:Schelhorn S-E, Fischer M, Tolosi L, Altmu¨ller J, Nu¨rnberg P, et al. (2013) Sensitive Detection of Viral Transcripts in Human Tumor Transcriptomes. PLoS Comput Biol 9(10): e1003228. doi:10.1371/journal.pcbi.1003228
Editor:Claus O. Wilke, University of Texas at Austin, United States of America ReceivedDecember 18, 2012;AcceptedJune 4, 2013;PublishedOctober 3, 2013
Copyright:ß2013 Schelhorn et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding:The work of SES was partially supported by DFG grant Klinische Forschergruppe KFO 129. The work of SES and TL was partially supported by grant No. 01GS08103 (Oncogene) of the German Federal Science Ministry. The Children’s Cancer Research Fund Neuroblastoma (Foerdergesellschaft Kinderkrebs-Neuroblastom-Forschung) supported the deep sequencing analysis (awarded to FB and MF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests:The authors have declared that no competing interests exist. * E-mail: [email protected]
Introduction
To date, pathogenic agents are known to be causally related to 20% of human cancer cases [1] and significantly affect the global health burden of this disease [2]. The majority of these agents comprise oncogenic viruses such as human papilloma virus (HPV), Epstein-Barr virus (EBV), hepatitis B virus (HBV), and hepatitis C virus (HCV) [3]. Characterizing the oncogenic potential of viral pathogens has important consequences for prevention, diagnosis, and treatment of malignant neoplasms [4,5]. Tumor viruses in particular have received renewed attention in the context of recent global efforts to characterize the etiology of cancer [6,7]. Consequently, viral cofactors for several idiopathic cancers are currently investigated [8] and epidemiological indicators suggest that additional human tumor viruses remain to be discovered [9]. Neuroblastoma is a heterogeneous embryonal tumor [10,11] that is accountable for 15% of deaths caused by malignant conditions in children [12]. The disease is associated with an
exceptionally low median age of presentation of17months [13] and is often diagnosedin utero. Metastatic neuroblastoma has two biologically divergent subtypes. Stage 4S is characterized by an age of presentation between in utero and 18 months, metastases confined to liver, skin, lymph nodes and bone marrow, and its ability to regress spontaneously [14,15]. In contrast, stage 4 tumors are presented at any age, demonstrate high infiltration rates in bone marrow and bone, and are most often progressive [10,16]. While genes related to neuronal differentiation have been described to be upregulated in stage 4S in comparison to stage 4 neuroblastoma, thereby indicating distinct levels of neuronal differentiation [17], little is currently known about the differences between molecular etiologies of stage 4 and stage 4S neuroblas-toma.
susceptibility to viral infections and is reminiscent of acute lymphoblastic leukemia – another pediatric tumor with uncertain etiology for which an infective cofactor has long been suspected [18]. Furthermore, epidemiological studies have associated reduced neuroblastoma risk with immunologic indicators such as previous childhood infections, day care attendance, and breast feeding [19,20] that are suggestive of an infective cofactor [21]. While transforming polyomaviruses such as JCV and BKV were previously identified within neuroblastoma samples and other pediatric embryonal tumors [22–24], newer studies seem to render these associations inconclusive [25]. Therefore, the role of pathogenic cofactors of neuroblastoma oncogenesis remains unresolved.
In general, the search for suspected viral cofactors of idiopathic diseases requires systematic screening of human tissues for viral biomarkers such as virus-derived nucleotide sequences. Unfortu-nately, viruses are of polyphyletic origin and thus lack common universal marker genes as they are frequently exploited in metagenomics studies targeting cellular microorganisms. Conse-quently, it is not currently possible to specifically PCR-amplify viral nucleotide sequences within a given tissue without prior information about the infective agent being sought [26]. As a result, several systematic assays for pathogen detection have been developed that do not rely on targeted PCR-amplification of viral factors [27] and were employed to identify Kaposi’s sarcoma-associated herpes virus (KSHV) as a human tumor virus [28]. These systematic approaches were recently supplemented by sensitive deep sequencing technologies [27]. These technologies were recently applied to exclude several cancer-virus associations based on negative evidence [29,30] and aided in the identification of MCPyV, a human polyomavirus, as a cofactor of Merkel cell carcinoma [31].
Deep sequencing technologies have enabled detection of both known and novel viruses with unprecedented sensitivity [32]. However, the large numbers of sequence fragments (‘‘reads’’)
generated by these methods necessitate data reduction approaches for filtering and condensing the list of putative viral transcripts. Two such approaches are currently represented in the literature:
digital transcript subtractionthat discards human sequence homologs from the sequence data and considers the remaining transcripts as potential viral signatures [30,31,33–39], and de-novo sequence assembly that aims to reconstruct whole viral genomes from overlapping reads [40–43]. Recently, variants of these of two approaches have been implemented in several computational pipelines such as PathSeq [44], RINS [45], and CaPSID [46].
Identification of tumor viruses in particular poses several important challenges to existing computational pipelines. Con-founding factors such as loss of viral genetic material from progressed tumors as well as limited replication competence or latent replication strategies often result in low or selective transcription of tumor viruses [5]. In addition, viral oncogenes homologous to human factors and chimeric transcripts originating from proviral insertion sites may share significant sequence similarity with human transcripts [47], thus making unequivocal identification of viral factors difficult. Last, high rates of viral sequence divergence from 10{5
{10{8 (dsDNA viruses) up to 10{4(ssRNA viruses) substitutions per site and year [48,49] hinder recognition of known viruses based on known reference sequences. We have developed Virana, a novel computational approach specifically tailored to detecting low-abundance transcripts that diverge from known viral reference sequences or share significant sequence homology with human factors. In particular, our method maps sequence reads to a combined reference database compris-ing the human genome and all known viral reference sequences. The approach is configured to allow for high mismatch rates and mappings to multiple reference sequences (‘multimaps’). By using this combined and sensitive mapping strategy, our approach is especially well suited for detecting human-viral chimeric tran-scripts and viruses diverging from known references. In contrast to existing subtractive approaches for viral transcript discovery, our method abstains from discarding reads homologous to the human genome from further analysis. Instead, Virana exploits multimaps to assign sequence reads to a homologous context comprising human reference transcripts and viral reference genomes. These homologous regions retain the full, unfiltered information contained in the raw sequence data while also being amenable to further analyses by multiple sequence alignments, human-viral phylogenies, and orthogonal taxonomic annotations, thus greatly aiding in the interpretation of the results.
We applied our novel approach on an overall number of 14 deep sequencing transcriptomes of stage 4 and stage 4S metastatic neuroblastoma in order to identify putative viral cofactors associated with this idiopathic disease.
Materials and Methods
Clinical samples and experimental deep sequencing data Primary neuroblastoma samples from stage 4 (progressive) patients (n~7) and stage 4S (regressive) patients (n~7) were obtained prior to treatment from the central neuroblastoma tumor bank at the University Hospital of Cologne, Germany. None of the tumors harbored amplification of the MYCN proto-oncogene as determined by two independent laboratories for each case by fluorescence in situ hybridisation (FISH) and Southern blot [50]. Only neuroblastoma samples with a tumor cell content of above 60% as assessed by a pathologist were selected for deep sequencing. Integrity of RNA was evaluated using the Bioanalyzer 2100 (Agilent Technologies) and only samples with an RNA integrity number of at least 7:5 were considered for further Author Summary
processing. Quality of all neuroblastoma samples and related deep sequencing data was additionally confirmed by an orthogonal computational analysis focusing on human gene expression in the context of differential splicing [51].
All patients were enrolled in the German Neuroblastoma trials with informed consent. In order to validate our approach we additionally employed a positive control panel consisting of tumors with known viral cofactors. An EBV-positive B-cell-lymphoma (BCL) was received from the Pediatric Oncology and Hematology Department of the Hannover Medical School. Deep-sequencing reads obtained from full transcriptome libraries of two HPV18-positive HeLa samples (HeLa) and a HPV16-HPV18-positive primary cervical squamous cell carcinoma (ceSCC) were downloaded from the Short Read Archive (SRA) and preprocessed as specified in the original publication [30]. Transcriptome data of a HBV-positive hepatocellular carcinoma (HCC) HKCI-5a cell line with con-firmed HBV integration events was downloaded from the SRA based on information in the original publication [52]. A negative control panel consisting of a normal brain transcriptome generated as part of the Illumina BodyMap 2.0 project was obtained from the SRA at run accession number ERR030882.
Library preparation and sequencing
mRNA libraries of the EBV-positive B-cell lymphoma and14 neuroblastomas were prepared following the Illumina RNA Sample Preparation Kit and Guide (Part #1004898 Rev. A). For each sample,5mg high-quality total RNA was processed for mRNA purification, chemical fragmentation, first strand synthesis, second strand synthesis, end repair, 39-end adenylation, adapter
ligation, and PCR amplification. Validated libraries underwent gel size selection and final paired-end sequencing with an effective read length of 2|36bp on the Illumina Genome Analyzer IIx following Illumina standard protocols. Additionally, libraries for two of the 14 neuroblastoma samples were generated using the same protocols and sequenced with an effective paired-end read length of2|95bp on a Illumina HiSeq 2000. All libraries had insert size distributions approximating m~150bp, s~50bp as later confirmed by read mapping. The data were filtered according to signal purity by the Illumina Realtime Analysis (RTA) software.
Simulated sequencing data
In this study we employ simulated sequencing data from three viral genomes that are homologous to human factors. Reads originating from the ABL1-homologue of the Abelson murine leukemia virus (A-MuLV, GI:9626953, positions 1326{2605), from the thegagregion of HERVK22I (obtained from Repbase [53], positions1{1452), and from Bo17, a GCNT3-homolog of the bovine herpesvirus 4 (BoHV-4, GI:13095578, positions 107098{108748) were generated in silico by dwgsim, a read simulator based on wgsim [54]. In addition, we produced simulated chimeric transcripts by fusing each of the aforemen-tioned sequence regions to the human TP53 gene, a known proto-oncogene (UCSC build hg19, GRCh37, chr17, positions 7572926–7579569). These artificial fusion transcripts were gener-ated using Fusim [55] based on TP53 exon models obtained from the UCSC refGene database [56]. Fusion transcripts were then used as templates for generating simulated data sets with dwgsim. Table 1.Sequencing panel characteristics.
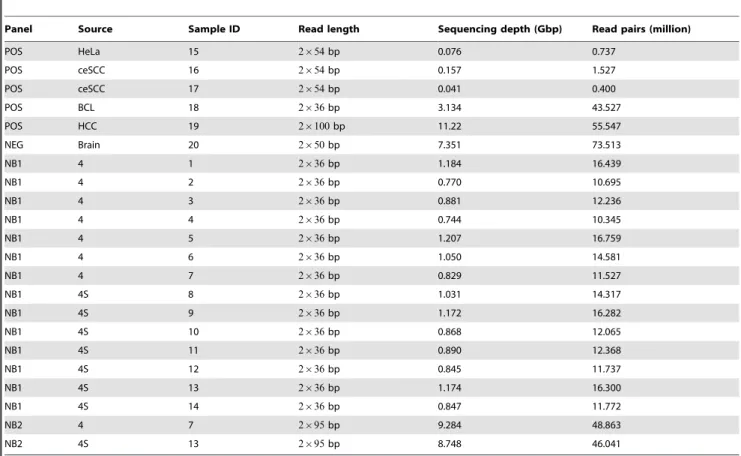
Panel Source Sample ID Read length Sequencing depth (Gbp) Read pairs (million)
POS HeLa 15 2|54bp 0.076 0.737
POS ceSCC 16 2|54bp 0.157 1.527
POS ceSCC 17 2|54bp 0.041 0.400
POS BCL 18 2|36bp 3.134 43.527
POS HCC 19 2|100bp 11.22 55.547
NEG Brain 20 2|50bp 7.351 73.513
NB1 4 1 2|36bp 1.184 16.439
NB1 4 2 2|36bp 0.770 10.695
NB1 4 3 2|36bp 0.881 12.236
NB1 4 4 2|36bp 0.744 10.345
NB1 4 5 2|36bp 1.207 16.759
NB1 4 6 2|36bp 1.050 14.581
NB1 4 7 2|36bp 0.829 11.527
NB1 4S 8 2|36bp 1.031 14.317
NB1 4S 9 2|36bp 1.172 16.282
NB1 4S 10 2|36bp 0.868 12.065
NB1 4S 11 2|36bp 0.890 12.368
NB1 4S 12 2|36bp 0.845 11.737
NB1 4S 13 2|36bp 1.174 16.300
NB1 4S 14 2|36bp 0.847 11.772
NB2 4 7 2|95bp 9.284 48.863
NB2 4S 13 2|95bp 8.748 46.041
In all cases, dwgsim was applied using the default empirical error model. Paired-end read lengths and insert size distributions were chosen according to the neuroblastoma sequencing data (see above). Additional simulated sequencing data generated by a related publication were analyzed as described in Section ‘‘Estimation of read mapping sensitivity’’.
Sample data notation
Sample panels containing neuroblastoma transcriptomes se-quenced at 2|36bp and 2|95bp effective read lengths are denoted as NB1 and NB2, respectively. While the NB1 panel contains seven transcriptomes of neuroblastoma stages 4 and 4S each, the NB2 panel contains one sample of stages 4 and 4S each (see Table 1). Positive control panels of human cancer transcrip-tomes with known viral cofactors (BCL, HeLa, ceSCC, and HCC) are denoted as POS. The negative control panel consisting of a normal human brain transcriptome is denoted as NEG.
Reference genomes
The current assembly of the human reference genome (UCSC build hg19, GRCh37) as well as corresponding refGene splice-site annotations were obtained from UCSC. Splice variant annotations and cDNA sequences for the human genome were downloaded from Ensembl [57]. A set of all 4,680 available complete viral reference genomes and their taxonomic lineages were obtained from NCBI via the E-utilities web service [58] and the database query: ‘‘Viruses[Organism] AND srcdb_refseq[PROP] NOT cellular organisms [ORGN]’’. In addition, we obtained consensus reference sequences for all human endogenous retroviruses (HERV-K/HML-2) represented in Repbase (Primate HERV,
HERVK11DI, HERVK11I, HERVK13I, HERVK22I,
HERVK3I, HERVK9I, HERVKC4)) [53]. All reference genomes were combined into a single human-viral reference database for Virana. Since RINS and CaPSID cannot use such a combined database, human and viral reference sequences were collected within two separate databases for these approaches.
Quality control, mapping, and assembly
Paired-end reads from the neuroblastoma panels and positive control panels were quality-controlled with an in-house sequence analysis framework in order to identify sample contamination, adapter contamination, and batch effects. After quality control, the sequence data consisted of13:494Gbp (NB1),18:032Gbp (NB2), 14:63Gbp (POS), and 7:351Gbp (NEG) of sequence reads, respectively (see Table 1).
All data were mapped against a combined human-viral reference database with the splicing-aware and gapped read mapper STAR [59] in paired-end mode. While Virana considers the read mapper to be a replaceable component, in principle, we decided to employ STAR due to its mapping speed, high sensitivity settings, and its consideration of putative chimeric transcripts. We configured the mapper for high sensitivity by following recommendations of the author of STAR (personal communication). In particular, we set the rate of acceptable mismatches to 0:3 times the length of each read and the
seedSearchStartLmax and winAnchorMultimapNmax parameters to 12 and 50, respectively. The minimum length of chimeric segments (chimSegmentMin) was reduced to 15 in order to detect fusion transcripts at short read lengths. Known splice sites from splice annotations of the human reference genome as well as canonical splice sites were considered in the mapping. For each read, multiple mapping locations with alignment score distances of up to 10 ranks relative to the best score were permitted (‘multimaps’). Read alignments were stored in standardized BAM files. STAR
supports detection of chimeric transcripts by reporting discordant read pairs whose ends map to different chromosomes. These discordant read pairs were employed in further analyses as detailed in the next section.
In order to identify putative new viral transcripts, read pairs with at least one unmapped read end were extracted from BAM files by the Samtools suite [54] and assembled into longer contigs by the de-novo transcriptome assemblers Trinity [60] and Oases [61] using default parameters. Oases was configured for using different k-mer values in order to facilitate reconstruction of low-abundance viral transcripts. Contigs of length less than 300bp were considered to be spurious assemblies and excluded from further processing.
Detection of chimeric transcripts
Virana supports detection of human-viral chimeric transcripts in two different manners. First, the read mapper employed in our study is able to partially align reads that contain a human-viral chimeric breakpoint to multiple reference sequences. Consequent-ly, these partially aligned reads can be detected by Virana within the generic analysis of homologous regions (see below). The second, more sensitive approach to detecting chimeric transcripts is based on paired-end read information. Since the STAR mapper assigns reads to a combined reference database comprising both human and viral reference sequences, ends of paired-end reads whose inserts span the breakpoint of a chimeric transcript will be aligned to different reference sequences. These discordant read pairs are reported by STAR during read mapping (see above) and can further be filtered by mismatch score or sequence complexity in order to yield a high-confidence list of chimeric transcripts.
Generation of homologous regions
A distinguishing feature of Virana is its ability to automatically reconstruct the homologous context of reads that map to both viral and human reference sequences. This homologous context is constructed in four steps:
(1) First, reads that map to at least one viral reference are extracted from the mapping together with their primary (highest alignment score) and secondary (up to ten ranks of alignment scores below the highest score) mapping positions (see Figure 1). Since viruses of the same taxonomic family often exhibit significant sequence similarity, reads that map to one family member often also map to related family members as well as to homologous loci in the human reference. Based on these primary and secondary mapping locations, Virana obtains overlapping human reference transcripts, viral genomic references, and viral taxonomic information pertain-ing to the location. For each sequence read, information obtained in this manner is collected in a data structure denoted as HIT. HITs originating from the same analysis panel are pooled for further analysis.
(2) Second, pooled HITs originating from the same analysis panel are assigned to viral taxonomic families based on the viral genomic references they refer to (see Box 1 Algorithm 1). Sets of HITs assigned to the same viral taxonomic family are denoted as the homologous group (HOG) of that family. The same HIT may, in principle, be assigned to several HOGs. (3) Third, since reads and references generally share local rather
approach to split HOGs into manageable and alignable clusters denoted as homologous regions (HORs, see Box 2 Algorithm 2):
(3a) The set of all reads within a HOG is re-aligned to the set of all references (human reference transcripts and viral reference genomes) within the HOG using a highly sensitive BLASTN [62] alignment (word size 7). Since all possible mapping locations are required for further processing, BLAST is configured for high permissiveness (E-value10). (3b) Each HIT is assigned to a singleton cluster. Clusters
containing reads that map to the same reference are merged if their reference mapping locations (as determined by BLASTN) are less or equal thanL~25basepairs apart (L -gaps). Optimal values forLare determined empirically, see Section ‘‘Estimation of required sequencing coverage for detection of a homologous region’’ for a robustness analysis. Merging continues until the number of clusters converges. Subsequently, all clusters with fewer than an empirically chosen cutoff oft~5reads are discarded in order to remove spurious hits. After filtering, each remaining cluster repre-sents a candidate HOR. Since cluster membership is defined by reads mapping to common references, each pair of references within the candidate HOR shares one or more regions of high local sequence similarity (e.g., the loci the read mapped to) connected byL-gaps.
(3c) For each HOR, parts of reference sequences that are neither covered by a read mapping location nor by an L-gap between read mapping locations are trimmed.
(4) Last, due to the high mutual similarity of sequences within trimmed HORs, sequences within each HOR are now amenable to sequence alignment against the longest reference sequence within that HOR using LASTZ, the successor of BLASTZ [63]. The resulting star-shaped multiple sequence alignment is then used for construction of per-sample (for reads) and per-gene (for human reference transcripts) consensus sequences. Aligned consensus sequences retain information on non-consensus nucleotides due to the usage of
IUPAC ambiguous nucleotide codes. Consensus sequences can then be manually inspected in order to determine single nucleotide permutations and indels up to length L that distinguish sequence reads, viral references, and human reference transcripts.
Consensus sequences can be further processed by phylogenetic analyses. For generating phylogenies, Virana employs the software PhyML [64] following the maximum likelihood approach and using default parameters recommended by the HIV sequence database (http://hiv.lanl.gov, GTR model of nucleotide substitu-tion, transition/transversion ratio: 4, gamma shape parameter: 1, number of substation rate categories: 4, approximate Likelihood Ratio Test (aLRT) using SH-like supports where applicable). We note that the topology of the phylogenetic trees constructed in this manner is stable with regard to the model choice; while more complex model parameters may yield better likelihoods in some instances, these differences do not influence interpretation of our results.
Taxonomic annotation
In this study, we additionally compare consensus sequences of aligned HOGs as well as de-novo assembled sequence contigs to nucleotide (NCBI NT) and protein (NCBI NR) reference archives Algorithm 1.Construction of homologous groups.
Data: Reads
Result: Homologous groups Initialise: homologous_groups;
forread in mappingdo
obtain all mapped references of read;
ifmapped against viral referencethen
Initialize: read_hit; add read to read_hit;
forviral reference the read maps todo
add alignment positions of viral reference to read_hit;
end
forhuman cDNA reference the read maps todo
add alignment positions of cDNA to read_hit;
end
forviral reference of a viral taxonomic family thus addeddo
add read_hit to the homologous group for that family;
end end end
Algorithm 2.Construction of homologous regions.
Data:Homologous groups
Result: Homologous regions Initialise: homologous_regions;
forhomologous_group in homologous_groupsdo
Initialize: homologous_regions;
forread_hit in homologous_groupdo
forhomologous_region in homologous_regionsdo ifread_hit and homologous_region share a reference
then
if alignment positions of shared reference are within l basepairsthen
merge read_hit into homologous_region; extend all reference aligment positions within homologous_region;
end end end end
ifread_hit not mergedthen
Initialize: homologous_region;
merge read_hit into homologous_region; add homologous_region to homologous_regions;
end end
whilemergeable_pair_of_homologous_regions existsdo forpair_of_homologous_regions in homologous_regions
do
if pair_of_homologous_regions shares a reference
then
ifalignment positions of shared reference are within l basepairsthen
merge pair_of_homologous_regions;
extend all reference aligment positions within new homologous_region;
in order to assign transcripts to a taxonomic origin. To this end, we employ several BLAST [58] search strategies (BLASTN, BLASTX, and TBLASTX) with sensitive word sizes (4,3, and3, respectively). TBLASTX bypasses synonymous mutations during similarity search and is particularly suited for detecting function-ally conserved homologs. This approach is therefore recom-mended for discovering remote similarities [65] and is widely used in environmental metagenomics [66]. A permissive E-value threshold of 0:1 is used for all comparisons in order to reduce the possibility of missing true viral hits. For each query transcript and search strategy, the three highest-scoring reference sequences
are extracted from the BLAST results. Subsequently, descriptions, taxonomic information, and available gene annotations for high-scoring reference hits are pooled and query transcripts are assigned a putative viral, human, or ambiguous origin based on the pooled information. In order to limit the search space of the computationally intensive TBLASTX procedure, we constrain the allowed taxonomic origin of reference sequences to only viral (NCBI taxon ID 10239) or human (NCBI taxon ID9606) hits while excluding artificial sequences (NCBI taxon ID81077) using the NCBI database query ‘‘(((txid10239 [ORGN]) OR (txid9606 [ORGN]) OR (human [ORGN])) NOT (txid81077 [ORGN]))’’.
Estimation of read mapping sensitivity
We quantify the ability of our novel method Virana and the related methods RINS [45] and CaPSID [46] at detecting diverged viral transcripts among human sequence data by employing a recently published validation data set [46]. This data set consists of a negative control background set of reads simulated from the human reference genome that is spiked with four sets of 10,000 reads simulated from 10 viral reference genomes. Nucleotide positions within reads of each of the four viral spike-in data sets are mutated randomly independently and uniformly with a set-specific probabilityh[f0, 0:05, 0:1, 0:25g before being merged with the background data set. The set of viral reference sequences represents10different viral families that infect plants (Cherry green ring mottle virus, Cestrum yellow leaf curling virus, Elm mottle virus, East African cassava mosaic virus), birds (Gallid herpesvirus 1), insects (Cotesia congregata bracovirus), bacteria (Guinea pig Chlamydia phage), amphibians (Frog adenovirus 1), and mammals (Rat coronavirus Parker, Banna virus).
All five data sets (non-spiked human negative control and four human-viral spike-in sets) are analyzed by Virana, RINS, and CaPSID using identical reference sequences as described in Section ‘‘Reference genomes’’. Sensitivity (fraction of correctly identified viral reads among all viral reads) and specificity (1{ fraction of falsely identified human reads among all human reads) of viral read detection are determined for each method and data set. Analyses are performed with either default parameters (Virana), parameters published in the original validation data set (CaPSID), or settings adapted by us in order to maximize sensitivity (RINS: minimal contig length decreased to 100, read lengths and insert size distributions according to input data).
Since all methods map to the same complete viral reference set, reads from a particular viral genome of the validation data set may be distributed across several closely related reference genomes, all of which may be considered valid mappings. For this reason, we added post-processing steps to CaPSID and RINS and performed this validation on the level of viral taxonomic families rather than on the level of single viral species. We note, however, that results of all tested methods including Virana retain information on single viral species throughout the analysis. In particular, sensitivity and specificity of the methods change only minimally if data is analyzed on the single species level.
Analysis of human-viral homologous and chimeric transcripts
Analysis of the human-viral homologous regions and chimeric transcripts based on simulated read data (see Section ‘‘Simulated sequencing data’’) was conducted by configuring CaPSID, RINS, and Virana analogous to the previous section. For the validation of fusion transcript detection, the number of true positives is set to the number of all reads originating from the human-viral fusion
Figure 1. Virana’s approach to identifying viral transcripts in
human tumors.a) Transcriptome sequence samples are first mapped
to a combined set of human and viral reference sequences in a splicing-aware fashion. b) Unmapped or discordantly mapped read pairs are further processed by assembly methods to detect novel viruses or transcript chimeras that may indicate proviral integration events. c) Reads mapping to one or more viral genomes (HITs) are analyzed in an integrated fashion by considering human homologous mapping locations and viral taxonomies. This process results in a number of homologous regions (HOR) for each viral family. HORs are represented as multiple sequence alignments incorporating a wealth of sequence information. Alignments are further enriched by taxonomic annotations and phylogenetic analyses.
transcript. Since all detection methods in this validation are configured to only report reads mapping to the viral part of the fusion transcript, sensitivity estimates are scaled down equally for all methods in this particular validation. Analysis of discordant read ends in order to detect the origins of chimeric transcripts was performed as described before (see Section ‘‘Detection of chimeric transcripts’’).
Estimation of required sequencing depth
Expanding on related work [34,35], we quantify the theoretical sensitivity of Virana by estimating the number of viral transcripts per cell that are required for achieving a certain minimal sequencing coverage at a probability of at least 95%. Based on human genome annotations obtained from UCSC, we determined an average length of human coding sequences (CDS) ofl~1,634bp. By conservatively assuming that an idealized cell contains200,000mRNAs [34] of average lengthlfragmented at f~500bp as a result of library preparation, an expected number ofm~653,600cDNA fragments are generated per cell. For a given viral transcript of length r and a viral transcript abundance x per cell, we expect a number of v~x r=f viral transcript fragments. Assuming a theoretical, unbiased sequenc-ing process, the probability of sequencsequenc-ing a viral transcript fragment among the overall m transcript fragments is
pviral~v=m. Given a single-end read length of j, a number
k~rc=(2j) reads are required to achieve a sequence coveragec
of that viral transcript. The probabilitypk
viralof observing at least kreads during sequencing with a sequencing depthnis specified by the cumulative binomial distribution function with
parame-ters k, nand pviral. Due to numerical instabilities of computing
the cumulative binomial distribution for large values n, we exploit the Central Limit Theorem and estimate pviral by the Camp-Paulson normal approximation to the binomial distribu-tion. This approach has a negligible approximation error of v0:007=pffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffin pviralq
, where q~1{pviral [67]. Our approach further depends on successfully reconstructed homologous regions, each requiring an empirically determined minimum number of t~5 transcripts separated by no more than L~25 base pairs.
Although the probabilitypregion of a homologous region being successfully constructed from viral transcripts at a given sequence coverage can be derived analytically for a special case [68], this solution neither considers edge effects occurring for small transcripts nor takes into account the distribution of insert sizes of paired-end reads. We therefore approach the problem empirically by in silico simulation of paired-end reads that are assigned randomly independently and uniformly to transcripts of different lengths and at varying coverages. This simulation process addresses the aforementioned confounding factors by considering transcript boundaries and sampling insert sizes from a normal distribution parametrized according to neuroblastoma sequence data employed in this study (see Section ‘‘Library preparation and sequencing’’). An mean estimator forpregionand its standard error
SEpregionwere derived by averaging the success rates of homologous
region constructions across1,000simulations for each transcript length, read length, region linkage, and read coverage.
Availability
All sequence data generated in this study are publicly available in the European Nucleotide Archive (ENA) at study accession number PRJEB4441. Software implementations of our method and all validation procedures are available at http://mpi-inf.mpg.de/,sven/virana.
Results
This study presents a novel approach to detecting viral transcripts in human tumor transcriptomes. In contrast to related approaches such as RINS and CaPSID that rely on subtracting reads homologous to human transcripts from the analysis, our novel method Virana assigns sequence reads to a combined human-viral reference database without discarding homology information (see Figure 1). By employing a particularly fast and sensitive read mapper, Virana gains sensitivity at discovering highly divergent and chimeric viral transcripts. In addition, this configuration allows for exploitation of multimaps (e.g., sequence reads mapping to several reference genomes with varying mismatch rates) to discover the homologous context of sequence reads with regard to viral and human reference sequences. Last, Virana employs chimeric alignments as well asde-novoassembly of unmapped sequence reads followed by taxonomic annotation in order to discover proviral integration events and novel viruses, respectively.
Detection of divergent viruses
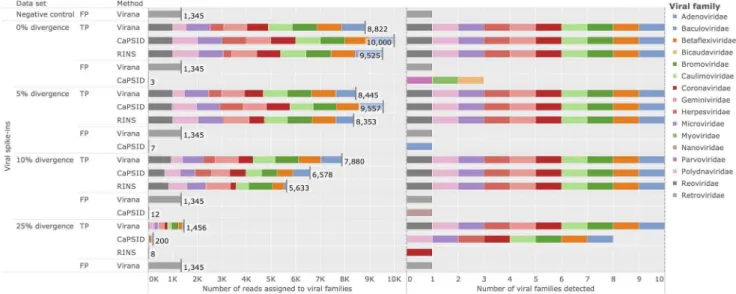
In order to compare Virana and the two subtractive approaches CaPSID and RINS in a controlled environment we rely on a previously published simulated data set consisting of a negative control data set free of viral reads, here denoted as background set. The background set is used to construct four additional validation data sets spiked with viral reads at increasing rates of sequence divergence (0%, 5%, 10%, 25%, see Materials and Methods). Performance is quantified in terms of sensitivity and specificity (see Materials and Methods). Applying all three viral detection methods on the validation data sets reveals comparatively high rates of correctly detected viral reads for CaPSID and RINS at low sequence divergences between 0% and 5%. Specifically, the two subtractive methods achieve0:99{1:13 fold higher sensitivities compared to Virana (sensitivities of 0:835{1:0 versus 0:844{0:882for subtractive approaches and Virana, respectively, see Figure 2). In contrast, Virana substantially surpasses subtrac-tive approaches at higher rates of viral sequence divergence (10– 25%), offering comparatively stable sensitivities between 7-fold and 182-fold higher than Capsid and RINS, respectively (sensitivities of 0:0008{0:6578 versus 0:1456{0:7880 for sub-tractive approaches and Virana, respectively, see Figure 2, left panel). Notably, while subtractive approaches fail to identify 20– 90% of viruses in settings of high sequence divergence, Virana is the only approach able to reliably detect the full set of viruses in all validation scenarios (see Figure 2, right panel). As a result of Virana’s ability to detect human-viral transcript homologs, reads originating from several human endogenous retroviruses (HERVs) that are part of the human reference genome but technically also belong to the viral familyRetroviridaeare detected in validation data at all levels of sequence divergence. Since the detected HERV reads originate from the human rather than from the viral part of the validation data, these reads classified as false positive (FP) hits for the purpose of this validation. As a result of this artifact, Virana exhibits a slightly lowered specificity compared to subtractive approaches (0.99985 versus 1.0 for Virana and CaPSID/RINS, respectively). However, we note that HERV reads are correctly classified by Virana during homologous region construction and by optional BLAST-based taxonomic annotation. These reads can therefore be safely and automatically ignored in subsequent analyses if HERV expression is of no interest to the researcher.
Figure 2. Detection of divergent viruses.Performance comparison of Virana, CaPSID, and RINS at detecting viral reads at different rates of simulated sequence divergence among a background set comprising human genomic reads. The background set without any spike-ins of viral reads serves as negative control. Left panel: stacked bars represent absolute numbers of detected reads grouped by sequence divergence, correctness of classification (TP: true positive, FP: false positive), and detection method. Falsely classified reads not assigned to any of the viral families present in the validation are labeled as false positives (FP). Colored segments indicate to which viral families the reads were assigned. Each condition allowed for the correct detection of up to10,000reads. Right panel: color coded markers for each condition and detection method indicating which viral families were identified. A maximum number of10viral families could be correctly identified in each condition.
doi:10.1371/journal.pcbi.1003228.g002
Figure 3. Time required for data analysis.Cumulative time in minutes required for analysis of the divergence validation set. Times are reported
for the negative control without viral spike-ins as well as for four mixed data sets consisting of negative control background set with viral spike-ins at different divergence rates. Segments within bar plots represent different analysis processes employed by the three viral detection methods Virana, CaPSID, and RINS. All measurements are based on a single CPU Intel(R) Xeon(R) E5-4640 clocked at 2.40 GHz.
analyzed. In contrast, RINS and CaPSID require two to17times longer per sample, respectively (see Figure 3). Interestingly, the majority of time spend by CaPSID is lost on subtraction, indicating that this step is a limiting factor of subtractive approaches. We note than reported times are based on analyses using a single compute core. Since all evaluated methods benefit from multithreading, dedicating additional compute cores to the analysis allows for further reduction in processing time.
Detection of low-coverage, homologous, and chimeric viral transcripts
Having established Virana’s ability to detect reads sampled at comparatively high coverage from viral genomes with low or no human-viral sequence similarity, we next test the sensitivity of the viral detection methods in a more challenging scenario involving gene regions of animal viruses that have close human homologs and are sampled at low sequencing coverages. Three such human-viral homologs are used in the analysis: V-ABL of the acutely transforming retrovirus A-MuLV, Bo17 of herpesvirus BoHV-4 (a model virus for oncogenic gammaherpesviruses such as EBV and KSHV and implied in several animal cancers [69]) and gag of HERV-K(HML2)22I, a class of human endogenous retroviruses associated with some forms of breast cancer [70]). Validation is based on simulated sequencing data and split into two scenarios
(see Materials and Methods for details). Within the first scenario, simulated sequencing reads are sampled directly from human-viral homologs while in the second scenario reads are generated from artificial fusion transcripts that each involve one of the three homologs fused to the human TP53 proto-oncogene. The resulting human-viral fusion transcripts mimic transcriptional signals indicating retroviral integration or homologous recombination of viral DNA next to a human gene which may result in activation of the latter by insertional mutagenesis.
We apply the viral detection methods Virana, CaPSID, and RINS on these two validation data sets in order to evaluate sensitivity at detecting viral genes that are similar to human factors either due to natural sequence homology or due to gene fusions. Performance is quantified by detection sensitivity, specificity, as well as by the absolute number of reads correctly detected. While all methods performed at a perfect specificity of1:0, only Virana detects viral transcripts at all coverages and with two to three-fold higher sensitivities compared to competing methods (Figure 4). In particular, sequence reads originating from endogenous retrovi-ruses were almost always subtracted from the analysis by RINS and CaPSID. In addition, RINS seemed to be confounded by low sequencing coverage, a fact most probably resulting from its heavy reliance on de-novo transcript assembly. Subsequent analysis of discordantly mapped read pairs by Virana (see Materials and
Figure 4. Detection of low-coverage, homologous, and chimeric viral transcripts.Displayed are performances of Virana, CaPSID, and RINS
at detecting the three human-viral homologous gene regions Bo17, gag, and vABL. Performance is quantified in terms of sensitivity (right panel) and absolute number of reads correctly identified (left panel) at differing sequencing coverages (2{60fold). Methods are validated at detecting both isolated gene regions (upper part) as well as at detecting human-viral fusion transcripts involving each of the three gene regions fused to the human TP53 proto-oncogene (lower part). Specificity of detection is 1.0 (100%) for all detection methods (not displayed).
Methods) correctly identified the TP53 gene as fusion partner of both V-ABL and Bo17, indicating that detection of human-viral chimeras is reliable even at low twofold coverage. Due to the repeat nature of the HERV-K sequence in the human genome and the resulting re-occurrence of HERV-K homologs at multiple loci in the human reference it was not possible to unambiguously identify the fusion partner of the HERV-Kgaggene.
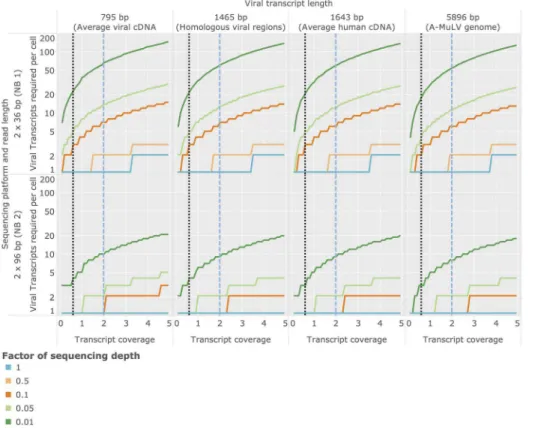
Estimation of optimal sequencing depth
Due to a variety of factors (see Discussion) human tumor viruses often replicate at very low levels within the infected cell. Determining the required sequencing depth for detecting viral transcripts present at specific cellular abundances is therefore crucial for planning transcriptome experiments designed to identify tumor viruses. Based on statistical arguments and average mRNA sizes (see Materials and Methods), we inferred the minimal abundances of viral transcripts required in an average cell required for detection depending (1) on the length of the transcript being sought and (2) on the sequencing depth employed in the experiment. Here we report results for an average viral cDNA-transcript (795bp), an average viral transcript region analyzed in the validation of human-viral homologs (Bo17 and vABL, 1,465bp, see previous section), an average length human CDS (1,634bp), and the genome size of a small tumor virus (A-MuLV,5,896bp). Based on these estimates and given an average sequencing depth as employed in the NB1 analysis panel, Virana requires a minimum twofold sequence coverage of an average viral cDNA transcript in order to detect the transcript within a homologous region with 99:9% probability
(Figure 5, upper left quadrant, dashed blue vertical line). This sequence coverage is produced with95% probability if at least one viral transcript is present per cell, on average (Figure 6, upper left quadrant, dashed blue vertical line). The number of viral transcripts per cell required for detection is inversely related to transcript length and sequencing depth, in principle: at a transcript length corresponding to a small viral genome (5,896bp) and a per-sample sequencing depth of1% of the sequencing depth generated in the NB1 panel, a transcript coverage of 0:6 and at least 55 viral transcripts per cell are required for reliable detection (Figure 6, upper right panel, dotted black vertical line).
Analysis of positive and negative experimental controls In order to evaluate Virana on experimental data we conducted an analysis of several positive and negative control samples with a cumulative size of21:982Gbp. The negative control sequencing data originates from a normal brain transcriptome that is suitable as a control for neuroblastoma data. Positive controls span a range of cancer transcriptomes that are associated with several viral cofactors such as a hepatocellular carcinoma (HCC) cell line with proviral integration of Hepatitis B virus, a cervical squamous cell carcinoma (ceSCC) and two HeLa cell line samples with associated human papillomavirus (HPV), and a Ebstein-Barr virus (EBV) positive B-cell lymphoma (BCL).
As displayed in Figure 7 (upper part), analysis of the brain negative control sample demonstrates that viral transcription is ubiquitous even in normal (non-cancerous) samples. Specifically, several bacteriophages of the taxonomic families Microvirodae,
Figure 5. Estimation of required sequencing coverage for detection of a homologous region. Probability of successful region
Myoviridae,Podoviridae, andSiphovoridaeindicate sample contamina-tion with bacteria as well as technical spike-ins (http://res.illumina. com/documents/products/technotes/technote_phixcontrolv3.pdf). Remarkably, the Coliphage phi-X174 genome of the family
Microvirodaecould be fully assembled by Virana’s homologous region construction, yielding a single fragment of 99% sequence identity and 100% coverage compared to the phi-x174 reference genome. In addition, several retroviral and flaviviral hits at low abundances of
1{28reads per million reads mapped (RPMM) highlight human factors such as HERV-Ks (endogenous retroviruses) as well as human proto-oncogenes SRC/ABL and DNAJC14/RP11 that have close homologs in the viral familiesRetroviridaeandFlaviviridae, respectively. The taxonomic ambiguity of these regions is automatically identified during Virana’s homologous region construction and confirmed by optional BLAST-based annotation compared to NCBI nt and nr databases (as indicated by thinner bars in Figure 7).
Figure 6. Estimation of required cellular transcript abundances for achieving a given transcript coverage.Sequencing coverage of viral
transcripts is depending on the average number of transcript copies per cell in the sequenced sample, on the length of the viral transcript being sought, and on characteristics of the sequencing process. In order to better visualize the optimal sequencing depth required for detection of viral factors, we estimated the required number of transcript copies per cell for different sequencing depths. These sequencing depths are expressed as factors relative to the depths employed for the NB1/NB2 panel generated in this study (which are here reported as a relative sequencing depth of 1). doi:10.1371/journal.pcbi.1003228.g006
Figure 7. Overview of identified homologous regions in positive and negative experimental controls.Left panel: cumulative numbers
of reads assigned to viral taxonomic families (log-scale). Each bar represents a homologous group (HOG) colored according to viral taxonomic family. Bars comprise several segments, each representing a homologous region (HOR). Heights of segments indicate the putative origin of reads assigned to this region (human, viral, or ambiguous). Viral families of bacteriophages are marked accordingly. Right panel: Analogous to left panel, but the lengths of bars represent relative rather than absolute abundances quantified in cumulative reads per million reads mapped (RPMM).
Analysis of positive control samples resulted in41homologous regions (HORs)spanning five viral families (see Figure 7, lower part). Viral cofactors associated with each of the cancer samples are correctly recovered at a high dynamic range of read abundances between 3 RPMM (HCC with integrated HBV provirus) and 1,628 RPMM (HeLa cell line associated with HPV18). In addition, several viral fragments were successfully reconstructed within HORs of the positive control samples, such as a 9,550bp long EBV segment containing latency-associated
factors EBNA 3b, 3c, and 4a (80% sequence identity with the wild type genome) as well as a1,693bp long HBV fragment containing the oncogenic HBV-X gene (98% sequence identity compared with Hepatitis B virus isolate HK1476). Similar to results on the negative control brain sample, several HORs with lower abundances assigned to the taxonomic families Retroviridae and
Flaviviridae represent human-viral sequence homologies that are automatically flagged to be of ambiguous taxonomic status by Virana.
Figure 8. Overview of identified homologous regions in neuroblastoma samples.Left panel: cumulative numbers of reads assigned to viral
taxonomic families (log-scale). Each bar represents a homologous group (HOG) colored according to viral taxonomic family. Bars comprise several segments, each representing a homologous region (HOR). Heights of segments indicate the putative origin of reads assigned to this region (human, viral, or ambiguous). Viral families of bacteriophages are marked accordingly. Right panel: Analogous to left panel, but the lengths of bars represent relative rather than absolute abundances quantified in cumulative reads per million mapped (RPMM).
doi:10.1371/journal.pcbi.1003228.g008
Table 2.Mapping rates.
Panel Source Sample ID Pairs mapped Both ends mapped Uniquely mapped Depth (Gbp)
POS HeLa 15 94.900% 94.900% 68.422% 0.127
POS ceSCC 16 90.803% 90.803% 69.561% 0.264
POS ceSCC 17 96.629% 96.629% 73.921% 0.075
POS BCL 18 91.612% 91.612% 63.528% 6.424
POS HCC 19 94.693% 94.693% 73.500% 14.924
NEG Brain 20 95.481% 95.481% 72.515% 11.234
NB1 4 1 95.878% 95.878% 69.422% 2.275
NB1 4 2 96.062% 96.062% 74.342% 1.43
NB1 4 3 96.385% 96.385% 75.938% 1.641
NB1 4 4 95.749% 95.749% 71.012% 1.503
NB1 4 5 95.057% 95.057% 69.203% 2.652
NB1 4 6 94.819% 94.819% 69.856% 2.39
NB1 4 7 96.597% 96.597% 72.107% 1.635
NB1 4S 8 95.952% 95.952% 70.681% 2.093
NB1 4S 9 95.242% 95.242% 74.009% 2.223
NB1 4S 10 96.854% 96.854% 74.756% 1.651
NB1 4S 11 96.819% 96.819% 75.256% 1.668
NB1 4S 12 96.710% 96.710% 74.899% 1.539
NB1 4S 13 95.344% 95.344% 72.326% 2.35
NB1 4S 14 97.110% 97.110% 74.829% 1.65
NB2 4 7 86.225% 86.225% 69.552% 12.243
NB2 4S 13 86.280% 86.280% 72.538% 11.517
Mapping ratios and depths of neuroblastoma (NB), positive control (POS), and negative control (NEG) panels. Mapped reads are relative to the number of sequenced read pairs that have passed quality control. Depths include reads with multiple mapping locations (‘multimaps’).
Interestingly, the HCC sample was also investigated in recent work focusing on detecting viral integration events [52]. In this recent study, the authors confirmed one integration event by Sanger sequencing while alluding to two additional events still awaiting experimental validation. By analyzing discordantly mapped read ends, Virana could correctly identify all three HBV fusion events involving human genes TRRAP (11 read pairs), ZNF48 (11 read pairs), and PLB1 (6 read pairs) as part of the primary mapping procedure.
Analysis of neuroblastoma samples
Deep-sequencing of14neuroblastoma samples on two sequenc-ing platforms yielded26:700Gbp (NB1) and23:760Gbp (NB2) of mapped read pairs (including multimaps), respectively (see Table 2). While samples were sequenced independently and marked with unique identifiers to allow for sample tracking at each step of the analysis, reads from each sample panel and each tumor stage (4 or 4S) were pooled for analysis. Processing the pooled sample panels with Virana resulted in 46 homologous regions representing four viral families (see Figure 8). All HORs were associated with low relative read abundances of 1{67 RPMM compared to confirmed viral signatures of experimental positive
controls (3{1,628 RPMM, see Figure 7). Several homologous regions assigned to bacteriophage viral families Baculoviridae and
Myoviridaeare attributable to sample contamination.
Reads assigned to viral familiesRetroviridaeandFlaviviridaewere determined to originate from either endogenous elements (HERVs) or from human proto-oncogenes that have close homologs in pestiviruses and acutely transforming retroviruses. HORs associated with these viral families were automatically assigned human or ambiguous taxonomic origin by Virana, as indicated by narrower bars in Figure 8. We undertook manual investigation of homologous relationships within each ambiguous HOR by analyzing multiple sequence alignments and phyloge-netic trees of the respective regions. These analyses revealed unambiguous clusterings of neuroblastoma sequence reads near human or endogenous factors in all cases (see Figure 9 for an example phylogeny).
No significant differences in viral expression signatures between neuroblastoma 4 and 4S stages could be detected except for HERV-K endogenous retroviruses which display36{86%higher abundances in stage 4S (NB1: 56 RPMM, NB2: 28 RPMM) than in stage 4 (NB1: 41 RPMM, NB2: 15 RPMM) neuroblastomas. All reads assigned to homologous regions were further analyzed for
Figure 9. Human-viral phylogeny based on a HOR.Phylogenetic tree of HOR#16 of the NB1 stage 4 panel. Viral reference sequences are
indicated with red branches and associated tip labels (‘Virus’) while human factors are labeled with green branches. Blue branches represent consensus sequences of neuroblastoma reads (‘Sample’). The tree was generated by the maximum likelihood approach PhyML using the multiple sequence alignment of the HOR as input (see Materials and Methods). Distances between nodes are quantified as substitutions per site. As can be derived from the tree, neuroblastoma consensus sequences are tightly clustered in close proximity to the endogenous retrovirus HERVK9I and two human factors, thereby unambiguously indicating the human origin of these neuroblastoma reads. Clusters of other sequences represent well known sequence homologies, as for example between human ABL1/SRC genes and acutely transforming retroviruses.
evidence of chimeric transcription (see Materials and Methods). While several read pairs with putative chimeric mappings could be identified, all viral chimeric read ends were clustered within low-complexity regions of the viral genomes. Analyses revealed that these putative chimeric mappings represent sequencing errors and low-complexity templates that non-specifically attracted reads of similarly low sequence complexity. No cluster of chimeric reads located at a specifically viral genome location and representing a human-viral breakpoint could be identified.
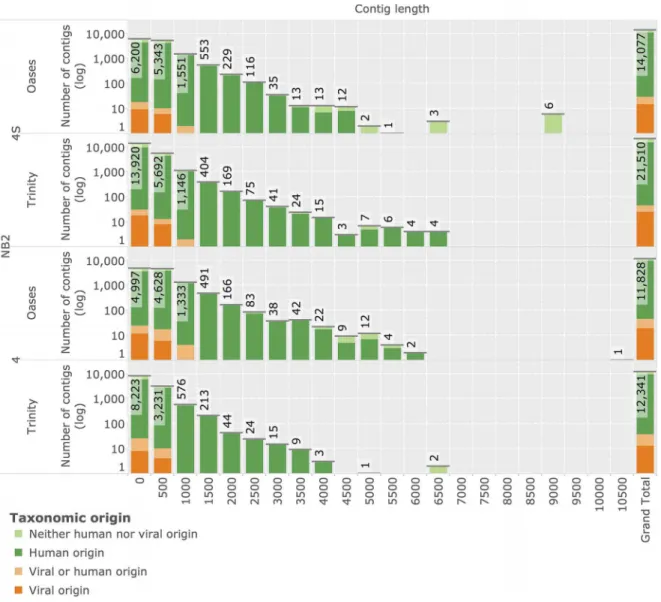
Reconstruction of novel transcripts byde-novoassembly In order to identify transcripts of novel viruses that do not map to known references, we generated de-novo transcriptome assemblies of all unmapped reads. We applied the twode Bruijn
graph based assembly methods Oases[61] and Trinity[60] that demonstrated best-in-class performance in recent evaluations [71] on sequencing data of the NB2 panel. This sequencing data
is especially amenable to assembly due to its long read length (see Table 1). Assembly resulted in 14,077 and 21,510 reconstructed neuroblastoma 4S contigs for Oases, and Trinity, respectively (see Figure 10). Assembly of the neuroblastoma 4 sample yielded 11,828 and 12,341 contigs from the same methods. Results of Oases and Trinity assemblies are compa-rable in terms of contig length. All contigs were subjected to taxonomic annotation using high-sensitivity TBLASTX annota-tion based on human and viral content of the NCBI nt and nr databases (see Materials and Methods). Overall, 72 contigs (0:1{0:16%of contigs of any specific assembly) were identified to be of putative viral origin. 26 contigs were assigned to bacteriophage references and excluded from further analysis. Based on searches against the full NCBI nr and nt databases followed by manual inspection, all remaining 46 contigs were determined to display higher similarities to bacterial or human sequences than to any viral reference.
Figure 10. Reconstruction of novel transcripts by de-novo assembly. Histograms display lengths of reconstructed sequence contigs
assembled from unmapped reads of NB2 stage 4 and stage 4S samples (y-axis in log-space). Two independent assembly methods, Trinity and Oases, were used in the reconstruction. The grand total number of contigs reconstructed within each assembly is displayed in the rightmost column. Reconstructed contigs are annotated with their putative taxonomic origin as inferred by comparison with NCBI nucleotide (nt) and protein (nr) archives using TBLASTX database searches.
Discussion
Neuroblastoma is a pediatric tumor of the sympathetic nervous system that represents the most common form of cancer in infancy. It is characterized by a striking diversity in biology and clinical behaviour of its subtypes. This heterogeneity as well as supporting epidemiological findings are highly suggestive of infectious cofactors involved in genesis and maintenance of the disease [19,20]. While several studies utilizing technologies with lower sensitivity compared to our approach have identified human polyomaviruses in neuroblastoma and pediatric embryonal tumors [22–24], newer investigations seem to render these associations inconclusive [25]. However, viral commensals of the families
polyomaviridaeandadenoviridaeare indeed suspected to acquire rare transforming properties as a consequence of viral latency or defective replication [72] and to encode oncogenes [73,74] whose carcinogenic potential in human is currently investigated [8,75]. We undertook the first systematic search for known and unknown viruses in transcriptomes of metastatic neuroblastoma by analyzing deep sequencing RNA-Seq data of14 metastatic neuroblastomas from two tumor stages as well as positive and negative experimental controls.
Several high-throughput methods for detecting viral sequence reads among human RNA-Seq data have been developed. Among these methods, PathSeq, CaPSID and RINS are most prominent due to their design as reusable computational pipelines. In this study we selected CaPSID and RINS due to their high performance and public availability and compared their detection performance with that of our novel method Virana. Both CaPSID and RINS follow a subtractive approach, e.g. they separately map input data to viral and human reference sequences and subtract viral read mappings that are similar to the human genome from the analysis. While CaPSID is conceptualised as a generalised framework that supports the subtraction process by means of a database and a web server, RINS features an integrated pipeline that splits input reads into shorter fragments in order to increase mapping sensitivity, followed by transcriptome assembly of putative viral reads into full length transcripts.
Both RNA and DNA viruses may share considerable sequence homology to human factors due to reasons such as lateral gene transfer, oncogene capture, ancestral endogenization, or insertional mutagenesis leading to chimeric transcripts [47]. Such homologous transcripts may display human-viral sequence similarities of 86% (Bovine Herpes virus) and up to 92% (acutely transforming retroviruses). Subtractive approaches silently discard these tran-script from the analysis due to their similarity to the human reference genome. In contrast, our novel method Virana follows a radically different approach. Instead of separate mapping to viral and human reference database followed by digital subtraction, Virana undertakes a particularly sensitive read mapping to a combined set of human and viral references. By allowing for multimaps, this mapping strategy facilitates discovery of viral transcripts regardless of their similarity to human factors. Apart from being conceptually simpler by relying on only one mapping step and discarding the subtraction procedure that is both possibly erroneous and computationally costly, this approach empowers the mapper to make informed decisions about relative alignment quality by weighing different human and viral reference positions against each other. As a direct consequence of this increased mapping quality, paired-end reads can be mapped across human and viral references, allowing for detection of human-viral chimeric tran-scription and proviral integration events.
We quantitatively validated Virana’s approach both in settings involving simulated reads as well as in real-world scenarios
involving experimental positive and negative controls. In these validations, Virana displays significantly higher detection sensitiv-ities than competing approaches especially at high rates of viral sequence divergence exceeding5% that are common for tumor viruses [76–78]. As a consequence, Virana was the only method able to detect all viral families independent of sequence divergence in the validation data set. In spite of the additional processing undertaken by our method, Virana features between and two and three times faster execution speeds compared to related methods. Interestingly, viral reads analyzed in the sequence divergence validation originate from a broad array of viral species, only two of which infect mammalian hosts and none of which display significant human-viral sequence homology. As a consequence, this validation favors subtractive approaches by reducing the danger of erroneous subtraction of viral reads that are similar to the human genome. In addition, the sequence divergence validation contained reads sampled at high coverage. However, transcripts of tumor viruses are often expressed at only low cellular abundances and are thus expected to have low sequence coverage. We therefore next validated the ability of viral detection approaches to detect viral transcripts homologous to human factors at varying levels of sequence coverage. Virana, by virtue of not relying on digital subtraction, demonstrated superior sensitivity at this validation both in settings of natural sequence homology as well as in cases of human-viral chimeric transcription. Specifically, Virana was the only method able to detect evidence for all viruses even at low twofold coverages. We observed that both RINS and CaPSID discarded a substantial amount of human-viral homol-ogous transcripts due to their high similarity to the human reference genome, a fact that explains the lower performance of these methods in this validation scenario.
Analysis of positive and negative experimental controls further reveals that Virana is able to detect viral transcripts associated with four types of cancer at a high dynamic range of relative abundances. While Virana displays a slightly reduced specificity in simulated and experimental evaluations, these false positive hits are limited to only two viral families (Flaviviridae and Retroviridae) that display high sequence similarity to human factors. These hits are additionally annotated with an ambiguous taxonomic origin by Virana. In addition, Virana provides extensive support for investigating such ambiguous viral hits by analyzing the homol-ogous context of putative viral reads in a context of multiple sequence alignments and phylogenies.
concerning Virana’s homologous region construction process and the sequencing depth of our experimental data, we can conclude that our approach requires minimal abundances of only two average-length viral transcripts per cell even under adverse conditions such as high viral divergence or extensive human-viral sequence homology. While representing a theoretical sensitivity that may be altered by sequencing biases [93], these copy numbers compare very favorably with related estimates reporting minimal abundances of one to several complete viral genomes per cell [27,34,35].
After applying Virana to several positive control panels of human cancers with known viral cofactors and accurately reconstructing large fragments of viruses that are causally related to the respective tumors, we analyzed neuroblastoma transcrip-tomes at high sequencing depth and using two different sequencing platforms. Analyses of neuroblastoma transcriptomes resulted in the detection of putative viral transcripts with high local sequence similarity to several viral families. However, automatic taxonomic annotation as well as detailed manual inspection of homologous regions pertaining to these families revealed the human or bacteriophage origin of all transcripts. While we could
find differences in the abundance of HERV-K transcripts between neuroblastoma stages 4 and 4S, the causative role of HERV transcription with regard to oncogenesis is currently unclear [94] and, as to our knowledge, only tentative associations with specific cancers have been made as to date [70]. Apart from these tentative differences in HERV-K abundances, no quantitative difference between neuroblastoma stages 4 and 4S could be identified with regard to viral transcription.
In conclusion, our observations provide negative evidence regarding the contested question of putative viral cofactors of metastatic neuroblastoma by suggesting that viruses are unlikely to be frequent cofactors in the maintenance of metastatic neuroblas-toma.
Author Contributions
Conceived and designed the experiments: SES LT. Performed the experiments: MF JA PN. Analyzed the data: SES. Contributed reagents/ materials/analysis tools: MF JA PN. Wrote the paper: SES. Designed the software used in analysis: SES. Provided guidance: HP TL FB.
References
1. Moore PS, Chang Y (2010) Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer 10: 878–889.
2. Parkin DM (2006) The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118: 3030–3044.
3. Sarid R, Gao SJ (2011) Viruses and human cancer: from detection to causality. Cancer Lett 305: 218–227.
4. Schiller JT, Lowy DR (2010) Vaccines to prevent infections by oncoviruses. Annu Rev Microbiol 64: 23–41.
5. zur Hausen H (2006) Infections Causing Human Cancer. Weinheim: Wiley-VCH.
6. zur Hausen H (2012) Red meat consumption and cancer: reasons to suspect involvement of bovine infectious factors in colorectal cancer. Int J Cancer 130: 2475–2483.
7. International Cancer Genome Consortium, Hudson TJ, Anderson W, Artez A, Barker AD, et al. (2010) International network of cancer genome projects. Nature 464: 993–998.
8. zur Hausen H (2009) The search for infectious causes of human cancers: where and why. Virology 392: 1–10.
9. Javier RT, Butel JS (2008) The History of Tumor Virology. Cancer Research 68: 7693–7706.
10. Brodeur GM (2003) Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3: 203–216.
11. Maris JM, Hogarty MD, Bagatell R, Cohn SL (2007) Neuroblastoma. The Lancet 369: 2106–2120.
12. Janoueix-Lerosey I, Schleiermacher G, Delattre O (2010) Molecular pathogen-esis of peripheral neuroblastic tumors. Oncogene 29: 1566–1579.
13. Kaatsch P (2010) Epidemiology of childhood cancer. Cancer Treat Rev 36: 277– 285.
14. D’Angio GJ, Evans AE, Koop CE (1971) Special pattern of widespread neuroblastoma with a favourable prognosis. Lancet 1: 1046–1049.
15. Shuangshoti S, Shuangshoti S, Nuchprayoon I, Kanjanapongkul S, Marrano P, et al. (2012) Natural course of low risk neuroblastoma. Pediatr Blood Cancer 58: 690–694.
16. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, et al. (2009) Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27: 1026–1033.
17. Fischer M, Oberthuer A, Brors B, Kahlert Y, Skowron M, et al. (2006) Differential expression of neuronal genes defines subtypes of disseminated neuroblastoma with favorable and unfavorable outcome. Clin Cancer Res 12: 5118–5128.
18. Roman E, Simpson J, Ansell P, Kinsey S, Mitchell CD, et al. (2007) Childhood acute lymphoblastic leukemia and infections in the first year of life: a report from the United Kingdom Childhood Cancer Study. Am J Epidemiol 165: 496–504. 19. Menegaux F, Olshan AF, Neglia JP, Pollock BH, Bondy ML (2004) Day care, childhood infections, and risk of neuroblastoma. Am J Epidemiol 159: 843–851. 20. Heck JE, Ritz B, Hung RJ, Hashibe M, Boffetta P (2009) The epidemiology of
neuroblastoma: a review. Paediatr Perinat Epidemiol 23: 125–143.
21. zur Hausen H (2009) Childhood leukemias and other hematopoietic malignancies: interdependence between an infectious event and chromosomal modifications. Int J Cancer 125: 1764–1770.
22. Jørgensen GE, Johnsen JI, Ponthan F, Kogner P, Flaegstad T, et al. (2000) Human polyomavirus BK (BKV) and neuroblastoma: mechanisms of oncogenic
action and possible strategy for novel treatment. Med Pediatr Oncol 35: 593– 596.
23. Krynska B, Del Valle L, Croul S, Gordon J, Katsetos CD, et al. (1999) Detection of human neurotropic JC virus DNA sequence and expression of the viral oncogenic protein in pediatric medulloblastomas. Proc Natl Acad Sci USA 96: 11519–11524.
24. Flaegstad T, Andresen PA, Johnsen JI, Asomani SK, Jørgensen GE, et al. (1999) A possible contributory role of BK virus infection in neuroblastoma development. Cancer Research 59: 1160–1163.
25. Stolt A, Kjellin M, Sasnauskas K, Luostarinen T, Koskela P, et al. (2005) Maternal human polyomavirus infection and risk of neuroblastoma in the child. Int J Cancer 113: 393–396.
26. Rohwer F, Edwards R (2002) The Phage Proteomic Tree: a genome-based taxonomy for phage. J Bacteriol 184: 4529–4535.
27. Bexfield N, Kellam P (2011) Metagenomics and the molecular identification of novel viruses. Vet J 190: 191–198.
28. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, et al. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266: 1865–1869.
29. Feldhahn M, Menzel M, Weide B, Bauer P, Meckbach D, et al. (2011) No evidence of viral genomes in whole-transcriptome sequencing of three melanoma metastases. Exp Dermatol 20: 766–768.
30. Arron ST, Ruby JG, Dybbro E, Ganem D, DeRisi JL (2011) Transcriptome sequencing demonstrates that human papillomavirus is not active in cutaneous squamous cell carcinoma. J Invest Dermatol 131: 1745–1753.
31. Feng H, Shuda M, Chang Y, Moore PS (2008) Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319: 1096–1100. 32. Lipkin WI (2010) Microbe hunting. Microbiol Mol Biol Rev 74: 363–377. 33. Duncan CG, Leary RJ, Lin JCH, Cummins J, Di C, et al. (2009) Identification
of microbial DNA in human cancer. BMC medical genomics 2: 22. 34. Feng H, Taylor JL, Benos PV, Newton R, Waddell K, et al. (2007) Human
transcriptome subtraction by using short sequence tags to search for tumor viruses in conjunctival carcinoma. J Virol 81: 11332–11340.
35. Moore RA, Warren RL, Freeman JD, Gustavsen JA, Che´nard C, et al. (2011) The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS ONE 6: e19838.
36. Xu Y, Stange-Thomann N, Weber G, Bo R, Dodge S, et al. (2003) Pathogen discovery from human tissue by sequence-based computational subtraction. Genomics 81: 329–335.
37. Weber G, Shendure J, Tanenbaum DM, Church GM, Meyerson M(2002) Identification of foreign gene sequences by transcript filtering against the human genome. Nat Genet 30: 141–142.
38. Isakov O, Modai S, Shomron N (2011) Pathogen detection using short-RNA deep sequencing subtraction and assembly. Bioinformatics 27: 2027–2030. 39. Patowary A, Chauhan RK, Singh M, Kv S, Periwal V, et al. (2012) De novo
identification of viral pathogens from cell culture hologenomes. BMC Res Notes 5: 11.
40. Ma M, Huang Y, Gong Z, Zhuang L, Li C, et al. (2011) Discovery of DNA Viruses in Wild-Caught Mosquitoes Using Small RNA High throughput Sequencing. PLoS ONE 6: e24758.