Centro de Tecnologia
Programa de P´
os-Gradua¸
c˜
ao em Engenharia El´
etrica
Aplica¸
c˜
ao de T´
ecnicas de Aprendizado de M´
aquina no
Reconhecimento de Classes Estruturais de Prote´ınas
Valnaide Gomes Bittencourt
Universidade Federal do Rio Grande do Norte
Centro de Tecnologia
Programa de P´
os-Gradua¸c˜
ao em Engenharia El´
etrica
APLICAC
¸ ˜
AO DE T´
ECNICAS DE APRENDIZADO DE M ´
AQUINA NO
RECONHECIMENTO DE CLASSES ESTRUTURAIS DE PROTE´INAS
Valnaide Gomes Bittencourt
Disserta¸c˜ao submetida ao Programa de P´
os-Gradua¸c˜ao em Engenharia El´etrica do Centro
de Tecnologia da Universidade Federal do Rio
Grande do Norte, como parte dos requisitos
necess´arios para obten¸c˜ao do grau de Mestre em
Ciˆencias.
Orientador: Prof. Dr. Jos´e Alfredo Ferreira Costa
Co-orientador: Prof. Dr. Marc´ılio Carlos Pereira de Souto
Natal, Novembro de 2005
PROGRAMA DE P ´OS-GRADUAC¸ ˜AO EM ENGENHARIA EL´ETRICA
Aprovada em 25 de Novembro de 2005 pela comiss˜ao examinadora,
formada pelos seguintes membros:
Prof. Dr. Jos´e Alfredo Ferreira Costa (Orientador) Departamento de Engenharia El´etrica - UFRN
Prof. Dr. Marc´ılio Carlos Pereira de Souto (Co-Orientador) Departamento de Inform´atica e Matem´atica Aplicada - UFRN
Prof. Dr. Adri˜ao Duarte D´oria Neto (Examinador Interno) Departamento de Engenharia de Computa¸c˜ao e Automa¸c˜ao - UFRN
Profa. Dra. Teresa Bernarda Ludermir (Examinador Externo) Centro de Inform´atica - UFPE
Agradecimentos
A Deus, por mais esta oportunidade em minha vida, acompanhando bem de perto todos os meus passos; e a Nossa Senhora, por sempre me cobrir com seu manto de prote¸c˜ao, dando-me paciˆencia e esperan¸ca necess´arias para a conclus˜ao deste trabalho.
Ao professor Jos´e Alfredo, pela confian¸ca em mim depositada quando aceitou ser o meu orien-tador, pela liberdade que me deu na escolha do tema abordado nesta disserta¸c˜ao, pelo entusiasmo constantemente demonstrado em nossas conversas e pelo apoio sempre concedido.
Ao professor Marc´ılio, por ter se mostrado dispon´ıvel para me ajudar mesmo antes de se tornar meu co-orientador, pela defini¸c˜ao da abordagem do trabalho, pelos constantes ensinamentos, dis-cuss˜oes e direcionamentos, pela dedica¸c˜ao e grande exemplo de ´etica, responsabilidade e competˆencia. Aos meus pais, S´ergio e Lourdinha, pelo incentivo e dedica¸c˜ao, pela maneira carinhosa e com-preensiva com que sempre me ap´oiam e pela educa¸c˜ao e amor incondicional; irm˜aos, Hegel e H´elcio, pela forte amizade e amor, pelo est´ımulo e apoio sempre a mim proporcionados; e cunhada, Greicy, pelo carinho, incentivo e por partilhar comigo a experiˆencia de se fazer uma p´os-gradua¸c˜ao.
Ao meu noivo, Silvio, pelo constante companheirismo, lealdade e amor, pela disponibilidade irrestrita, apesar de sua agenda lotada, e pela ativa presen¸ca ao longo da elabora¸c˜ao desta disserta¸c˜ao. Aos meus amigos mais pr´oximos, cuja cita¸c˜ao sabem ser para eles direcionada, pela afei¸c˜ao e considera¸c˜ao de sempre.
Aos novos colegas e amigos do mestrado, pelos momentos agrad´aveis juntos, pela freq¨uente companhia em almo¸cos, pela descontra¸c˜ao e partilha de sentimentos.
`
A CAPES, pelo apoio financeiro; ao PROMETH, na pessoa do professor Dario, a quem tamb´em agrade¸co pela constante preocupa¸c˜ao e interesse em minha vida n˜ao apenas profissional, e ao LA-BILIC, pelo apoio t´ecnico.
Por fim, agrade¸co a todas as pessoas do meu conv´ıvio, que, de uma forma ou de outra, con-tribu´ıram para o desenvolvimento deste trabalho.
Rm 12,12
Resumo
Atualmente, a classifica¸c˜ao estrutural de prote´ınas, que diz respeito `a inferˆencia de padr˜oes
em sua conforma¸c˜ao 3D, ´e um dos principais problemas em aberto da Biologia Molecular.
Esse problema vem recebendo a aten¸c˜ao de muitos pesquisadores na ´area de Bioinform´atica
pelo fato de as fun¸c˜oes das prote´ınas estarem intrinsecamente relacionadas `as suas diferentes
conforma¸c˜oes espaciais, que s˜ao de dif´ıcil obten¸c˜ao experimental em laborat´orio.
Considerando a grande diferen¸ca entre o n´umero de seq¨uˆencias de prote´ınas conhecidas e
o n´umero de estruturas tridimensionais determinadas experimentalmente, ´e alta a demanda
por t´ecnicas automatizadas de classifica¸c˜ao estrutural de prote´ınas. Nesse contexto, as
ferramentas computacionais, principalmente as t´ecnicas de Aprendizado de M´aquina (AM),
tornaram-se alternativas essenciais para tratar esse problema.
Neste trabalho, t´ecnicas de AM s˜ao empregadas no reconhecimento de classes
estrutu-rais de prote´ınas: ´Arvore de Decis˜ao, k-Vizinhos Mais Pr´oximos, Na¨ıve Bayes, M´aquinas de Vetores Suporte e Redes Neurais Artificiais. Esses m´etodos foram escolhidos por
re-presentarem diferentes paradigmas de aprendizado e serem bastante citados na literatura.
Visando conseguir uma melhoria de desempenho na solu¸c˜ao do problema abordado, sistemas
de multiclassifica¸c˜ao homogˆenea (Bagging e Boosting) e heterogˆenea (Voting, Stacking e StackingC) s˜ao aplicados nesta pesquisa, usando como base as t´ecnicas de AM anterior-mente mencionadas. Al´em disso, pelo fato de a base de dados de prote´ınas considerada
neste trabalho apresentar o problema de classes desbalanceadas, t´ecnicas artificiais de
ba-lanceamento de classes (Under-sampling Aleat´orio, Tomek Links, CNN, NCL e OSS) s˜ao utilizadas a fim de minimizar esse problema e melhorar o desempenho dos classificadores.
Para a avalia¸c˜ao dos m´etodos de AM, um procedimento de valida¸c˜ao cruzada ´e
em-pregado, em que a acur´acia dos classificadores ´e medida atrav´es das m´edias da taxa de
classifica¸c˜ao incorreta nos conjuntos de testes independentes. Essas m´edias s˜ao
compara-das duas a duas pelo teste de hip´otese a fim de avaliar se h´a diferen¸ca estatisticamente
significativa entre elas.
Com os resultados obtidos, pode-se observar, entre os classificadores base, o
desempe-nho superior do m´etodo M´aquinas de Vetores Suporte. Os sistemas de multiclassifica¸c˜ao
(homogˆenea e heterogˆenea), por sua vez, apresentaram, em geral, uma acur´acia superior ou
similar a dos classificadores usados como base, destacando-se oBoosting que usou ´Arvore de Decis˜ao em sua forma¸c˜ao e oStackingC tendo como meta classificador a Regress˜ao Linear. O m´etodoVoting, apesar de sua simplicidade, tamb´em mostrou-se adequado para a solu¸c˜ao do problema considerado nesta disserta¸c˜ao. Em rela¸c˜ao `as t´ecnicas de balanceamento de
classes, n˜ao foram alcan¸cados melhores resultados de classifica¸c˜ao global com as bases de
dados obtidas com a aplica¸c˜ao de tais t´ecnicas. No entanto, foi poss´ıvel uma melhor
classi-fica¸c˜ao espec´ıfica da classe minorit´aria, de dif´ıcil aprendizado. A t´ecnica NCL foi a que se
Abstract
Nowadays, classifying proteins in structural classes, which concerns the inference of
pat-terns in their 3D conformation, is one of the most important open problems in Molecular
Biology. The main reason for this is that the function of a protein is intrinsically related to
its spatial conformation. However, such conformations are very difficult to be obtained
ex-perimentally in laboratory. Thus, this problem has drawn the attention of many researchers
in Bioinformatics.
Considering the great difference between the number of protein sequences already known
and the number of three-dimensional structures determined experimentally, the demand of
automated techniques for structural classification of proteins is very high. In this context,
computational tools, especially Machine Learning (ML) techniques, have become essential
to deal with this problem.
In this work, ML techniques are used in the recognition of protein structural classes:
Decision Trees,k-Nearest Neighbor, Na¨ıve Bayes, Support Vector Machine and Neural Net-works. These methods have been chosen because they represent different paradigms of
learning and have been widely used in the Bioinfornmatics literature. Aiming to obtain
an improvment in the performance of these techniques (individual classifiers),
homoge-neous (Bagging and Boosting) and heterogehomoge-neous (Voting, Stacking and StackingC)
multi-classification systems are used. Moreover, since the protein database used in this work
presents the problem of imbalanced classes, artificial techniques for class balance
(Under-sampling Random, Tomek Links, CNN, NCL and OSS) are used to minimize such a problem.
In order to evaluate the ML methods, a cross-validation procedure is applied, where
the accuracy of the classifiers is measured using the mean of classification error rate, on
independent test sets. These means are compared, two by two, by the hypothesis test
aiming to evaluate if there is, statistically, a significant difference between them.
With respect to the results obtained with the individual classifiers, Support Vector
Machine presented the best accuracy. In terms of the multi-classification systems
(homoge-neous and heteroge(homoge-neous), they showed, in general, a superior or similar performance when
compared to the one achieved by the individual classifiers used - especially Boosting with
Decision Tree and the StackingC with Linear Regression as meta classifier. The Voting
method, despite of its simplicity, has shown to be adequate for solving the problem
pre-sented in this work. The techniques for class balance, on the other hand, have not produced
a significant improvement in the global classification error. Nevertheless, the use of such
techniques did improve the classification error for the minority class. In this context, the
Sum´
ario
1 Introdu¸c˜ao 1
1.1 Motiva¸c˜ao . . . 1
1.2 Objetivos . . . 5
1.3 Organiza¸c˜ao da Disserta¸c˜ao . . . 6
2 Prote´ınas 7 2.1 Introdu¸c˜ao . . . 7
2.2 Estruturas de Prote´ına . . . 8
2.2.1 Estrutura Prim´aria . . . 8
2.2.2 Estrutura Secund´aria . . . 12
2.2.3 Estrutura Terci´aria . . . 14
2.2.4 Estrutura Quatern´aria . . . 17
2.3 Classifica¸c˜ao Estrutural . . . 18
2.3.1 Bancos de Dados Biol´ogicos . . . 19
2.3.2 Classe all-α . . . 20
2.3.3 Classe all-β . . . 20
2.3.4 Classe α/β . . . 21
2.3.5 Classe α+β . . . 22
2.4 An´alise Computacional . . . 22
2.5 Trabalhos Relacionados . . . 25
3 Aprendizado de M´aquina 30
3.1 Introdu¸c˜ao . . . 30
3.1.1 Paradigmas de AM . . . 31
3.1.2 Aprendizado Supervisionado . . . 32
3.2 Sistemas de Multiclassifica¸c˜ao . . . 34
3.2.1 Multiclassificadores Homogˆeneos . . . 36
3.2.2 Multiclassificadores Heterogˆeneos . . . 41
3.3 Pr´e-processamento de Dados . . . 46
4 M´etodos e Experimentos 54 4.1 Base de Dados . . . 54
4.2 Metodologia de Valida¸c˜ao . . . 57
4.2.1 Valida¸c˜ao Cruzada . . . 58
4.2.2 Teste de Hip´otese . . . 59
4.3 Experimentos . . . 61
5 Resultados 66 5.1 Classificadores base . . . 66
5.2 Multiclassificadores . . . 67
5.2.1 Multiclassificadores Homogˆeneos . . . 67
5.2.2 Multiclassificadores Heterogˆeneos . . . 68
5.2.3 Discuss˜oes . . . 70
5.3 Pr´e-processamento de Dados . . . 75
5.4 Considera¸c˜oes finais . . . 85
Lista de Figuras
2.1 Estrutura prim´aria de uma prote´ına . . . 8
2.2 Estrutura geral de um amino´acido . . . 9
2.3 Forma¸c˜ao de liga¸c˜oes pept´ıdicas e cadeia polipept´ıdica resultante . . . 11
2.4 Representa¸c˜oes da α-h´elice . . . 13
2.5 Representa¸c˜oes da β-folha mista . . . 14
2.6 Estrutura terci´aria de uma prote´ına da Gram-negativa . . . 16
2.7 Estrutura terci´aria, em estereoscopia . . . 16
2.8 Estruturas secund´aria e terci´aria, em estereoscopia . . . 17
2.9 Estrutura quatern´aria da hemoglobina humana . . . 18
2.10 Prote´ına da classeall-β, em estereoscopia . . . 21
2.11 Prote´ına da classeα/β, em estereoscopia . . . 22
3.1 Estrutura de sistemas de multiclassifica¸c˜ao . . . 35
3.2 Representa¸c˜ao do m´etodo Bagging . . . 37
3.3 Representa¸c˜ao do m´etodo AdaBoosting . . . 39
3.4 Representa¸c˜ao do m´etodo Stacking . . . 42
3.5 Representa¸c˜ao do m´etodo Stacking eStackingC . . . 45
3.6 Exemplo de dados com duas classes desbalanceadas . . . 49
5.1 Resumo dos resultados obtidos com os multiclassificadores homogˆeneos e seus respectivos classificadores base . . . 70
5.2 Resumo dos resultados obtidos com os multiclassificadores heterogˆeneos e
seus classificadores base . . . 71
5.3 Melhores resultados dos multiclassificadores homogˆeneos e heterogˆeneos . . 72
5.4 Resultados obtidos com a base de dados gerada com a t´ecnica NCL . . . 78
5.5 Resultados obtidos com as bases de dados original e as geradas com as t´ecnicas deunder-sampling . . . 80
5.6 Resultados obtidos para a classe minorit´aria . . . 81
5.7 Resultados obtidos para a classe majorit´aria . . . 81
5.8 M´edia de redu¸c˜ao erro na classe minorit´aria . . . 82
Lista de Tabelas
2.1 Nomenclatura e simbologia dos 20 amino´acidos que podem formar as prote´ınas 10
2.2 C´odigo gen´etico . . . 11
3.1 Representa¸c˜ao de uma matriz de confus˜ao . . . 33
4.1 As seis propriedades extra´ıdas da seq¨uˆencia de prote´ına . . . 55
4.2 Distribui¸c˜ao das prote´ınas nas classes estruturais . . . 55
4.3 Distribui¸c˜ao da quantidade de prote´ınas nas dobras e nas classes estruturais 57 5.1 Taxa de erro dos classificadores base (%) . . . 67
5.2 Taxa de erro dos m´etodos Bagging (%) . . . 67
5.3 Taxa de erro dos m´etodos Boosting (%) . . . 68
5.4 Taxa de erro do m´etodoStacking (%) . . . 68
5.5 Taxa de erro do m´etodoStackingC (%) . . . 69
5.6 Taxa de erro do m´etodoVoting (%) . . . 69
5.7 Matrizes de confus˜ao dos classificadores base . . . 74
5.8 Taxa de erro, por classe (%) . . . 75
5.9 Taxa de erro global para cada t´ecnica de under-samplig (%) . . . 75
5.10 Taxa de erro da classeα+β para cada t´ecnica deunder-samplig (%) . . . 76
5.11 Taxa de erro da classeα/β para cada t´ecnica de under-samplig (%) . . . . 76
5.12 Taxa de erro dos multiclassificadores com a base de dados originada pela t´ecnica NCL (%) . . . 77
5.13 Taxa de erro, por classe, com a t´ecnica NCL (%) . . . 78
5.14 Taxa de erro global e das classes minorit´aria (α+β) e majorit´aria (α/β) para a base de dados original (%) . . . 79
5.15 Matrizes de confus˜ao dos classificadores base com a base de dados gerada
Cap´ıtulo 1
Introdu¸
c˜
ao
1.1
Motiva¸
c˜
ao
A utiliza¸c˜ao de novas e eficientes t´ecnicas na an´alise de seq¨uˆencias de genomas vem sendo
respons´avel pelo consider´avel crescimento dos bancos de dados biol´ogicos dispon´ıveis
(Wa-terman 1995; Meidanis and Set´ubal 1997; Baldi and Brunak 2001). Esses dados necessitam
de m´etodos adequados de manipula¸c˜ao e an´alise para que possam ser utilizados de forma
mais efetiva pelos bi´ologos. A investiga¸c˜ao e aperfei¸coamento desses m´etodos ´e o grande
de-safio de uma nova ´area de pesquisa que surgiu na d´ecada de 90, acompanhando os projetos
genoma, chamada de Bioinform´atica ou Biologia Computacional (Souto et al. 2003).
A Bioinform´atica tem por objetivo o estudo e aplica¸c˜ao de t´ecnicas computacionais a
tarefas das mais diversas ´areas da Biologia, dentre elas, a Biologia Molecular (Lorena et al.
2002; Set´ubal 2003; Souto et al. 2003). Nesse contexto, a computa¸c˜ao pode ser aplicada na
resolu¸c˜ao de uma s´erie de problemas, tais como: compara¸c˜ao de seq¨uˆencias (DNA, RNA
e prote´ınas), montagem de fragmentos, reconhecimento de genes, identifica¸c˜ao e an´alise da
express˜ao de genes, reconstru¸c˜ao de ´arvores filogen´eticas e determina¸c˜ao da estrutura de
prote´ınas (Baldi and Brunak 2001; Set´ubal 2003; Souto et al. 2003).
Dentre esses problemas, a predi¸c˜ao te´orica da estrutura de prote´ınas ´e, atualmente, o
grande desafio da Bioinform´atica. O estudo relativo a este assunto vem caracterizando uma
nova fase para as pesquisas gen´eticas, denominada Proteˆomica (Guimar˜aes and Melo 2003),
que surgiu ap´os a finaliza¸c˜ao do seq¨uenciamento do genoma humano (Consortium 2001) e
de diversos outros organismos. O termo “Proteˆomica” envolve a identifica¸c˜ao de todas as
prote´ınas expressas pelo genoma bem como a determina¸c˜ao de suas fun¸c˜oes fisiol´ogicas e
patol´ogicas.
A fun¸c˜ao da prote´ına ´e intrinsecamente relacionada `a conforma¸c˜ao espacial (estrutura
tridimensional - 3D) que ela apresenta (Guimar˜aes and Melo 2003). Al´em disso, o
conhe-cimento da estrutura 3D da prote´ına ´e essencial para, por exemplo, o desenvolvimento de
novos medicamentos e m´etodos de diagn´ostico. Por´em, a identifica¸c˜ao dessa conforma¸c˜ao
espacial n˜ao ´e uma tarefa simples. Por exemplo, os m´etodos tradicionais de obten¸c˜ao da
es-trutura 3D de prote´ınas, tais como cristalografia de raiosX(Abola et al. 2000) e Ressonˆancia Magn´etica Nuclear (RMN) (G¨untert 2003), s˜ao muito caros, trabalhosos, demorados e tˆem
limita¸c˜oes pr´oprias (Chinnasamy, Sung, and Mittal 2004).
De fato, determinar a seq¨uˆencia de uma prote´ına ´e relativamente mais f´acil do que
determinar a sua estrutura 3D, o que leva a uma grande diferen¸ca entre o n´umero de
seq¨uˆencias e o n´umero de estruturas e fun¸c˜oes prot´eicas conhecidas (Nelson and Cox 2000).
A fim de diminuir esta disparidade, m´etodos computacionais s˜ao cada vez mais empregados.
Dessa forma, a identifica¸c˜ao automatizada da estrutura 3D da prote´ına tornou-se o foco mais
recente de pesquisa na ´area de Bioinform´atica (Mirkin and Ritter 1999).
Computacionalmente, destacam-se dois tipos de pesquisas na identifica¸c˜ao da estrutura
3D de uma prote´ına: a an´alise de seq¨uˆencia e a an´alise de estrutura (Guimar˜aes and Melo
2003). A modelagem de uma prote´ına atrav´es da an´alise de seq¨uencia baseia-se no conceito
de evolu¸c˜ao molecular. Isto ´e, parte-se do princ´ıpio de que a similaridade entre a seq¨uencia
de amino´acidos (estrutura prim´aria) de uma prote´ına da qual n˜ao se sabe a fun¸c˜ao e a
seq¨uencia de uma prote´ına cuja estrutura tridimensional ´e conhecida implica em similaridade
estrutural entre elas. Contudo, embora muitas das prote´ınas possuam um alto grau de
similaridade em termos de seq¨uencia de amino´acidos, elas podem n˜ao compartilhar da
mesma fun¸c˜ao. Por outro lado, duas prote´ınas podem ter um baixo grau de similaridade
e, entretanto, apresentarem fun¸c˜oes semelhantes (Okun 2004). A an´alise de estrutura, por
sua vez, diz respeito `a predi¸c˜ao da estrutura tridimensional de uma prote´ına a partir de sua
1.1. MOTIVAC¸ ˜AO 3
A inferˆencia da estrutura 3D da prote´ına com base apenas em sua seq¨uˆencia foi
demons-trado ser um problema NP-dif´ıcil (Guimar˜aes and Melo 2003). Devido a alta complexidade,
tal procedimento ´e dividido em uma s´erie de passos intermedi´arios, como por exemplo:
a predi¸c˜ao de estrutura secund´aria (Jones 1999; Petersen et al. 2000; Baldi and Brunak
2001; Pollastri et al. 2002; Guimar˜aes, Melo, and Cavalcanti 2003) e a classifica¸c˜ao
estru-tural (Bologna and Appel 2002; Ding and Dubchak 2001; Tan, Gilbert, and Deville 2003;
Huang et al. 2003; Okun 2004; Chinnasamy, Sung, and Mittal 2004). A predi¸c˜ao de
estru-tura secund´aria de prote´ınas consiste na localiza¸c˜ao, na seq¨uˆencia, de subestruturas comuns,
comoα-h´elices eβ-folhas.
A classifica¸c˜ao estrutural de prote´ınas, por sua vez, diz respeito `a identifica¸c˜ao de
padr˜oes estruturais na conforma¸c˜ao 3D. Esta classifica¸c˜ao pode ser decomposta em dois
n´ıveis hier´arquicos. No primeiro n´ıvel, as prote´ınas s˜ao classificadas em dobras (problema
conhecido como reconhecimento de dobras de prote´ınas). No segundo n´ıvel, as prote´ınas s˜ao
classificadas em classes estruturais, de acordo com os tipos de dobras por elas apresentados.
Este problema ´e conhecido como reconhecimento de classes estruturais de prote´ınas, que
ser´a foco desta disserta¸c˜ao.
Atualmente, mais de 700 tipos diferentes de dobras de prote´ınas j´a foram
identifica-dos (Lo Conte et al. 2000), classificaidentifica-dos normalmente em sete classes estruturais distintas.
As similaridades estruturais das prote´ınas com mesmas dobras derivam das propriedades
f´ısicas e qu´ımicas que favorecem determinados arranjos e topologias. Considerando a grande
diferen¸ca entre o n´umero de seq¨uˆencias conhecidas e o n´umero de estruturas 3D
determi-nadas experimentalmente (a rela¸c˜ao ´e mais de 100 para 1), a demanda por t´ecnicas
auto-matizadas de reconhecimento (ou predi¸c˜ao) de dobras ou classes estruturais de prote´ınas ´e
bastante alta (Okun 2004).
As ferramentas que usam computa¸c˜ao convencional, no entanto, s˜ao limitadas para
abordar problemas biol´ogicos complexos, como este em quest˜ao. Isto ocorre, entre outras
raz˜oes, devido `a ausˆencia de uma teoria fundamental em n´ıvel molecular e `a ineficiˆencia
das ferramentas convencionais em lidar com grandes quantidades de dados (Souto et al.
aprender automaticamente a partir de grandes volumes de dados e produzir hip´oteses ´uteis,
s˜ao, ent˜ao, cada vez mais empregadas para tratar problemas em Biologia Molecular (Baldi
and Brunak 2001). De fato, o uso de t´ecnicas de AM tem sido consideravelmente explorado
na predi¸c˜ao autom´atica de estruturas de prote´ınas (Selbig and Argos 1998; Baldi et al.
2000; Turcotte, Muggleton, and Sternberg 2001; Ding and Dubchak 2001; Tan, Gilbert,
and Deville 2003).
Neste trabalho, t´ecnicas de AM s˜ao empregadas para realizar a predi¸c˜ao de classe
estru-turais de prote´ınas: ´Arvore de Decis˜ao (AD), k-Vizinhos Mais Pr´oximos (k-NN, do inglˆes k-Nearest Neighbor), Na¨ıve Bayes (NB), M´aquinas de Vetores Suporte (SVM, do inglˆes Support Vector machine) e Redes Neurais Artificiais (RNA). Tais m´etodos foram escolhi-dos por representarem diferentes paradigmas de aprendizado e serem bastante citaescolhi-dos na
literatura, inclusive na abordagem espec´ıfica deste problema (Ding and Dubchak 2001; Tan,
Gilbert, and Deville 2003; Chung et al. 2003).
Visando conseguir uma melhoria de desempenho na predi¸c˜ao de classes estruturais de
prote´ınas, sistemas de multiclassifica¸c˜ao (homogˆenea e heterogˆenea), ouensembles, s˜ao em-pregados nesta pesquisa, usando como base as t´ecnicas de AM anteriormente mencionadas.
Recentemente, m´etodos multiclassificadores aplicados a problemas de Bioinform´atica foram
investigados (Dudoit, Fridlyand, and Speed 2002; Long and Vega 2003; Tan and Gilbert
2003). Freq¨uentemente, o erro de um sistema de multiclassifica¸c˜ao ´e menor do que o erro de
um ´unico classificador (Bologna and Appel 2002). Neste trabalho, s˜ao usados os seguintes
multiclassificadores: Bagging, Boosting, Voting, Stacking e StackingC.
Al´em disso, a base de dados de prote´ınas usada neste trabalho apresenta o problema
de classes desbalanceadas (Batista 2003). Com o objetivo de minimizar esse problema e
melhorar o desempenho dos classificadores empregados, s˜ao utilizadas algumas t´ecnicas de
pr´e-processamento de dados encontradas na literatura e at´e ent˜ao n˜ao aplicadas a esta base
1.2. OBJETIVOS 5
1.2
Objetivos
O objetivo principal desta disserta¸c˜ao ´e a investiga¸c˜ao e aplica¸c˜ao de t´ecnicas de
Apren-dizado de M´aquina capazes de garantir uma alta precis˜ao na classifica¸c˜ao de prote´ınas em
classes estruturais. De modo geral, a abrangˆencia deste trabalho pode ser resumida da
seguinte maneira:
• Problema: classifica¸c˜ao de prote´ınas em classes estruturais (all-α, all-β, α/β, α+β e small).
• Dados de entrada: prote´ınas representantes de cada uma das cinco classes
estrutu-rais consideradas. Cada prote´ına ´e apresentada como sendo um vetor den atributos cont´ınuos obtidos de propriedades derivadas de sua estrutura prim´aria (seq¨uencia de
amino´acidos).
• Objetivo: usar diferentes t´ecnicas de AM para gerar classificadores capazes de
clas-sificar adequadamente as prote´ınas em classes estruturais.
Inicialmente, ´e realizada uma an´alise de desempenho das seguintes t´ecnicas de AM,
esco-lhidas por motivos previamente apresentados: AD,k-NN, NB, SVM e RNA do tipo Multi-Layer Perceptron (MLP). Tomando como base os resultados obtidos com essas t´ecnicas, s˜ao gerados multiclassificadores homogˆeneos (Bagging eBoosting) e heterogˆeneos (Voting, Stacking e StackingC), sendo o desempenho destes comparados entre si e entre os dos classificadores usados em sua forma¸c˜ao, a fim de se identificar o m´etodo que seja capaz
de realizar uma melhor classifica¸c˜ao das prote´ınas. Desse modo, al´em da avalia¸c˜ao dos
classificadores usados como base, este trabalho faz um estudo da viabilidade de um
am-biente multiclassificador aplicado ao problema do reconhecimento de classes estruturais de
prote´ınas.
Na busca por melhores desempenhos dos classificadores e diante do n´ıtido problema de
desbalanceamento de classes na base de dados empregada neste trabalho, s˜ao tamb´em
mais especificamente t´ecnicas de under-sampling, tais como: Under-sampling Aleat´orio, Tomek Links, CNN, NCL e OSS.
Para avaliar os m´etodos de classifica¸c˜ao usados neste trabalho, ´e empregada uma
metodo-logia de valida¸c˜ao, baseada na utiliza¸c˜ao dok-fold cross validation(Costa Filho, Carvalho, and Souto 2003), que estima o erro m´edio de generaliza¸c˜ao de um certo modelo. Tamb´em ´e
aplicado o teste de hip´otese (Dietterich 1998), a fim de detectar diferen¸cas estatisticamente
significativas entre os resultados obtidos com os diferentes m´etodos de classifica¸c˜ao, inclusive
com o emprego das diferentes bases de dados geradas com as t´ecnicas de under-sampling.
1.3
Organiza¸
c˜
ao da Disserta¸
c˜
ao
Esta disserta¸c˜ao est´a dividida em seis cap´ıtulos, organizados da seguinte maneira:
• Cap´ıtulo 2: s˜ao apresentadas algumas defini¸c˜oes importantes no que diz respeito
`
as prote´ınas, de modo a se entender melhor o problema de classifica¸c˜ao estrutural
de prote´ınas, e feita uma breve revis˜ao na literatura sobre trabalhos relacionados ao
assunto abordado nesta pesquisa.
• Cap´ıtulo 3: s˜ao explicitados alguns conceitos b´asicos da ´area de AM e descritos os
sistemas de multiclassifica¸c˜ao e de pr´e-processamento de dados (t´ecnicas de under-sampling de balanceamento de classes) que s˜ao utilizados neste trabalho.
• Cap´ıtulo 4: s˜ao descritas a base de dados, a metodologia de valida¸c˜ao dos resultados
e a maneira pela qual os experimentos s˜ao realizados.
• Cap´ıtulo 5: s˜ao mostrados e analisados os resultados obtidos nos experimentos.
• Cap´ıtulo 6: s˜ao apresentadas, por fim, algumas conclus˜oes e perspectivas futuras
Cap´ıtulo 2
Prote´ınas
Este cap´ıtulo descreve alguns aspectos importantes em rela¸c˜ao `as prote´ınas, necess´arios `a
compreens˜ao do problema abordado nesta disserta¸c˜ao. A Se¸c˜ao 2.1 introduz concep¸c˜oes
b´asicas sobre prote´ınas. A Se¸c˜ao 2.2 aborda a constitui¸c˜ao e forma¸c˜ao da estrutura de
prote´ınas para se compreender melhor a classifica¸c˜ao empregada neste trabalho, apresentada
na Se¸c˜ao 2.3. A Se¸c˜ao 2.4 mostra alguns enfoques computacionais relativos `a solu¸c˜ao
de problemas na ´area de Proteˆomica, principalmente sobre a classifica¸c˜ao estrutural de
prote´ınas. Uma breve revis˜ao na literatura sobre trabalhos relacionados ao assunto abordado
nesta pesquisa ´e apresentada na Se¸c˜ao 2.5.
2.1
Introdu¸
c˜
ao
Genoma ´e o conjunto completo de informa¸c˜oes necess´arias para o desenvolvimento de um
organismo, que se encontra armazenado nos cromossomos (Guimar˜aes and Melo 2003). Os
cromossomos s˜ao formados essencialmente por cadeias de ´acido desoxirribonucl´eico (DNA,
do inglˆes DeoxyriboNucleic Acid). Essas cadeias contˆem milhares de genes, a partir dos quais s˜ao fabricadas todas as prote´ınas de um organismo atrav´es do processo de express˜ao
gˆenica (Nelson and Cox 2000).
As prote´ınas1s˜ao macromol´eculas complexas, compostas de amino´acidos, necess´arias em todos os processos qu´ımicos que ocorrem nos organismos vivos (Lewis 2001): de regula¸c˜ao
1
A palavra “prote´ına” vem do grego “proteios”, que significa “em primeiro lugar” (Tsunoda 2005).
(como as enzimas que catalisam as rea¸c˜oes qu´ımicas das c´elulas), de imuniza¸c˜ao (como os
anticorpos que protegem o organismo contra corpos estranhos) e de constru¸c˜ao de outras
mol´eculas (como na produ¸c˜ao de ´acidos nucl´eicos, carboidratos e lip´ıdios) (Lewis 2001;
Souto et al. 2003). Por essa raz˜ao, as prote´ınas s˜ao consideradas os constituintes b´asicos da
vida. De fato, elas tˆem um papel essencial no metabolismo, participando praticamente de
todas as atividades celulares (Nelson and Cox 2000).
2.2
Estruturas de Prote´ına
As prote´ınas apresentam estruturas complexas que podem ser organizadas em quatro n´ıveis:
estrutura prim´aria (Se¸c˜ao 2.2.1), secund´aria (Se¸c˜ao 2.2.2), terci´aria (Se¸c˜ao 2.2.3) e quatern´
a-ria (Se¸c˜ao 2.2.4). Os trˆes ´ultimos est˜ao relacionados a estrutura espacial da prote´ına (Silva
1999).
2.2.1
Estrutura Prim´
aria
A estrutura prim´aria de uma prote´ına (apresentada na Figura 2.1, retirada de (Silva 1999))
´e simplesmente a seq¨uˆencia linear dos amino´acidos ligados entre si por liga¸c˜oes pept´ıdicas
que constituem essa prote´ına. A seguir, ser˜ao esclarecidos alguns aspectos a respeito dos
amino´acidos e da cadeia polipept´ıdica formada por eles.
Figura 2.1: Estrutura prim´aria de uma prote´ına
Amino´acidos - Composi¸c˜ao e Estrutura
Os amino´acidos (cuja estrutura geral pode ser vista na Figura 2.2) s˜ao formados por um
2.2. ESTRUTURAS DE PROTE´INA 9
e um grupo am´ınico (-NH2), comuns a todos os amino´acidos, e um grupo R, ou cadeia lateral, que os distingue entre si (Nelson and Cox 2000). Essas cadeias laterais podem diferir
bastante em rela¸c˜ao ao tamanho, estrutura, forma e propriedades qu´ımicas. Elas exercem
influˆencia em muitas caracter´ısticas dos amino´acidos, como por exemplo, na solubilidade
do amino´acido em ´agua (Guimar˜aes and Melo 2003).
Figura 2.2: Estrutura geral de um amino´acido
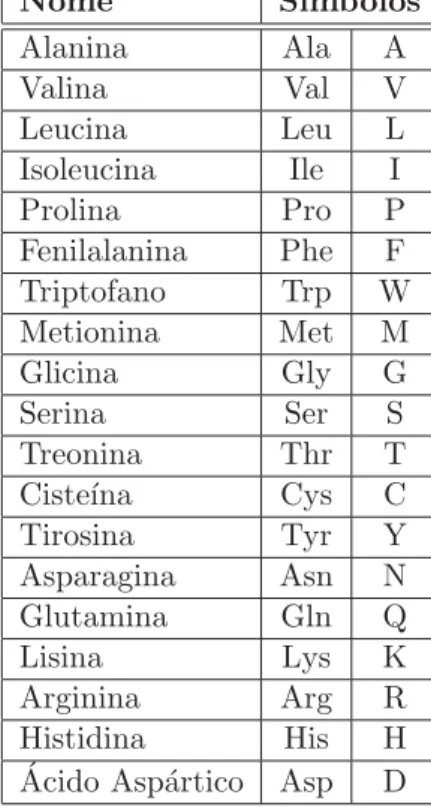
Existem apenas 20 amino´acidos diferentes (listados na Tabela 2.1) codificados, no
pro-cesso de express˜ao gˆenica, pelos c´odons (grupo de trˆes nucleot´ıdeos do ´acido ribonucl´eico
mensageiro2). Por´em, h´a 64 poss´ıveis combina¸c˜oes de triplas de nucleot´ıdeos, ou seja, 64
c´odons. Portanto, muitos dos amino´acidos s˜ao mapeados por mais de um c´odon. Desses 64
c´odons, trˆes s˜ao respons´aveis por indicar o final da tradu¸c˜ao, sendo denominados c´odons de
parada (Souto et al. 2003). As diferentes codifica¸c˜oes, que podem ser visualizadas na Tabela
2.2, ´e o que representa o c´odigo gen´etico. O primeiro, o segundo e o terceiro nucleot´ıdeo dos
c´odons s˜ao representados, respectivamente, pela coluna mais a esquerda, a primeira linha
e a coluna mais a direita da tabela (Esquerda-Topo-Direita) (Lewis 2001). Exemplo: ATG
codifica Metionina (Met). Na Tabela 2.2, o c´odonTer ´e o c´odon de parada.
2
Tabela 2.1: Nomenclatura e simbologia dos 20 amino´acidos que podem formar as prote´ınas
Nome S´ımbolos
Alanina Ala A
Valina Val V
Leucina Leu L
Isoleucina Ile I
Prolina Pro P
Fenilalanina Phe F Triptofano Trp W Metionina Met M
Glicina Gly G
Serina Ser S
Treonina Thr T Ciste´ına Cys C Tirosina Tyr Y Asparagina Asn N Glutamina Gln Q
Lisina Lys K
Arginina Arg R Histidina His H
´
Acido Asp´artico Asp D
Cadeia Polipept´ıdica
Durante a s´ıntese de prote´ına, o grupo carbox´ılico de um amino´acido e o grupo am´ınico
de outro liberam uma mol´ecula de ´agua e formam uma liga¸c˜ao covalente3 denominada liga¸c˜ao pept´ıdica. Ap´os a incorpora¸c˜ao `a cadeia polipept´ıdica, os amino´acidos individuais
s˜ao chamados de res´ıduos de amino´acidos4. A cadeia polipept´ıdica cont´em de algumas
dezenas a v´arias centenas de res´ıduos de amino´acidos (ou simplesmente amino´acidos) que,
ligados deste modo, formam uma estrutura em zig-zag de onde sobressaem as v´arias cadeias
laterais. A Figura 2.3 (retirada de (Silva 1999)) ilustra o processo de forma¸c˜ao de liga¸c˜oes
pept´ıdicas e a cadeia polipept´ıdica resultante.
3
Liga¸c˜ao entre dois ´atomos com partilha de dois ou mais el´etrons.
4
2.2. ESTRUTURAS DE PROTE´INA 11
Tabela 2.2: C´odigo gen´etico
T C A G
T Phe Ser Tyr Cys T
C
Leu Ter Ter A
Trp G
C Leu Pro His Arg T
C
Gin A
G
A IIe Thr Asn Ser T
C
Lys Arg A
Met G
G Val Ala Asp Gly T
C
Glu A
G
O primeiro amino´acido da cadeia, que tem o grupo am´ınico livre, ´e chamado de
ex-tremidade N-terminal, ou aminoterminal; e o ´ultimo, que tem o grupo carbox´ılico livre,
extremidade C-terminal, ou carboxiterminal. A estrutura prim´aria de uma prote´ına
con-siste na seq¨uˆencia de amino´acidos da sua cadeia polipept´ıdica, representada no sentido da
extremidade N-terminal para a extremidade C- terminal. Caso a prote´ına seja formada
por v´arias cadeias, a estrutura prim´aria consiste nas respectivas seq¨uˆencias. A Figura
2.1 mostrada anteriormente representa a estrutura prim´aria de uma prote´ına, denominada
prote´ına G.
2.2.2
Estrutura Secund´
aria
A estrutura secund´aria diz respeito `a rela¸c˜ao espacial entre amino´acidos pr´oximos (Creighton
1993). Essa estrutura ´e caracterizada por padr˜oes tridimensionais regulares e repetitivos que
ocorrem localmente no dobramento da prote´ına. Dois dos padr˜oes de estrutura secund´aria
mais comuns s˜ao asα-h´elices e asβ-folhas (Chothia et al. 1997).
1. α-h´elices
Uma α-h´elice surge a partir da forma¸c˜ao de pontes de hidrogˆenio entre o ´atomo de hidrogˆenio do grupo am´ınico e o ´atomo de oxigˆenio do grupo carbox´ılico das liga¸c˜oes
pept´ıdicas, formando uma estrutura helicoidal com 3,6 res´ıduos de amino´acidos em
cada volta. ´E uma estrutura consideravelmente est´avel tendo em vista que todos os
res´ıduos participam das pontes de hidrogˆenio (com exce¸c˜ao dos res´ıduos das
extremi-dades da h´elice) (Nelson and Cox 2000). Nela, todas as cadeias lateraisRprojetam-se para fora da h´elice. A Figura 2.4 (retirada de (Silva 1999)) mostra trˆes representa¸c˜oes
diferentes daα-h´elice: Ball & stick, sticks e cartoon5.
Al´em das α-h´elice, existem outros tipos de h´elice, como as π h´elice e a h´elice 310, menos est´aveis e muito menos comuns do que aα-h´elice.
5
2.2. ESTRUTURAS DE PROTE´INA 13
Figura 2.4: Representa¸c˜oes daα-h´elice
2. β-folhas
A estrutura de uma β-folha resulta da forma¸c˜ao de pontes de hidrogˆenio entre duas ou mais cadeias polipept´ıdicas adjacentes, formando uma estrutura planar onde as
cadeias laterais se encontram viradas para cima e para baixo, e nunca interagem
umas com as outras.
De acordo com a orienta¸c˜ao relativa dos segmentos da β-folha (orienta¸c˜ao amino-carboxila), esta recebe a classifica¸c˜ao de paralela (segmentos todos orientados na
mesma dire¸c˜ao), antiparalela (segmentos adjacentes orientados em dire¸c˜oes opostas)
(Nelson and Cox 2000). A Figura 2.5 (retirada de (Silva 1999)) mostra duas
repre-senta¸c˜oes diferentes daβ-folha mista.
Figura 2.5: Representa¸c˜oes daβ-folha mista
2.2.3
Estrutura Terci´
aria
Essencialmente, a estrutura terci´aria (ou tridimensional) de uma prote´ına consiste na
con-forma¸c˜ao tridimensional dos elementos da estrutura secund´aria em uma ´unica cadeia
polipep-t´ıdica, que resulta em uma estrutura compacta onde os ´atomos ocupam posi¸c˜oes espec´ıficas. ´
E, portanto, o arranjo tridimensional de todos os ´atomos que comp˜oem uma prote´ına (Levitt
and Chothia 1976), sendo estabilizado por intera¸c˜oes qu´ımicas entre as cadeias laterais dos
amino´acidos como, dentre outras, for¸cas de van der Waals, oxida¸c˜ao de ciste´ına, liga¸c˜oes
2.2. ESTRUTURAS DE PROTE´INA 15
A estrutura terci´aria relaciona-se com os loopings e dobramentos da cadeia prot´eica sobre ela mesma. Loopings e dobramentos s˜ao processos nos quais uma mol´ecula n˜ao organizada, que acabou de ser sintetizada, adquire uma estrutura altamente organizada
como conseq¨uˆencia de intera¸c˜oes entre as cadeias laterais dos amino´acidos (Guimar˜aes and
Melo 2003).
A estrutura tridimensional de uma prote´ına apresenta v´arias caracter´ısticas
importan-tes (Motta 2003), como:
• Muitas prote´ınas dobram-se de modo que os res´ıduos de amino´acidos que est˜ao
dis-tantes uns dos outros na estrutura prim´aria podem estar pr´oximos na estrutura
terci´aria.
• Algumas prote´ınas dobram-se em duas ou mais regi˜oes compactas conectadas por
um segmento flex´ıvel de cadeia polipept´ıdica. Essas unidades compactas, chamadas
dom´ınios, s˜ao formadas por 40 a 400 res´ıduos de amino´acidos. Dom´ınios s˜ao segmentos
estruturalmente independentes que tˆem fun¸c˜oes espec´ıficas. As pequenas prote´ınas
possuem, geralmente, apenas um dom´ınio.
A Figura 2.6 (retirada de (Guimar˜aes and Melo 2003)) mostra uma estrutura
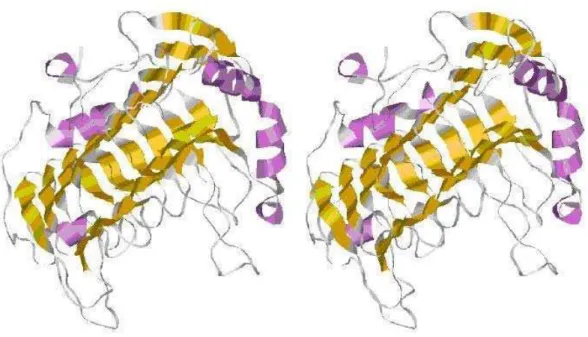
tridimen-sional de uma prote´ına da bact´eriaGram-negativae a Figura 2.7 (retirada de (Silva 1999)) mostra, em estereoscopia6, uma representa¸c˜ao do dom´ınio B1 da prote´ına G, cuja estrutura
prim´aria foi apresentada na Figura 2.1.
Muitas vezes ´e extremamente ´util visualizar as estruturas secund´aria e terci´aria no
mesmo modelo. Para isso, utilizam-se as representa¸c˜oes pict´oricas daα-h´elice e daβ-folhas, como as apresentadas nas Figuras 2.4 e 2.5 (Se¸c˜ao 2.2.2), respectivamente. O resultado da
visualiza¸c˜ao conjunta das duas estruturas em um ´unico modelo encontra-se ilustrado na
Figura 2.8 (retirada de (Silva 1999)), que representa o dom´ınio B1 da prote´ına G (cuja
estrutura terci´aria foi mostrada na Figura 2.6, em estereoscopia). Esta ´e a forma mais
comum de representa¸c˜ao das estruturas secund´aria e terci´aria de prote´ınas.
6
Figura 2.6: Estrutura terci´aria de uma prote´ına daGram-negativa
2.2. ESTRUTURAS DE PROTE´INA 17
Figura 2.8: Estruturas secund´aria e terci´aria, em estereoscopia
2.2.4
Estrutura Quatern´
aria
A estrutura quatern´aria consiste nas rela¸c˜oes e disposi¸c˜oes relativas das cadeias
polipep-t´ıdicas presentes nas prote´ınas multim´ericas, ou seja, prote´ınas que apresentam mais de
uma cadeia polipept´ıdica.
As cadeias polipept´ıdicas das prote´ınas multim´ericas s˜ao associadas por intera¸c˜oes
n˜ao-covalentes entre os grupos desprotegidos que n˜ao participam do dobramento de cada uma
das cadeias. O arranjo espacial que as subcadeias da prote´ına passam a ter ´e conhecido
como estrutura quatern´aria.
Dependendo da estrutura quatern´aria da prote´ına, ela pode ser classificada como fibrosa
(cadeias polipept´ıdicas dispostas ao longo de um eixo, formando uma estrutura alongada)
ou globular (cadeias polipept´ıdicas muito compactas, formando uma estrutura esf´erica).
A Figura 2.9 (retirada de (Silva 1999)) mostra a conforma¸c˜ao da hemoglobina humana,
Figura 2.9: Estrutura quatern´aria da hemoglobina humana
2.3
Classifica¸
c˜
ao Estrutural
A maioria dos trabalhos dedicados ao estudo da estrutura de prote´ınas empregam uma das
classifica¸c˜oes estruturais cl´assicas definidas por bancos de dados de prote´ınas mundialmente
difundidos. Muitas dessas pesquisas abordam a classifica¸c˜ao estrutural de prote´ınas
princi-palmente em classes e dobras, de acordo com o banco de dados SCOP (do inglˆes Structure Classification of Protein) (Murzin et al. 1995). Esta disserta¸c˜ao trata a classifica¸c˜ao de prote´ınas em classes estruturais, conforme o SCOP.
De acordo com a classifica¸c˜ao do banco de dados SCOP e com a estrutura espacial
apresentada pelas prote´ınas, elas podem ser catalogadas em sete diferentes classes,
repre-sentadas por all-α, all-β, α/β, α+β, small, multi-domain proteins (α and β) e membrane and cell surface proteins and peptides. As principais classes estruturais que est˜ao presentes em todos os trabalhos de classifica¸c˜ao estrutural de prote´ınas de acordo com o banco de
dados SCOP (all-α,all-β, α/β e α+β) ser˜ao apresentadas mais adiante.
Essa classifica¸c˜ao, como j´a comentado, ´e espec´ıfica do banco de dados SCOP. Portanto,
antes de se entrar em detalhes em rela¸c˜ao `as principais classes estruturais de prote´ınas, ser´a
2.3. CLASSIFICAC¸ ˜AO ESTRUTURAL 19
especificamente, sobre banco de dados de prote´ınas, dentre eles, o SCOP, cuja classifica¸c˜ao
hier´arquica ´e usada n˜ao apenas neste trabalho, mas na maioria dos trabalhos dedicados `a
classifica¸c˜ao estrutural de prote´ınas (Bologna and Appel 2002; Ding and Dubchak 2001; Pal
and Chakraborty 2003; Huang et al. 2003; Chinnasamy, Sung, and Mittal 2004).
2.3.1
Bancos de Dados Biol´
ogicos
De maneira geral, os bancos de dados biol´ogicos podem ser classificados em prim´arios,
se-cund´arios e compostos (Tsunoda 2005). Um banco de dados prim´ario cont´em informa¸c˜ao
sobre a seq¨uˆencia de amino´acidos (ou bases nucl´eicas) componentes da prote´ına (ou DNA).
Exemplos desses s˜ao: PDB,Swiss-ProteGenBank. Um banco de dados secund´ario cont´em dados derivados do banco de dados prim´ario acrescidos de outras informa¸c˜oes, como por
exemplo: seq¨uˆencia conservada, classifica¸c˜oes e s´ıtios ativos. Exemplos desses bancos s˜ao:
SCOP, CATH, PROSITE e eMOTIF. Os bancos de dados compostos, por sua vez,
ar-mazenam dados de v´arias fontes prim´arias diferentes.
O PDB (do inglˆes Protein Data Bank) (Abola et al. 1997) ´e um reposit´orio interna-cional de dados p´ublicos de estruturas tridimensionais de macromol´eculas biol´ogicas. Seu
conte´udo ´e originado de cristalografia de raios X e experimentos de RMN. Os principais objetivos do PDB s˜ao (Tsunoda 2005): permitir a localiza¸c˜ao de estruturas de interesse,
fornecer um portal para informa¸c˜oes adicionais sobre macromol´eculas na Internet, manter
uma base de dados atualizada para pesquisadores e divulgar gratuitamente informa¸c˜oes
sobre as macromol´eculas biol´ogicas.
Um dos bancos de dados secund´arios originados do PDB ´e o CATH (do inglˆes Class, Architecture, Topology, and Homologous superfamily) (Orengo et al. 1997), que permite a an´alise das estruturas prot´eicas atrav´es de uma classifica¸c˜ao hier´arquica em dom´ınios
realizada sobre a estrutura secund´aria. Esta base de dados faz a classifica¸c˜ao da estrutura
das prote´ınas em quatro n´ıveis: classe, arquitetura, topologia e super-fam´ılias. No n´ıvel
hier´arquico “classe”, as prote´ınas s˜ao classificadas em trˆes grandes categorias: mainly-α, mainly-β e α-β. Nesta ´ultima classe, s˜ao inclu´ıdas as estruturasα/β e α+β.
et al. 1995), que fornece uma descri¸c˜ao detalhada de rela¸c˜oes evolucion´arias e estruturais
entre todas as prote´ınas com estruturas conhecidas. O objetivo do SCOP ´e realizar uma
classifica¸c˜ao estrutural e n˜ao funcional. A classifica¸c˜ao de prote´ınas no SCOP ´e, em sua
maioria, executada manualmente ou de modo semi-autom´atico, tornando-se uma tarefa
complexa e que leva uma grande quantidade de tempo. Em sua ´ultima vers˜ao, SCOP
introduziu caracter´ısticas que permitiram um gerenciamento mais r´ıgido e um aumento do
n´umero de estruturas experimentais de projetos de genoma estrutural (Tsunoda 2005).
O banco de dados SCOP ´e dividido em quatro n´ıveis hier´arquicos: classe, dobra (fold),
super-fam´ılia e fam´ılia. No n´ıvel “classe”, as prote´ınas podem ser catalogadas, como j´a
mencionado anteriormente, em sete diferentes categorias: all-α, all-β, α/β, α+β, small, multi-domain proteins (αandβ) emembrane and cell surface proteins and peptides. Cada uma dessas classes apresenta um grande conjunto de dobras que as caracteriza e permite
classificar as prote´ınas com um maior n´ıvel de detalhes em rela¸c˜ao `as estruturas que
apre-sentam. As dobras, por sua vez, podem ser classificadas em diferentes super-fam´ılias e estas,
em v´arias fam´ılias.
2.3.2
Classe
all-
α
As prote´ınas pertencentes `a classe estruturalall-αs˜ao formadas quase exclusivamente porα -h´elices, com eventuaisβ-folhas localizadas na periferia da prote´ına. A hemoglobina humana, apresentada na Figura 2.9 (Se¸c˜ao 2.2.4), ´e um exemplo de uma prote´ına da classeall-α.
S˜ao classificadas como da classeall-αaproximadamente 220 dobras diferentes de prote´ı-nas, dentre as quais podem ser citadas: Globin-like, Cytochrome c, DNA-binding 3-helical bundle, 4-helical up and down bundle, 4-helical cytokines eEF hand-like.
2.3.3
Classe
all-
β
2.3. CLASSIFICAC¸ ˜AO ESTRUTURAL 21
Figura 2.10: Prote´ına da classeall-β, em estereoscopia
Como exemplo de dobras de prote´ınas que pertencem `a classeall-β, dentre as 144 exis-tentes, podem-se citar: Immunoglobin-likeβ-sandwich,Cupredoxin-like,Viral coat & capsid proteins, Concanavalin A like lectins/glucanases, SH3-like barrel, OB-fold eβ-trefoil.
2.3.4
Classe
α
/
β
As prote´ınas pertencentes `a classeα/βapresentam uma alternˆancia acentuada deα-h´elices e β-folhas ao longo da seq¨uˆencia, dispostas de tal forma que asβ-folhas, tipicamente paralelas, formam um aglomerado central rodeado porα-h´elices. A Figura 2.11 (retirada de (Silva 1999)) representa uma prote´ına da classe α/β.
Alguns exemplos de dobras de prote´ınas (dentre, aproximadamente, 135 dobras
Figura 2.11: Prote´ına da classeα/β, em estereoscopia
2.3.5
Classe
α
+
β
A classe α+β inclui as prote´ınas que, sendo formadas por um n´umero significativo de α-h´elices e β-folhas, n˜ao s˜ao dominadas por nenhum desses padr˜oes, nem apresentam a alternˆancia observada na classeα/β. O dom´ınio B1 da prote´ına G, apresentado na Figura 2.7 (Se¸c˜ao 2.2.3), pertence `a classeα+β.
Constituem exemplos de dobras de prote´ınas (dentre as, aproximadamente, 270 dobras
diferentes) pertencentes `a classeα+β: β-grasp(Ubiquitin-like),Ferrodoxin-like, Lysozyme-like, Cysteine proteinases,LysM domain eDodecin subunit-like.
2.4
An´
alise Computacional
Como j´a comentado no Cap´ıtulo 1, o grande desafio da Bioinform´atica em Proteˆomica diz
respeito `a predi¸c˜ao te´orica da estrutura 3D de prote´ınas. Do ponto de vista computacional, a
predi¸c˜ao da estrutura tridimensional de prote´ınas pode ser realizada de diferentes maneiras.
Dentre as quais, podem ser citadas:
• Homologia (Higgins and Taylor 2001): conhecida tamb´em por modelagem
compa-rativa, trata-se da compara¸c˜ao entre seq¨uencias de prote´ınas. Este m´etodo usa uma
2.4. AN ´ALISE COMPUTACIONAL 23
prote´ına com seq¨uˆencia similar. Estas duas prote´ınas com alto n´ıvel de similaridade
entre suas seq¨uencias s˜ao consideradas hom´ologas, ou seja, prote´ınas que derivam de
uma prote´ına ancestral comum e que sofreram mudan¸cas devido a fatores relacionados
`
a evolu¸c˜ao. Elas podem exercer a mesma fun¸c˜ao, ainda que encontradas em esp´ecies
diferentes, ou podem exercer fun¸c˜oes diferentes, embora mantenham informa¸c˜oes
so-bre o relacionamento original.
• Threading (Akutsu et al. 2003): t´ecnica baseada na compara¸c˜ao de prote´ına, para a
qual se deseja obter a estrutura, com modelos descritivos dos dobramentos de prote´ınas
hom´ologas. Nesses modelos, s˜ao descritas, por exemplo: a distˆancia entre os res´ıduos
de amino´acidos, a estrutura secund´aria de cada fragmento e as caracter´ısticas
f´ısico-qu´ımicas de cada res´ıduo de amino´acido.
• Ab Initio (Bonneau et al. 2001): m´etodo para a previs˜ao da estrutura 3D de uma
prote´ına sem a utiliza¸c˜ao de informa¸c˜ao estrutural de outras prote´ınas para
com-para¸c˜ao. Os poucos programas atualmente existentes que utilizam este m´etodo tˆem
muito a melhorar para que possam ser aceitos pela comunidade cient´ıfica. Por esta
raz˜ao, prever uma estrutura tridimensional de prote´ına na total ausˆencia de homologia
continua sendo um problema considerado sem solu¸c˜ao.
Esses m´etodos destinam-se a encontrar diretamente a estrutura tridimensional da
prote´ı-na. No entanto, devido a elevada complexidade desta tarefa, ela vem sendo, normalmente,
realizada em etapas:
• 1a¯ etapa: o passo inicial na predi¸c˜ao da estrutura 3D de uma prote´ına ´e, em geral,
a predi¸c˜ao da estrutura secund´aria, ou seja, a localiza¸c˜ao na seq¨uˆencia prot´eica de
subestruturas comuns, como as α-h´elices eβ-folhas (Pollastri et al. 2002).
• 2a¯ etapa: outro passo importante ´e a descoberta de semelhan¸cas entre conforma¸c˜oes
prot´eicas (classifica¸c˜ao estrutural de prote´ınas). Tais similaridades podem ser
eviden-ciadas atrav´es da busca por padr˜oes estruturais que podem, por exemplo, caracterizar
O foco deste trabalho ´e a abordagem da 2a¯ etapa, ou seja, a an´alise estrutural das
prote´ınas para se buscar padr˜oes comuns na estrutura prot´eica, possibilitando a classifica¸c˜ao
da prote´ına no grupo com um padr˜ao caracter´ıstico. Nesta disserta¸c˜ao, isso ´e feito atrav´es
da aplica¸c˜ao de m´etodos de AM.
Como n˜ao existe uma rela¸c˜ao direta entre a seq¨uˆencia e a estrutura espacial da prote´ına,
muita aten¸c˜ao tem-se dado a t´ecnicas de AM, capazes de aprender automaticamente a partir
de grandes volumes de dados, para classificar as prote´ınas a partir de propriedades (obtidas
das estruturas prim´arias) previamente conhecidas. Recentemente, as ferramentas de AM
vˆem sendo usadas pela maior parte das pesquisas para a classifica¸c˜ao de prote´ınas atrav´es
da an´alise estrutural (Tan, Gilbert, and Deville 2003).
No tipo de abordagem apresentado na 2a¯ etapa, busca-se encontrar, freq¨uentemente,
representa¸c˜oes de padr˜oes de dobramento que facilitem a determina¸c˜ao da rela¸c˜ao entre as
topologias e fun¸c˜oes das prote´ınas (Guimar˜aes and Melo 2003). A solu¸c˜ao do problema de
classifica¸c˜ao estrutural de prote´ınas ´e feita normalmente em dois n´ıveis diferentes, tendo em
vista que dobras de prote´ınas s˜ao uma subclassifica¸c˜ao das classes estruturais, e a maioria
dos trabalhos de classifica¸c˜ao estrutural se restringe a essas duas poss´ıveis categorias (classe
e dobra) (Okun 2004).
No n´ıvel um, no reconhecimento de dobras de prote´ınas, uma prote´ına ´e classificada
em uma das poss´ıveis dobras, enquanto que no n´ıvel dois, no reconhecimento de classes
estruturais de prote´ınas, ela ´e atribu´ıda a uma das classes estruturais em que as dobras s˜ao
catalogadas. A classifica¸c˜ao pode ser feita em um desses n´ıveis ou em ambos os n´ıveis. Neste
´
ultimo caso, a classifica¸c˜ao pode ou n˜ao ser hier´arquica. Sendo hier´arquica, os classificadores
de n´ıvel um empregam as sa´ıdas dos classificadores de n´ıvel dois. Isso significa que os
classificadores de n´ıvel um n˜ao s˜ao treinados para predizer todas as dobras poss´ıveis de
prote´ınas, mas somente aquelas que pertencem `a classe estrutural predita no segundo n´ıvel
de classifica¸c˜ao (Huang, Lin, and Pal 2003).
Cada um desses classificadores pode ser obtido com a aplica¸c˜ao de um dos v´arios
2.5. TRABALHOS RELACIONADOS 25
das tendˆencias atuais na classifica¸c˜ao estrutural de prote´ınas ´e o uso de Redes Neurais
Arti-ficiais (RNA) e M´aquinas de Vetores Suporte (SVM) como algoritmos de aprendizado (Ding
and Dubchak 2001; Cai et al. 2001; Huang et al. 2003; Markowetz, Edler, and Vingron 2003;
Chung et al. 2003; Pal and Chakraborty 2003). Al´em disso, esses classificadores podem ser
resultado da combina¸c˜ao de m classificadores independentes, usando ou n˜ao o mesmo al-goritmo de AM, originando os chamados sistemas de multiclassifica¸c˜ao, que ´e atualmente
uma das principais ´areas de pesquisa em AM (Tan and Gilbert 2003).
A classifica¸c˜ao autom´atica de prote´ınas em dobras ou classes estruturais com t´ecnicas
de AM ´e feita atrav´es de propriedades derivadas da estrutura prim´aria das prote´ınas.
Nor-malmente, s˜ao usadas as seguintes seis propriedades extra´ıdas de suas seq¨uˆencias (Ding and
Dubchak 2001): composi¸c˜ao de amino´acidos, predi¸c˜ao da estrutura secund´aria, polaridade,
volume de van der Walls, polarizabilidade e hidrofobicidade. Cada propriedade ´e
represen-tada por um vetor denatributos cont´ınuos (n≥1). Uma vez que a eficiˆencia de t´ecnicas de AM depende, em grande parte, da qualidade dos dados considerados durante o treinamento,
h´a pesquisas dedicadas ao estudo para se determinar quais das propriedades anteriormente
citadas favorecem mais o desempenho dos m´etodos utilizados (Huang, Lin, and Pal 2003;
Pal and Chakraborty 2003; Huang et al. 2003).
2.5
Trabalhos Relacionados
Levitt and Chothia (1976) foram uns dos primeiros pesquisadores a realizar uma
classi-fica¸c˜ao de prote´ınas baseada em sua estrutura. Com base na observa¸c˜ao visual da sucess˜ao
de padr˜oes estruturais na cadeia polipept´ıdica, dividiram um conjunto de prote´ınas
globu-lares em quatro classes estruturais distintas, com designa¸c˜oes semelhantes, mas defini¸c˜oes
ligeiramente diferentes das que se tem hoje. Com o passar do tempo, esse assunto foi se
tornando foco da aten¸c˜ao de muitos pesquisadores (Nakashima, Nishikawa, and Ooi 1986;
Chou 1989; Kneller, Cohen, and Langridge 1990; Mitchie, Oregon, and Thomton 1996),
que contribu´ıram para a forma¸c˜ao das bases de dados que atualmente s˜ao usadas para a
Duas d´ecadas depois, Dubchak et al. (1999) formaram uma base de dados, a partir dos
bancos de dados PDB e SCOP, para a classifica¸c˜ao estrutural de prote´ınas, e empregaram
RNA na classifica¸c˜ao de prote´ınas em dobras. Ding and Dubchak (2001) realizaram uma
modifica¸c˜ao na base de dados de Dubchak et al. (1999) e formaram uma nova base de dados,
que se popularizou e passou a ser usada pela maioria dos trabalhos seguintes relacionados ao
problema de classifica¸c˜ao estrutural de prote´ınas (dispon´ıvel em http://www.nersc.gov/
~cding/protein/). Esta base de dados foi formada com 698 prote´ınas, representadas por
vetores de propriedades apresentando um total de 126 atributos. Nesta base, duas prote´ınas
n˜ao tˆem mais do que 35% de similaridade entre suas seq¨uˆencias.
Assim como Dubchak et al. (1999), v´arios outros pesquisadores consideraram o problema
de classifica¸c˜ao estrutural de prote´ınas apenas no n´ıvel um, no reconhecimento de dobras de
prote´ınas. Ding and Dubchak (2001) classificaram as prote´ınas em 27 dobras usando SVM e
RNA com trˆes m´etodos de multiclassifica¸c˜ao de duas classes (“um-contra-outros”, “unique
-um-contra-outros” e “todos-contra-todos”). Nesse trabalho, foi utilizado um grande n´umero
de classificadores e introduzido o m´etodo SVM ao problema de classifica¸c˜ao de
prote´ı-nas. Bologna and Appel (2002) usaram um sistema multiclassificador de redesPerceptrons Multi-Layer Interpretable Discretized four-layer (DIMLP), em que cada rede, ao contr´ario de Ding and Dubchak (2001), aprende todas as dobras de prote´ınas simultaneamente. Para
combinar as sa´ıdas das redes, os sistemas de multiclassifica¸c˜ao Bagging e Arcing foram empregados. Edler, Grassmann, and Suhai (2001), por sua vez, com uma base de dados
diferente da utilizada nos trabalhos anteriores, contendo apenas 268 prote´ınas, realizaram
um estudo estat´ıstico baseado em regress˜ao log´ıstica e modelos aditivos na predi¸c˜ao de
dobras de prote´ınas.
Diferentemente desses pesquisadores, outros trabalhos se detiveram `a classifica¸c˜ao de
prote´ınas em classes estruturais, ou seja, ao n´ıvel dois do problema de classifica¸c˜ao estrutural
de prote´ınas. Dentre eles, destaca-se Tan, Gilbert, and Deville (2003), que motivados pela
discuss˜ao apresentada no trabalho de Ding and Dubchak (2001) sobre o desempenho dos
classificadores em fun¸c˜ao do desbalanceamento de classes e da elevada quantidade de
2.5. TRABALHOS RELACIONADOS 27
Knowledge for Imbalance Sample Sets) e o aplicaram a uma vers˜ao modificada da base de dados de Ding and Dubchak (2001) com o objetivo de classificar as prote´ınas em cinco
classes estruturais (all-α, all-β, α/β, α+β esmall). A id´eia do eKISS ´e combinar, atrav´es de regras, a sa´ıda dos classificadores gerados pelo PART (Frank and Witten 1998), com as
abordagens “um-contra-outros” e “todos-contra-todos”. Eles compararam os resultados do
eKISS com os do PART e verificaram uma boa aplicabilidade do sistema proposto.
Tamb´em classificando as prote´ınas apenas em classes estruturais, podem ser citados os
trabalhos de: Cai et al. (2001), que usaram SVM para classificar as prote´ınas nas quatro
classes estruturais principais (all-α, all-β, α/β eα+β) e compararam os resultados obtidos com os da RNA de um de seus trabalhos anteriores (Cai and Zhou 2000); e Markowetz, Edler,
and Vingron (2003), que usaram SVM com kernel gaussiano e v´arioskernels polinomiais para classificar as prote´ınas nas mesmas classes. Markowetz, Edler, and Vingron (2003)
usaram, no entanto, uma base de dados diferente, elaborada por eles pr´oprios (dispon´ıvel
em http://www.dkfz.de/biostatistics/protein/gsme97.html).
A maioria das pesquisas abordam, na verdade, a classifica¸c˜ao estrutural de prote´ınas
em sua plenitude, isto ´e, nos dois n´ıveis em que o problema ´e comumente dividido. Pal
and Chakraborty (2003) realizaram uma classifica¸c˜ao independente em cada um dos dois
n´ıveis de classifica¸c˜ao. Treinaram RNA do tipo MLP e Radial Basis Function Network (RBFN) com uma base de dados que continha vetores com 400 atributos, obtidos apenas
da propriedade de hidrofobicidade dos amino´acidos. Tamb´em preocupados em usar na base
de dados apenas as propriedades que mais favorecem o desempenho dos m´etodos de AM
(do total de seis propriedades normalmente utilizadas), Huang et al. (2003) usaram uma
gate functionpara selecionar as propriedades prot´eicas consideradas relevantes que deveriam integrar a base de dados para o aprendizado dasgated RNAempregadas. Tamb´em foi usada, assim como no trabalho de Pal and Chakraborty (2003), uma classifica¸c˜ao independente em
cada n´ıvel.
Chinnasamy, Sung, and Mittal (2004) realizaram um pr´e-processamento na base de
dados de Markowetz, Edler, and Vingron (2003) e de Ding and Dubchak (2001) atrav´es
formadas pela sele¸c˜ao apenas das propriedades composi¸c˜ao de amino´acidos e predi¸c˜ao da
estrutura secund´aria. Apresentaram o sistema BAYESPROT, que se baseia no uso das
Tree-Augmented Networks (TAN), criadas a partir da teoria da aprendizagem bayesiana, para a classifica¸c˜ao estrutural de prote´ınas. Assim como Okun (2004), que empregou o
algoritmoK-Local Hyperplane Distance Nearest Neighbor algorithm(HKNN), duas tarefas foram realizadas independentemente: 1) a classifica¸c˜ao de prote´ınas em 27 dobras mais
significativas e 2) a classifica¸c˜ao de prote´ınas nas quatro classes estruturais principais.
De modo um pouco diferente das abordagens anteriores, Huang, Lin, and Pal (2003) e
Chung et al. (2003) realizaram uma classifica¸c˜ao hier´arquica entre os n´ıveis um e dois da
classifica¸c˜ao estrutural de prote´ınas. Huang, Lin, and Pal (2003) propuseram uma Hierar-chical Learning Architecture (HLA). A arquitetura proposta permite que tanto o m´etodo RNA como o SVM sejam empregados. Chung et al. (2003) usaram RNA e SVM como
clas-sificadores de um sistema de dois n´ıveis. Modelos simples de RNA (MLP, RBFN eGeneral Regression Neural Network - GRNN) com uma ´unica camada escondida foram usados.
Discuss˜oes
Pode-se observar que a maioria dos trabalhos faz uso da base de dados criada por Ding
and Dubchak (2001), com algumas exce¸c˜oes em rela¸c˜ao `a dimensionalidade dos vetores
de atributos e `a quantidade de prote´ınas empregadas. Tan, Gilbert, and Deville (2003),
por sua vez, sugeriram uma mudan¸ca maior nesta base de dados, realizando nela alguns
aperfei¸coamentos (maiores detalhes s˜ao expostos na Se¸c˜ao 4.1). Nesta disserta¸c˜ao, ´e usada
a base de dados de Tan, Gilbert, and Deville (2003), por ela apresentar, aparentemente,
menos imprecis˜oes.
As t´ecnicas de AM est˜ao presentes em quase todas as cita¸c˜oes encontradas na literatura
relativas `a classifica¸c˜ao estrutural de prote´ınas. Dentre os trabalhos encontrados, ressalta-se
a maior freq¨uˆencia do emprego de RNA e SVM, sendo, inclusive, utilizados na composi¸c˜ao
de sistemas de multiclassifica¸c˜ao homogˆenea. O ´unico trabalho encontrado que faz uso da
multiclassifica¸c˜ao heterogˆenea ´e o de Tan, Gilbert, and Deville (2003), que combina os
2.5. TRABALHOS RELACIONADOS 29
de AM, sem se restringir a RNA e SVM. Ao contr´ario, busca-se empregar algoritmos de
AM variados e at´e mesmo de que n˜ao se tem conhecimento de j´a terem sido usados na
solu¸c˜ao deste problema, como ´Arvore de Decis˜ao (AD) e diferentes m´etodos de
multiclassi-fica¸c˜ao heterogˆenea. Outro estudo feito neste trabalho, sem referˆencia anterior, diz respeito
`a an´alise da aplica¸c˜ao de t´ecnicas de pr´e-processamento de dados, direcionadas `a redu¸c˜ao
do desbalanceamento da quantidade de instˆancias nas classes, com o intuito de aumentar o
desempenho dos m´etodos de AM na classifica¸c˜ao estrutural de prote´ınas.
Como mencionado anteriormente, o problema pode ser abordado em dois n´ıveis
diferen-tes. No n´ıvel um, as prote´ınas s˜ao classificadas em dobras e no outro, em classes estruturais.
A maioria dos trabalhos considerou, em seus experimentos, esses dois n´ıveis citados, seja
atrav´es de uma classifica¸c˜ao hier´arquica ou independente entre os n´ıveis. Por´em, alguns
pesquisadores, como Ding and Dubchak (2001) e Markowetz, Edler, and Vingron (2003),
focaram suas pesquisas apenas no n´ıvel um e outros, como Cai et al. (2001) e Tan, Gilbert,
and Deville (2003) apenas no n´ıvel dois. Neste trabalho, assim como nos ´ultimos citados,
a pesquisa ´e restrita ao n´ıvel dois (reconhecimento de classes estruturais), com o intuito
de abordar mais profundamente este filtro de classifica¸c˜ao e garantir poss´ıveis melhores
resultados em trabalhos futuros de classifica¸c˜ao de prote´ınas no n´ıvel um.
A acur´acia global dos classificadores foi usada como m´etrica em quase todos os trabalhos,
a menos de Tan, Gilbert, and Deville (2003) que apresentou os resultados em termos de F-measure (Rijsbergen 1979) e de curvas ROC (Provost, Fawcett, and Kohavi 1998). Apesar de a mesma m´etrica ser usada pela maioria das pesquisas, houve uma grande variabilidade
na metodologia aplicada durante a realiza¸c˜ao dos experimentos, como por exemplo: alguns
trabalhos empregaram, como forma de valida¸c˜ao, o k-fold cross validation (com valores diferentes parak) enquanto que outros, oholdout. Por causa dessas diferentes metodologias empregadas, n˜ao ´e poss´ıvel realizar uma justa compara¸c˜ao entre os resultados obtidos nos
Aprendizado de M´
aquina
Este cap´ıtulo descreve caracter´ısticas e funcionalidades b´asicas de m´etodos computacionais
utilizados nesta disserta¸c˜ao. A Se¸c˜ao 3.1 apresenta conceitos gerais de Aprendizado de
M´aquina (AM). A Se¸c˜ao 3.2 descreve os diferentes sistemas de multiclassifica¸c˜ao homogˆenea
(Bagging eBoosting) e heterogˆenea (Voting,Stacking eStackingC) empregados neste tra-balho. Na Se¸c˜ao 3.3, ´e discutida a importˆancia de pr´e-processamento de dados e s˜ao descritas
as principais t´ecnicas de under-sampling.
3.1
Introdu¸
c˜
ao
Aprendizado de M´aquina (AM) estuda como construir algoritmos que melhoram o pr´oprio
desempenho em alguma tarefa por meio de experiˆencia (Mitchell 1997). Aprender, nesse
contexto, pode ser definido (para situa¸c˜oes em que o desempenho em alguma tarefa pode
ser medido) como a seguir: diz-se que um programa computacional aprende a partir da
experiˆenciaE, em rela¸c˜ao a uma classe de tarefasT, com medida de desempenhoD, se seu desempenho nas tarefasT, medida por D, melhora com a experiˆencia E (Mitchell 1997).
A maioria dos m´etodos de AM adquirem experiˆencia estritamente a partir dos dados
conhecidos do problema. Assim, melhor ser´a o desempenho dos m´etodos de AM quanto
melhor for a qualidade dos dados (Batista 2003). Devido aos dados dispon´ıveis, diversos
aspectos podem influenciar na performance de um sistema de aprendizado. Em bases de
dados reais, dois desses aspectos est˜ao relacionados com a presen¸ca de valores desconhecidos,
3.1. INTRODUC¸ ˜AO 31
pois distor¸c˜oes podem ser introduzidas no conhecimento induzido, e com a diferen¸ca entre o
n´umero de instˆancias que pertencem a diferentes classes, uma vez que quando esta diferen¸ca
´e grande, sistemas de AM podem ter dificuldade em aprender o conceito relacionado a classe
minorit´aria (Batista, Prati, and Monard 2004). Tais problemas podem ser minimizados, por
exemplo, com a aplica¸c˜ao de t´ecnicas de pr´e-processamento de dados, vistas na Se¸c˜ao 3.3.
3.1.1
Paradigmas de AM
T´ecnicas de AM podem ser divididas em aprendizado supervisionado e aprendizado n˜ao
supervisionado, de acordo com os dados dispon´ıveis para a realiza¸c˜ao do processo de indu¸c˜ao.
No aprendizado supervisionado, o indutor recebe um conjunto de instˆancias, cada
instˆan-cia sendo formada por um conjunto de atributos de entrada e um conjunto de atributos
de sa´ıda (r´otulos) antes do processo de aprendizado (Souto et al. 2003). O objetivo do
algoritmo de aprendizado ´e construir um classificador que possa determinar corretamente a
classe de novas instˆancias ainda n˜ao rotuladas. Como exemplos de t´ecnicas de aprendizado
supervisionado podem ser citados, dentre outros: redes neurais artificiais do tipomultilayer perceptroncombackpropagation, m´aquinas de vetores de suporte e ´arvores de decis˜ao.
Por outro lado, o aprendizado n˜ao supervisionado ´e realizado quando, para cada instˆ
an-cia, apenas os atributos de entrada est˜ao dispon´ıveis. Esse tipo de aprendizado ´e utilizado
quando o objetivo ´e encontrar, em um conjunto de dados, padr˜oes ou tendˆencias
(agru-pamentos ou clusters) que auxiliem o entendimento desses dados (Costa 1999). Como exemplos de t´ecnicas de aprendizado n˜ao supervisionado podem ser citados, dentre outros:
redes neurais do tipo mapa auto-organiz´aveis (SOM),k-m´edias e agrupamento hier´arquico. Para o reconhecimento de classes estruturais de prote´ınas, t´ecnicas de aprendizado
su-pervisionado s˜ao utilizadas neste trabalho. Na solu¸c˜ao deste problema, os indutores recebem
como entrada, no processo de aprendizado, instˆancias formadas por um conjunto de vetores
de atributos, derivados da estrutura prim´aria da prote´ına, e rotuladas pela classe estrutural
a que se referem.
Foram selecionados, para serem empregadas na gera¸c˜ao dos classificadores base dos