FERNANDA MARQUES DA CUNHA
PEPTÍ DEOS I NTRACELULARES COMO NOVOS
MODULADORES DA TRANSDUÇÃO DE SI NAL DE
RECEPTORES ACOPLADOS À PROTEÍ NA G
Tese apresentada à Universidade
Federal de São Paulo – Escola Paulista
de Medicina para obtenção do título de
Doutor em Ciências
São Paulo
Cunha, Fernanda Marques
Peptídeos intracelulares como novos moduladores da transdução de sinal de receptores acoplados à proteína G. / Fernanda Marques da Cunha.-- São Paulo, 2008.
Tese elaborada no Departamento de Biologia
Celular e Desenvolvimento durante o curso de pós-graduação em Biologia Molecular e
apresentada à Universidade Federal de São
Paulo – Escola Paulista de Medicina como requisito parcial para obtenção do título de
Doutor em Ciências.
“O correr da vida embrulha tudo, a vida é
assim: esquenta e esfria, aperta e daí
afrouxa, sossega e depois desinquieta. O que ela quer da gente é coragem.”
AGRADECI MENTOS
Aos meus queridos avós, José e Maria Marques cujo carinho e incentivo são tão
fortes que irradiam até mim.
Ao meu orientador Emer pela oportunidade de desenvolver um trabalho muito interessante e extremamente desafiador com as melhores ferramentas.
Aos meus colegas e amigos Denise, Lilian, Leandro e Sayami, não só pelas
discussões científicas e não científicas e ajuda de todos os tipos, mas também pela
cumplicidade e por terem dividido comigo risos e lágrimas, alegrias e angústias. Sempre sorrirei ao lembrar de vocês.
À Grazi e Maurício, minha família em São Paulo.
Ao Alex, meu melhor companheiro de todas as horas, pelo carinho, cuidado e
paciência.
Às novas colegas Carla e Camila pelo agradável convívio.
Ao Zeca e ao Fio, pelos momentos de descontração.
Às Profs. Regina P. Markus e Zulma F. Ferreira pelo carinho com que me
receberam em seu laboratório e pela orientação nos ensaios de microfisiometria.
À Andréa S. Heimann pelo auxílio nos ensaios com as colunas de afinidade.
Ao Clécio pelos ensaios de espectrometria de massa.
A todos aqueles que direta ou indiretamente contribuíram para a realização deste
Í NDI CE
LI STA DE FI GURAS... xi
LI STA DE TABELAS... xiii
LI STA DE ABREVI ATURAS... xiv
ABREVI ATURA DOS AMI NOÁCI DOS... xvi
RESUMO... xvii
1 I NTRODUÇÃO... 01
1.1 Módulos de interação... 01
1.2 Efeito de peptídeos intracelulares desenhados... 07
1.3 Geração de peptídeos pela célula... 08
1.4 Geração de peptídeos intracelulares... 08
1.5 Endo-oligopeptidase EC3.4.24.15... 11
1.6 Peptídeos intracelulares e sinalização... 17
2 OBJETI VO... 19
3 MATERI AI S E MÉTODOS... 20
3.1 I solamento de peptídeos de cérebro de rato... 20
3.6 Síntese dos peptídeos escolhidos acoplados ao TAT por pontes
dissulfeto... 24
3.7 Cultura de células... 24
3.8 Monitoramento da entrada do peptídeo na célula e do desligamento do TAT... 24
3.9 Ensaio microfisiométrico... 26
3.10 Ensaio de gene repórter: Ensaio da luciferase... 27
3.10.1 Construção do plasmídeo reporter... 28
3.10.1.1 I nserção do CRE/ VI P no pGL3 básico... 28
3.10.1.2 Clonagem dos MREs no pGL3 + CRE... 29
3.10.2 Transfecção celular e ensaio da luciferase... 31
3.11 Fosforilação dos peptídeos pela PKC... 32
3.12 Aumento da expressão da EP24.15 em células CHO-S e HEK293 e ensaio da luciferase... 32
3.13 Acoplamento dos peptídeos a colunas de purificação por afinidade e captura de ligantes... 33
3.14 Análise estatística... 35
4 RESULTADOS... 36
4.1 I dentificação de novos peptídeos de cérebro de rato... 36
4.2 Ensaios cinéticos com a EP24.15 recombinante ativa e determinação dos sítios de hidrólise... 38
citoplasmáticos de cérebro de ratos, células CHO-S e HEK293... 42
4.4 Monitoramento da entrada do peptídeo na célula e do desligamento do TAT... 42
4.5 Ensaio microfisiométrico... 45
4.6 Ensaio de gene repórter: Ensaio da luciferase... 47
4.6.1 Construção do plasmídeo reporter... 47
4.6.2 Ensaio da luciferase... 48
4.7 Fosforilação dos peptídeos pela PKC... 52
4.8 Aumento da expressão da EP24.15 em células CHO-S e HEK293 e ensaio da luciferase... 54
4.9 Acoplamento dos peptídeos a colunas de purificação por afinidade e captura de ligantes... 55
5 DI SCUSSÃO... 58
6 CONCLUSÕES... 69
7 REFERÊNCI AS BI BLI OGRÁFI CAS... 70
ANEXO I – Proteínas identificadas no eluato das colunas de afinidade
ANEXO I I - Artigos publicados durante o período de doutoramento
LI STA DE FI GURAS
FI GURA 1 Domínios de interação e organização protéica... 02
FIGURA 2 Natureza modular das proteínas envolvidas na sinalização... 03
FI GURA 3 Famílias de domínios de interação e seus ligantes... 05
FI GURA 4 Mecanismo de troca tiol-dissulfeto proposta para a multimerização da EP24.15 através da S-glutationilação... 13
FIGURA 5 Distribuição dos sítios encontrados em 39 peptídeos isolados com a EP24.15 inativa... 17
FI GURA 6 Esquema temporal do ensaio de microfisiometria... 27
FI GURA 7 Vetor reporter pGL3-MRE-CRE... 28
FI GURA 8 Vetores usados no ensaio da luciferase... 31
FI GURA 9 Esquema temporal do ensaio da luciferase... 32
FI GURA 10 Cinética enzimática da EP24.15 na presença dos peptídeos estudados... 39
FI GURA 11 Cromatogramas das análises por HPLC do produto de incubação dos peptídeos com a EP24.15... 40
FI GURA 12 Padrão de clivagem dos peptídeos 5A e FE2 pela EP24.15... 41
FI GURA 13 Cromatogramas das corridas de HPLC... 44
FI GURA 14 Ensaios microfisiométricos... 46
FI GURA 17 Ensaio da luciferase... 52
FIGURA 18 Fosforilação in vitro dos peptídeos pela PKC... 53 FIGURA 19 Efeito do aumento da expressão da EP24.15 sobre expressão de
LI STA DE TABELAS
TABELA 1 Peptídeos extraídos de cérebro de ratos com a metodologia de
captura pela EP24.15 inativa e identificados por espectrometria de massa... 36
TABELA 2 Peptídeos escolhidos para síntese e análise... 37
TABELA 3 Constantes de inibição da EP24.15 calculadas para os
peptídeos estudados... 38 TABELA 4 Concentração intracelular de peptídeos nas células CHO-S... 43
TABELA 5 Proteínas identificadas nos eluatos das colunas e que possuem
ABREVI ATURAS
AMPc monofosfato cíclico de adenosina
ANG I I angiotensina I I
AT1 receptor AT1 para a angiotensina
ATP trifosfato de adenosina
CHO-S células de ovário de hamster chinês
DNA ácido desoxirribonucléico
EP24.15 endo-oligopeptidase E.C. 3.4.24.15
GDP difosfato de guanosina
GTP trifosfato de guanosina
GPCR receptor acoplado à proteína G
HEK293 células de rim embrionário humano
HI V vírus da imunodeficiência humana
HPLC cromatografia líquida de alta performance
HSP65 heat shock protein 65
JNK c-Jun N-terminal kinase
PTB ligação a fosfotirosina
QFS quenched fluorescent substrate
RNA ácido ribonucléico
SH2 região de homologia à Src 2
SH3 região de homologia à Src 3
CKI caseína quinase I
CKI I caseína quinase I I
ABREVI ATURA DOS AMI NOÁCI DOS
Alanina Ala A
Asparagina Asn N
Ácido aspártico Asp D
Arginina Arg R
Cisteína Cys C
Fenilalanina Phe F
Glicina Gly G
Glutamina Gln Q
Ácido glutâmico Glu E
Histidina His H
I soleucina I le I
Leucina Leu L
Lisina Lys K
Metionina Met M
Prolina Pro P
Serina Ser S
Tirosina Tyr Y
Treonina Thr T
Triptofano Trp W
RESUMO
A degradação de proteínas pelo sistema ubiquitina-proteassoma gera uma
grande quantidade de oligopeptídeos dentro das células. Para investigar possíveis
efeitos desses oligopeptídeos, alguns deles foram isolados do cérebro de rato,
sintetizados acoplados à seqüência peptídica TAT através de pontes dissulfeto,
sendo então analisados nas vias de transdução de sinal de receptores acoplados à
proteína G. A mistura contendo os quatro peptídeos analisados (20-80 µM) inibiu de forma significativa o aumento da taxa de acidificação do meio extracelular
induzido pela angiotensina I I em células CHO-S que expressam o receptor AT1
(CHO-S-AT1). Adicionalmente, tanto sozinhos quanto em mistura, estes peptídeos
aumentaram a transcrição do gene repórter da luciferase induzida pela
angiotensina I I em células CHO-S-AT1, assim como aquela induzida pelo
isoproterenol em células HEK293. Os peptídeos sem TAT, incapazes de atravessar
a membrana das células, não alteraram as respostas à estimulação dos receptores,
sugerindo um efeito intracelular dos peptídeos nas cascatas de transdução de
sinal. Além disso, todos os peptídeos estudados inibiram competitivamente a
degradação de um substrato sintético da oligopeptidase EP24.15 in vitro. O aumento da expressão da EP24.15 em células CHO-S e HEK293 foi suficiente para
reduzir a atividade do gene repórter luciferase induzida pela angiotensina I I ou
pelo isoproterenol. Finalmente, a utilização dos peptídeos como “isca” em colunas
de afinidade revelou que diversas proteínas envolvidas na sinalização de
adaptina A-alfa e a dinamina-1. Estes resultados sugerem que antes de serem
completamente degradados, os peptídeos intracelulares semelhantes àqueles
gerados pelo proteassoma podem afetar ativamente a sinalização intracelular,
Introdução
1 I NTRODUÇÃO
Com a elucidação do genoma de diversos organismos, constatou-se que,
contrariamente ao que se acreditava na era pré-genômica, a complexidade dos
organismos não era diretamente relacionada ao número de genes, mas sim
definida pela interação dos produtos destes genes no tempo e no espaço (GAVI N
et al., 2002; STUMPF et al., 2008). De fato, o grande desafio da biologia pós-genômica tem sido entender como a informação genética resulta na ação
orquestrada dos produtos gênicos nos âmbitos temporal e espacial. I sto é refletido
pelo fato do número de doenças humanas exceder o número de genes do genoma
(ROSES et al., 2000). Além disso, o número total de genes em humanos não difere substancialmente do número de genes do verme nematóide Caenorhabtidis elegans, reforçando a idéia de que a complexidade reside na combinação contextual dos produtos gênicos (GAVI N et al., 2002).
1.1 Módulos de interação
As proteínas raramente agem sozinhas. Na maior parte das vezes, elas
interagem com outras proteínas, DNA, RNA ou ainda moléculas de outra natureza
para orquestrar uma tarefa celular determinada. Estes complexos proteicos
representam mais do que a soma das partes, adquirindo uma nova função (GAVI N
Introdução
Figura 1. Domínios de interação e organização protéica. Um vasto repertório de domínios de interação que contribuem para as propriedades bioquímicas e biofísicas das proteínas foi caracterizado. Os domínios são usados de forma combinatória para formar proteínas com diferentes funções bioquímicas. De forma semelhante, as proteínas são usadas para a formação de complexos
com diversos efeitos biológicos. Adapatado de Gavin et al. (2003).
As proteínas regulatórias são construídas de forma modular (Fig. 2), onde
cada módulo corresponde a um domínio de interação ou dotado de atividade
enzimática, possibilitando a previsão das funções biológicas de uma dada proteína
com base nos domínios que a compõem (COPLEY et al., 2002). Os domínios de interação possuem seqüências de aminoácido conservadas (COPLEY et al., 2002) e encontram-se presentes em centenas de cópias no proteoma humano. De fato, os
Domínios, ~ 20 Å
Proteínas, ~ 50 - 100 Å
Complexos proteicos, ~ 500 - 1000 Å Domínios, ~ 20 Å
Proteínas, ~ 50 - 100 Å
Introdução
Figura 2. Natureza modular das proteínas envolvidas na sinalização. Membros de várias famílias de proteínas e a organização dos domínios que as compõem. CH- homologia à calponina, C1- região conservada 1 da PKC (ligação ao PMA), C2- região conservada 2 da PKC (ligação aos fosfolipídeos),
DNA BD- domínio de ligação ao DNA, EF – mãos EF, FCH- domínio de homologia Fes/ CI P4, 4H- four
helix bundle, GAP- domínio ativador de GTPase, GEF- fator de troca GDP-GTP, PH- domínio de homologia à plecstrina, PLC- fosfolipase C, PTB- domínio de ligação à fosfotirosina, PTPc/ DSPc- tirosina fosfatase, SAM- sítio estéril alfa, SH2- domínio de homologia à Src 2, SH3- domínio de homologia à Src 3, SOCS supressor da sinalição de citocina, STAT- transdutor de sinal e ativador da transcrição, TA- domínio de ativação da transcrição, Uba- domínio associado à ubiquitina. Adaptado
de Pawson et al. (2001).
Os domínios possuem funções variadas, incluindo o endereçamento de
proteínas a um local subcelular específico, o reconhecimento de modificações
pós-Adaptador
Scaffold
Quinases
Fosfatase
Sinalização da Ras
Transcrição
Ubiquitinação
Regulação do citoesqueleto
Regulação da sinalização
Sinalização por fosfolipídeos
Adaptador
Scaffold
Quinases
Fosfatase
Sinalização da Ras
Transcrição
Ubiquitinação
Regulação do citoesqueleto
Regulação da sinalização
Introdução
traducionais em outras moléculas, a formação de complexos multiprotéicos de
sinalização, além do controle da conformação, atividade e especificidade de
substrato das enzimas (PAWSON & NASH, 2000). Para isso, eles reconhecem sítios
expostos em outras proteínas, incluindo sítios fosforilados, ricos em prolina e
regiões C-terminais, ou ainda se ligam às cabeças polares de fosfoinositídeos de
membrana (PAWSON & NASH, 2003 – Fig. 3), com constantes de dissociação que
variam de nano a micromolar. Embora alguns domínios sejam bastante específicos
a ponto de sua afinidade por um sítio peptídico ser suficiente para a interação
específica nas células, outros são muito versáteis em suas propriedades de ligação.
Um domínio determinado pode interagir com diversos ligantes distintos de forma
simultânea e seqüencial, nas etapas sucessivas da transdução de sinal. Além disso,
domínios de um mesmo grupo podem ter ligantes bastante diferentes. Nestes
casos, a especificidade da sinalização é garantida por fatores outros que somente
a afinidade por seus ligantes, incluindo interações terciárias, localização subcelular
e a organização estrutural das proteínas envolvidas, competição entre domínios e
interações multidomínios (PAWSON &NASH, 2003).
Durante a transdução de sinal, enzimas, como por exemplo, as quinases,
Introdução
Figura 3. Famílias de domínios de interação e seus ligantes. Os domínios de interação ligam proteínas, fosfolipídeos ou ácidos nucléicos. Um grupo destes domínios encontra-se ilustrado, com seus ligantes indicados por flechas (PAWSON & NASH, 2003).
Uma análise recente sugeriu que aproximadamente 10.000 tipos de
interações protéicas estruturalmente diferentes podem ser antecipadas com base
nos módulos que constituem cada uma das proteínas (ALOY et al., 2004). A vasta maioria destas interações pertence a um dos seguintes grupos: i) a interação entre
domínios, que envolve uma extensa superfície de interação física ou ii) interação
com sítios peptídicos curtos (Fig. 3). Muitas das interações onde um domínio se
liga a uma seqüência peptídica curta (sítio) são relativamente fracas (acontecem Peptídeo modificado
Peptídeo
Domínio/ domínio
Fosfolipídeo
Ácido nucleico Peptídeo modificado
Peptídeo
Domínio/ domínio
Fosfolipídeo
Ácido nucleico Peptídeo modificado
Peptídeo
Domínio/ domínio
Fosfolipídeo
Introdução
na faixa de concentração micromolar) e freqüentemente dependem de
modificações pós-traducionais do ligante peptídico. Estas modificações incluindo
fosforilação, hidroxilação, acetilação, metilação e ubiquitinação muitas vezes
completam os sítios de ligação para os domínios de interação funcionando como
pontos de controle para as interações protéicas (SEET et al., 2006). Em alguns casos, a interação domínio-sítio requer uma alteração conformacional do domínio
de interação para que sua superfície de ligação seja exposta. Desta forma, a
interação domínio-sítio tende a ser transitória, característica que a torna bastante
apropriada à regulação das vias de sinalização (PAWSON & LI NDI NG, 2005).
Exatamente por serem transitórias e condicionais, as interações entre domínio e
peptídeo são difíceis de serem estudadas. De fato, elas são sub-representadas em
ensaios de larga escala de purificação por afinidade e podem ser perdidas em
ensaios de duplo híbrido se necessitam, por exemplo, da fosforilação de uma
tirosina, evento que não acontece em leveduras (PAWSON & LI NDI NG, 2005).
Além disso, a identificação sistemática de sítios de interação funcionalmente
relevantes é complicada pelo fato de serem seqüências peptídicas bastante curtas
(4 a 8 resíduos de aminoácidos) que freqüentemente encontram-se em regiões
Introdução
1.2 Efeito de peptídeos intracelulares desenhados
Estudos demonstram que seqüências peptídicas curtas racionalmente
criadas são capazes de alterar a interação proteína-proteína normal, com efeitos
significativos em modelos animais. Peptídeos como o Ht31, derivado da proteína
associada à quinase de tireóide humana dependente de AMPc (AKAP) têm sido
usados para alterar a localização da PKA na célula (ROSENMUND et al., 1994). Da mesma forma, foi demonstrado que a quinase c-Jun NH2-terminal (JNK), membro do grupo das quinases ativadas por mitógenos (MAPKs), é inibida por um peptídeo
derivado da JNK interacting protein (JI P) que impede o acesso da quinase ao seu substrato, o c-Jun. Quando uma versão permeável à célula deste peptídeo foi
injetada em camundongos, observou-se uma melhora significativa da resistência à
insulina e por conseguinte da tolerância à glicose em um modelo de diabetes do
tipo 2 (KANETO et al., 2004). Além disso, Kheifets et al. (2006) mostraram que um peptídeo de 8 resíduos de aminoácidos derivado da anexina V era capaz de inibir a
translocação da δPKC após ativação e conseqüentemente sua ação, como confirmado por seu efeito protetor em um modelo animal de infarto cardíaco.
Diante destes e de outros dados, fica claro que peptídeos que mimetizam os sítios
de interação entre proteínas são capazes de interferir acentuadamente na
Introdução
1.3 Geração de peptídeos pela célula
Peptídeos são produzidos pelas células a partir de proteínas sintetizadas
especificamente para esta finalidade (pró-proteínas), ou como sub-produtos do
metabolismo protéico. No primeiro caso, os produtos formados são conhecidos
agentes moduladores da comunicação celular (neuropeptídeos) que agem através
da ligação a receptores localizados na membrana plasmática. No segundo caso,
peptídeos intermediários são gerados em compartimentos outros que não aqueles
especializados em degradação protéica pela digestão limitada de proteínas.
Enquanto que os neuropeptídeos já foram extensivamente estudados e
caracterizados, pouco se sabe a respeito dos intermediários peptídicos gerados
durante o metabolismo protéico.
1.4 Geração de peptídeos intracelulares
A proteólise é um processo indispensável à homeostase celular e
desenvolveu-se em seres eucarióticos muito antes do sistema imunológico. Quase
todas as proteínas nas células de mamíferos são continuamente degradadas e
substituídas por re-síntese. A taxa de degradação dos constituintes celulares varia
Introdução
célula. Além disso, muitos processos celulares como a progressão do ciclo celular,
a transcrição gênica e as vias metabólicas são controlados pela degradação rápida
de moléculas regulatórias chave (ROCK & GOLDBERG, 1999).
Diversas são as vias envolvidas na proteólise. Proteínas extracelulares
internalizadas por endocitose, como antígenos externos ou polipeptídeos
circulantes, assim como algumas proteínas celulares, como receptores de
membrana, são degradas nos lisossomos por proteases dependentes de pH (DI CE,
1987). A degradação lisossomal responde por uma pequena fração (< 10-20% ) do
turnover normal das proteínas. Por outro lado, a maior parte das proteínas celulares é degradada no citoplasma e no núcleo por um sistema proteolítico
(ROCK et al., 1994). Os primeiros trabalhos mostraram que este sistema requeria ATP (GRONOSTAJSKI et al., 1985), o que surpreendeu pelo fato da hidrólise dos peptídeos ser uma reação termodinamicamente favorável, e pelo fato de não se
conhecer proteases dependentes de ATP. Um sistema de degradação não
lisossomal foi então demonstrado na fração solúvel de extratos celulares
(ETLI NGER & GOLDBERG, 1977). A análise da dependência do ATP levou à
elucidação da via ubiquitina-proteassomo, na qual o ATP é essencial para a
marcação covalente dos substratos a serem degradados e para o funcionamento
do grande complexo proteolítico, o proteassomo 26S (HERSHKO & CI ECHANOVER,
1992).
A via da ubiquitina-proteassomo consiste nas ações orquestradas de
Introdução
degradação pelo proteassomo 26S. Este grande complexo multicatalítico degrada
proteínas poli-ubiquitinadas em pequenos peptídeos de 3 a 24 resíduos de
aminoácidos (SARI C et al., 2004), e está envolvido não só na degradação de proteínas velhas ou danificadas, mas também na regulação da transcrição gênica
(MURATANI & TANSEY, 2003), no controle de qualidade das proteínas recém
sintetizadas (LECKER et al., 2006), na disponibilização de aminoácidos para geração de energia (LECKER et al., 2006) além do funcionamento do sistema imune (LECKER & GOLDBERG, 2002). No citoplasma, núcleo e mitocôndrias há
geração constante de peptídeos pela ação do proteassomo. Foi demonstrado que
em eucariotos, 30% a 90% das proteínas recém-sintetizadas podem ser
degradadas pelos proteassomos minutos após sua síntese (LECKER & GOLDBERG,
2002; LI PPI NCOTT-SCHWARTZ et al., 1988). Desta forma, a ação do proteassomo gera e libera continuamente peptídeos livres nas células (GOLDBERG, 2003;
PI CKART, 2004).
Evidências sugerem que os peptídeos gerados pelo proteassomo no
citoplasma, mitocôndrias e núcleo (WOJCI K & DEMARTI NO, 2003) são
rapidamente hidrolisados em aminoácidos que são então utilizados na síntese de
Introdução
este mesmo estudo sugeriu que as principais enzimas envolvidas na degradação
destes peptídeos eram as aminopeptidases, uma vez que a proteção da porção
N-terminal destes mesmos peptídeos com o grupamento F-moc impediu o
aparecimento de fluorescência e logo, de hidrólise (REI TS et al., 2003). Embora este e outros trabalhos afirmem que peptídeos são rapidamente degradados em
células de mamíferos (REITS et al., 2003; ORLOWSKI et al., 1983; SARI C et al., 2004), é sabido que alguns peptídeos intracelulares gerados pelo proteassomo,
escapam da degradação completa e são apresentados na superfície celular ao
sistema imune, complexados a moléculas do complexo maior de
histocompatibilidade de classe I (MHC-I ; ROCK et al., 2002; GOLDBERG et al., 2002). De fato, estima-se que 10.000 peptídeos representantes do leque de
proteínas sintetizadas cubram a membrana celular de cada célula do organismo
humano (RAMMENSEE et al., 2002). Diversas peptidases estão envolvidas com o processamento de peptídeos antigênicos gerados pelo proteassomo (ROCK et al., 2004), dentre elas a endo-oligopeptidase EC 3.4.24.15.
1.5 Endo-oligopeptidase EC 3.4.24.15
A endopeptidase 24.15, também conhecida como timet-oligopeptidase (EC
3.4.24.15; EP24.15 ) é uma metaloendopeptidase exibindo o motivo característico
de ligação com zinco HEXXH, pertencente à família M3 de metalopeptidases (RAWLI NGS & BARRETT, 1995). A EP24.15 é amplamente distribuída em
Introdução
testículo, cérebro e hipófise (CHU & ORLOWSKI , 1985). A EP24.15 possui uma
especificidade restrita a oligopeptídeos de 6 a 17 resíduos de aminoácidos, tendo
preferência por ligações peptídicas no C-terminal de aminoácidos hidrofóbicos, não
sendo capaz de hidrolisar proteínas (OLIVEI RA et al., 2001). A EP24.15 foi originalmente isolada da fração citosólica do homogenato do cérebro de ratos por
Orlowski et al. (1983), como sendo uma metalopeptidase ativa em pH neutro e inibida por quelantes de cátions divalentes. A inibição provocada pela remoção do
metal do sítio catalítico com quelantes é revertida pela adição de zinco, mesmo em
baixas concentrações (TI SLJAR & BARRETT, 1990). A presença de Zn+ 2 em seu
sítio catalítico foi determinada por espectroscopia de absorção atômica (TI SLJAR &
BARRETT, 1990).
Através de mutações sítio dirigidas, Cummins et al. (1999) elucidaram quais eram os resíduos responsáveis pela coordenação do zinco no sítio ativo
(473HEXXH477), mostrando ainda que, além das histidinas, há um terceiro ligante
do zinco, o ácido glutâmico 502. A observação deste terceiro ligante a
aproximadamente 25 resíduos de aminoácido de distância é uma característica
Introdução
possibilitando o livre acesso do substrato ao centro catalítico (SHRI MPTON et al., 1997). Uma vez que inibidores naturais da enzima não foram descobertos, a
diferença entre o potencial redox do meio extracelular e intracelular foi proposta
como o modo pelo qual a atividade da EP24.15 é regulada (SHRI MPTON et al., 2002). Mais recentemente, Demasi et al. (2008) mostraram que dentro das células, a EP24.15 encontra-se naturalmente S-glutationilada em resíduos de
cisteína e que isto tem um profundo impacto sobre a atividade enzimática. Pelo
fato da enzima possuir vários resíduos de cisteína expostos, a quantidade de
glutationilação é variável. Assim quanto maior o grau de glutationilação, maior é a
quantidade de oligômeros da enzima e conseqüentemente menor a atividade
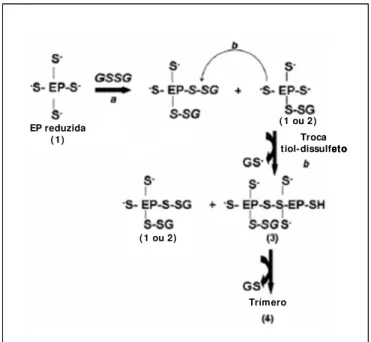
peptidásica (DEMASI et al., 2008 – Fig 4).
Figura 4. Mecanismo de troca tiol-dissulfeto proposta para a multimerização da EP24.15 através da S-glutationilação. Ânions tiolatos específicos de EP24.15 (Ep reduzida) são S-glutatiolados pelo GSSG (a). Os resíduos remanescentes de cisteína reduzida atacam a ponte dissulfeto da EP24.15 S-glutationilada (b), deslocando o GSH e formando uma ponte sulfúrica interprotéica (3). O processo avança dependendo da disponibilidade dos grupos reativos EP24.15-Cis-SH (1 e 2) ou se a concentração de GSSG for baixa, gerando formas multiméricas de EP24.15 (3, 4 ou mesmo multímeros maiores). Por outro lado, quando a concentração de GSSG é alta ou quando os ânions
EP reduzida ( 1)
( 1 ou 2) Troca tiol-dissulfeto
Trímero ( 1 ou 2)
EP reduzida ( 1)
( 1 ou 2) Troca tiol-dissulfeto
Trímero ( 1 ou 2)
EP reduzida ( 1)
( 1 ou 2) Troca tiol-dissulfeto
Introdução
glutationilação da EP24.15 prevalesce sobre a oligomerização inibindo a troca tiol-dissulfeto. Nesta
condição, a forma monomérica altamente S-glutationilada seria a mais abundante (DEMASI et al.,
2008).
Desde que a EP24.15 foi isolada por Orlowski et al. (1983), acredita-se que ela participe do metabolismo e/ ou processamento de uma série de
neuropeptídeos. Entre os neuropeptídeos clivados por esta enzima estão a
bradicinina, a neurotensina, o hormônio liberador de gonadotrofinas (GnRH), a
somatostatina, a substância P, assim como as angiotensinas I e I I (SHRI MPTON et al., 2002). Foi demonstrado ainda que a EP24.15 é capaz de gerar as respectivas encefalinas a partir de precursores opióides intermediários (CHU & ORLOWSKI ,
1985). Por esta razão a EP24.15 foi envolvida em diversos processos fisiológicos
como a percepção de dor (MOLI NEAUX & AYALLA, 1990; KEST et al., 1991; KEST
et al., 1992), a homeostase cardiovascular e renal (ORLOWSKI et al., 1983; CARDOZO & ORLOWSKI , 1993; TELFORD et al., 1995) e a reprodução (PI EROTTI
et al., 1991; LEW et al., 1997; WU et al., 1997; SMI TH et al., 2000).
Embora a EP24.15 seja secretada, sua presença em vesículas secretórias
contendo o marcador ß-endorfina não foi observada, e seu time-course de secreção é distinto daquele descrito para os neuropeptídeos. Mostrou-se então que
Introdução
anos mostram que a maior quantidade de EP24.15 concentra-se no meio
intracelular (FONTENELE-NETO et al., 2001), mais especificamente no citoplasma e no núcleo das células. Assim, é muito provável que além das funções já descritas,
a EP24.15 participe de processos intracelulares. De fato, diversos trabalhos
associam a peptidase em questão à apresentação de antígenos pelas moléculas de
MHC-I (SI LVA et al., 1999, PORTARO et al., 1999; YORK et al., 2003; KIM et al., 2003). Como anteriormente mencionado, os peptídeos apresentados na superfície
celular compartilham a característica de possuir nove a doze resíduos de
aminoácidos e, portanto, têm o tamanho ótimo dos substratos da EP24.15. A
manipulação da atividade intracelular da EP24.15 seguida da análise de antígenos
apresentados via MHC-I levou à primeira sugestão de sua função intracelular: o
metabolismo de peptídeos apresentados como antígenos ligados a MHC-I (SI LVA
et al., 1999; PORTARO et al., 1999). Em macrófagos, o inibidor da EP24.15 CFP-AAF-pAB reduz, enquanto a adição da enzima recombinante ativa aumenta a
apresentação de antígenos derivados da heat shock protein 65 (HSP65) do bacilo da tuberculose através do MHC-I . A degradação de vários peptídeos apresentados
como antígenos pelo MHC-I foi analisada, levando a sugestão de que in vitro a EP24.15 não seria capaz de clivar eficientemente peptídeos antigênicos (PORTARO
et al., 1999). Em conjunto, esses dois trabalhos sugeriam que a EP24.15 poderia degradar de forma mais eficiente aqueles peptídeos gerados pelo proteassomo não
destinados à apresentação pelo MHC-I , permitindo o acúmulo e o transporte
Introdução
retículo endoplasmático rugoso. Trabalhos posteriores, utilizando proteínas outras
que não a HSP65 como antígeno, demonstraram que a redução na atividade da
EP24.15 levava a um aumento na apresentação de peptídeos antigênicos pelo
MHC-I (YORK et al., 2003; KI M et al., 2003). Dessa forma, parece que a função da EP24.15 pode ser tanto a de prevenir quanto destruir peptídeos antigênicos
apresentados pelo MHC-I . Embora controversos, esses dados demonstram a
participação irrefutável da EP24.15 neste processo intracelular.
Com o intuito de aumentar o conhecimento acerca das funções
intracelulares da EP24.15, uma série de estudos cujo o objetivo de identificar
novos substratos ou inibidores competitivos intracelulares para a EP24.15 foi
realizada. Para tal, mutações pontuais que levavam à inativação catalítica da
EP24.15 foram geradas (RI OLI et al., 2003). O uso da forma inativa da EP24.15 permitiu o isolamento e identificação de vários peptídeos naturais anteriormente
desconhecidos, sendo que a maior parte deles era constituída por fragmentos de
proteínas intracelulares e apresentavam sítios de modificação pós-traducional,
Introdução
Figura 5. Distribuição dos sítios encontrados em 39 peptídeos isolados com a EP24.15 inativa. Todos os peptídeos correspondem a fragmentos de proteínas intracelulares. Em parênteses encontra-se o número de peptídeos identificados contendo o sítio indicado. PKC – proteína quinase C, CKII – caseína quinase I I, CK I – caseína quinase I, p38 MAPK – quinase ativada por mitógeno
proteína 38, no hit – nenhum sítio previsto (MACHADO et al., 2006).
Uma observação bastante interessante do trabalho de Machado et al.
(2006) é a de que a fosforilação de peptídeos substratos da EP24.15 altera
significativamente sua cinética de degradação, sugerindo que esta modificação
pós-traducional possa regular, pelo menos em parte, o metabolismo e a função
celular de peptídeos.
1.6 Peptídeos intracelulares e sinalização
Uma ampla gama de dados mostra que seqüências peptídicas curtas são
capazes de alterar significativamente as interações entre proteínas e,
conseqüentemente, a resposta celular a determinado sinal. Entretanto, na maior
parte dos estudos, os peptídeos foram racionalmente desenhados para a
modulação da interação proteína-proteína de interesse. Por outro lado, dentro das
células, muitos intermediários peptídicos são continuamente gerados. Embora
muito pouco se saiba a respeito destes intermediários, um estudo mostrou
pós-Introdução
traducional (MACHADO et al., 2006). Estes e outros fatos levaram nosso grupo a formular a hipótese de que peptídeos intracelulares gerados pelo proteassomo 26S
durante o turnover protéico, poderiam fisiologicamente regular interações entre proteínas dentro das células, além de estimular ou inibir enzimas proteolíticas e
não proteolíticas, modulando cascatas de sinalização a exemplo dos peptídeos
Objetivo
2 OBJETI VO
Materiais e Métodos
3 MATERI AI S E MÉTODOS
3.1 I solamento de peptídeos de cérebro de rato - O isolamento dos
peptídeos de cérebro de ratos Wistar machos adultos (250-300 g) foi realizado
conforme descrito anteriormente, com algumas alterações (RIOLI
et al.
, 2003).Com o intuito de inativar instantaneamente a proteólise inespecífica observada
logo após o sacrifício, o cérebro dos animais foi submetido a uma sessão de 10s de
microondas, logo após sua decapitação, sendo a temperatura no interior no
cérebro monitorada com um
probe
. A seguir, o cérebro foi homogeneizado emsolução de HCl 50 mM com o auxílio de um homogeneizador de tecidos (Tissue
Tearor, Biospec Products, Inc.) e aquecido a 70ºC por 15 min em banho-maria. Os
homogenatos foram centrifugados a 1500 x g a 4ºC e o sobrenandante foi
transferido para tubos de ultracentrífuga, sendo centrifugado a 100000 x g por 30
min a 4ºC. O sobrenadante foi filtrado em membrana Millipore que permite a
passagem de moléculas com peso molecular inferior a 5 kDa (
Centrifugal
Filter
Unit
, MILLIPORE, 5,000 NMCO) e o material não retido pela membrana (eluato -Materiais e Métodos
2003) por 1 h. Após este tempo, a enzima ligada aos peptídeos foi separada do
resto da solução por gelfiltração a seco (1000 x g, 2 min) em resina Sephadex
G-25. Novamente, as amostras resultantes foram secas em Speed Vac e estocadas
em freezer a -80ºC até o seqüenciamento.
3.2 Quantificação de peptídeos endógenos intracelulares– A
quantificação dos peptídeos endógenos presentes nos extratos intracelulares de
cérebro de ratos assim como de células CHO-S e HEK293 foi realizada utilizando a
fluorescamina (UDENFRIEND
et al.
, 1972). A reação ocorreu em pH 6.8 para quesomente os grupamentos amino terminais dos peptídeos e não dos resíduos de
aminoácidos, reagissem com a fluorescamina. Uma quantidade de 2,5 µL de
amostra foi misturada a 25 µL de tampão fosfato (0,2 M, pH 6.8) e 12,5 µL de
uma solução de fluorescamina 0,3 mg/mL em acetona. Após agitação vigorosa por
1 min, 110 µL de água foram adicionados, e a fluorescência emitida pela
fluorescamina ligada aos peptídeos foi mensurada em um fluorímetro SpectraMax
M2e (Molecular Devices, USA) nos comprimentos de onda de excitação de 370 ηm
e emissão de 480 ηm. Uma mistura de peptídeos de composição e concentração
conhecidas foi usada como padrão para determinação da concentração peptídica.
3.3 Seqüenciamento de peptídeos por LC-MS/ MS - Os
seqüenciamentos de peptídeos por LC-MS/MS foram realizados em um aparelho
Materiais e Métodos
(CapLC, Waters). A mistura peptídica obtida conforme descrito acima foi separada
por gradiente de eluição (0-85%) utilizando mistura água/acetonitrila contendo
0.1% ácido fórmico em coluna capilar de 75 µm empacotada com sílica C18. Os
dados foram adquiridos automaticamente em modo dependente (DDA) após
geração de múltiplos peptídeos protonados por ionização em eletron spray (ESI),
seguido de dissociação em MS/MS por colisão com argônio em intensidade de 10 a
30 eV. O spectro de massa do íon produzido foi processado usando MaxEnt3 e o
seqüenciamento dos peptídeos foi realizado manualmente com a ajuda do
programa PepSeq (Micromass). Condições típicas de LC e ESI foram: fluxo de 10
ηL/min, voltagem no capilar do nanofluxo de 3.5 kV, temperatura do bloco de
80oC, e voltagem do cone de 100 V.
3.4 Análise das seqüências peptídicas - Para identificar os prováveis
precursores protéicos dos peptídeos seqüenciados, toda a aquisição de dados foi
processada em formato de texto (.pkl) pelo programa ProteinLynx 2.0 e uma busca
no banco de dados do NCBI, foi realizada através do programa Mascot. A busca de
possíveis sítios de modificação pós-traducional foi realizada no site
Materiais e Métodos
produzida em bactérias foi determinada em um ensaio contínuo utilizando-se o
substrato com fluorescência dependente de clivagem (QFS –
7-metoxicoumarina-4-acetil-Pro-Leu-Gly-Pro-dLys-(2,4-dinitrofenil). Em cada poço de uma placa de 96
poços foram misturados: 50 ηg de EP24.15 recombinante, 2 µM de QFS e cada um
dos peptídeos (0 a 50 µM). Os ensaios foram realizados em triplicata e o aumento
da fluorescência derivada da hidrólise do QFS pela EP24.15 foi monitorada por 2 h
com a freqüência de uma leitura por minuto (SHRIMPTON
et al.
, 1997; TULLAIet
al.
, 2000). As constantes de inibição aparente (Kiap) dos peptídeos foramdeterminadas com a equação:
Ki
=Ki
ap/(1+[S]/Km
), assumindo-se queKi
=IC50 eonde [S] = concentração molar do substrato QFS,
Km
= constante deMichaelis-Menten (RIOLI
et al.
, 2003). Os valores de IC50 para os peptídeos foramdeterminados por regressão linear de uma plotagem do tipo XY onde X =
concentração logarítmica do peptídeo e Y = percentual de inibição obtido
experimentalmente na presença de cada peptídeo.
Para determinar o percentual de hidrólise de cada peptídeo, uma solução 100 µM
de peptídeo foi incubada com 0,16 ηg/µL de EP24.15 recombinante em uma
reação com cinética de ordem zero, após 15 min de incubação a temperatura
ambiente, as amostras foram analisadas por HPLC. A área dos picos dos peptídeos
antes e após a incubação com EP24.15 foi determinada e usada para o cálculo da
quantidade de peptídeo hidrolisado. O percentual de hidrólise obtido para cada
peptídeo de tempo foi normalizado ao da bradicinina, sendo os resultados
Materiais e Métodos
subseqüentemente analisado por HPLC para detecção da hidrólise dos peptídeos.
Nos casos em que a clivagem do peptídeo foi detectada, nova amostra foi
preparada incubando-se 100 µM de peptídeo com EP24.15 recombinante (0,16
ηg/µL), agora por 4 h a temperatura ambiente. As ligações peptídicas hidrolisadas
pela EP24.15 foram determinadas nestas amostras por espectrometria de massa
em um aparelho de MALDI-TOF (GE HealthCare, Uppsala, Sweden). As condições
de MALDI utilizadas nas análises foram: modo scan, 50 V de voltagem do cone e
uma janela de análise de 200 – 2000 m/z.
3.6 Síntese dos peptídeos escolhidos acoplados ao TAT por pontes
dissulfeto - Soughayer
et al.
(2004) demonstraram que substratos de quinaseligados ao TAT por pontes dissulfeto entram em células, separam-se do TAT pela
redução das pontes e tornam-se biodisponíveis no citoplasma. Mais ainda os
autores mostraram que os substratos eram fosforilados mediante a estimulação
das quinases com ferramentas farmacológicas. Com base neste artigo, os
peptídeos escolhidos para estudo foram encomendados para síntese.
Materiais e Métodos
3.8 Monitoramento da entrada do peptídeo na célula e do
desligamento do TAT - Células CHO-S foram semeadas em placas de 6 poços.
Os peptídeos ligados ao TAT por pontes dissulfeto foram incubados em uma
concentração de 20 µM por 30 min. Após este tempo, as células foram coletadas
em PBS gelado, centrifugadas por 800 x g, 4°C por 10 min e ressuspendidas em
água milli-Q (130 µL). Procedeu-se a três ciclos de
congelamento/descongelamento para lise celular, sendo o lisado centrifugado a
20000 x g por 30 min a 4°C. Cinqüenta microlitros do sobrenadante foram
injetados em um aparelho de HPLC (Shimadzu) consistindo de um controlador
(SCL-10A VP), duas bombas de pistão (LC-10 AD VP), um detector de UV (SPD-10
AV VP) e um detector de fluorescência (RF-10A XL). Para possibilitar a detecção de
fluorescência da fluoresceína apesar do baixo pH no qual a separação
cromatográfica normalmente ocorre, foi acoplada uma terceira bomba ao sistema,
para bombeamento de uma solução de NH4OH/água 2.5:100 (v/v), que se junta
ao fluxo que sai do detector de UV em direção ao detector de fluorescência,
elevando o pH da solução para 11 (BAECHLE
et al.
, 2005). As separaçõescromatográficas foram feitas em uma coluna XTerra® RP18 (Waters) corridas em
um gradiente binário linear (25% a 75% de B em 20 min, onde A=TFA 0.08% em
água e B= acetonitrila 90% em água e TFA 0.08%), na velocidade de 1mL/min.
A eluição dos peptídeos das amostras foi monitorada por fluorescência
utilizando os comprimentos de onda de excitação e emissão de 490 e 520 nm,
Materiais e Métodos
3.9 Ensaio microfisiométrico - Células CHO-S (Invitrogen) estavelmente
transfectadas com o receptor AT1 para angiotensina II (ang II) e gentilmente
cedidas pela Dr. Graciela C. Pignatari (PIGNATARI
et al.
, 2006) foram semeadasem
transwells
(Costar Corning) em uma densidade de 2x105 células/transwell
eacondicionadas por 20 h em incubadora de células. No dia dos experimentos, os
transwells
foram colocados nas câmaras sensoras do microfisiômetroCytosensor™
(Molecular Devices, USA), que possuem um sistema biossensor de silicone,
responsável pelo monitoramento contínuo do pH do meio extracelular, sendo que a
ativação de receptores/vias de transdução de sinal é detectada pelo aumento da
velocidade de acidificação do meio extracelular. O pH do meio a ser usado nos
experimentos foi cuidadosamente acertado para 7.4. A ang II usada para estimular
as células e as soluções de peptídeos foram diluídas no mesmo meio citado acima
tendo também seu pH monitorado e mantido em 7.4. As câmaras foram
perfundidas com meio DMEM sem bicarbonato, suplementado com NaCl 44 mM,
num fluxo de 100 µL/min. Cada ciclo de medição de pH consistiu em um período
de 1 min, onde a bomba de perfusão permaneceu ligada por 40 s e desligada nos
Materiais e Métodos
células. Após 30 min de contato com o(s) peptídeo(s), foi feito um segundo
estímulo com ang II nas mesmas concentrações (Fig. 6).
Figura 6. Esquema temporal do ensaio de microfisiometria. As células foram deixadas no aparelho por 60 min para estabilização da resposta de acidificação basal. A ang II foi incubada por 15 s
sendo posteriormente lavada dos transwells e o aumento da taxa de acidificação do meio
extracelular (ECAR) foi monitorado por 10 min. A incubação subseqüente com o(s) peptídeo(s) foi realizada por 30 min, tempo após o qual foi feito um novo estímulo com ang II.
3.10 Ensaio de gene repórter: Ensaio da luciferase - A ativação de
receptores de membrana acoplados a proteína G desencadeia a ativação de
cascatas de sinalização intracelular, levando à modulação da taxa transcricional de
genes específicos. Desta forma, ensaios de atividade farmacológica baseados em
genes repórteres são úteis no monitoramento das respostas induzidas via ativação
de GPCR, assim como também pode ser usado na identificação de agentes que
interfiram nestas cascatas, alterando-as de alguma forma. Fitzgerald
et al.
(1999)desenvolveram uma estratégia que permite a realização de ensaios funcionais em
larga escala para acompanhamento da ativação de vias transducionais. Este
sistema utiliza a expressão de um gene repórter (luciferase) controlado
simultaneamente por diferentes regiões promotoras (responsivas a AMPc, Ca2+ e
IP3, dentre outros) ligadas
in tanden
(FITZGERALDet al.
, 1999; Fig. 7). Com baseneste artigo, foi construído o plasmídeo usado nas análises.
60 min 15s 10 min 30 min 15s 10 min
DMEM Peptídeo
Ang II
60 min 15s 10 min 30 min 15s 10 min
Ang II
DMEM DMEM
60 min 15s 10 min 30 min 15s 10 min
DMEM Peptídeo
Ang II
60 min 15s 10 min 30 min 15s 10 min
Ang II
DMEM DMEM
60 min 15s 10 min 30 min 15s 10 min
DMEM Peptídeo
Ang II
60 min 15s 10 min 30 min 15s 10 min
Ang II
Materiais e Métodos
Figura 7. Vetor reporter pGL3-MRE-CRE. A produção da luciferase encontra-se sob o controle
transcricional de três MREs (multiple responsive element – derivado da região promotora da
interleucina-6) e do CRE (cAMP responsive element – derivado da região promotora do peptídeo
vasoativo intestinal).
3.10.1 Construção do plasmídeo repórter
3.10.1.1 Inserção do CRE/VIP no pGL3 básico: a região promotora do peptídeo
vasoativo intestinal (VIP) dos nucleotídeos -94 a 152 (região que contém o CRE)
foi amplificada e incorporada aos sítios para das enzimas de restrição XhoI e Hind
III (nas porções 5´e 3´, respectivamente) por reação de PCR, a partir do DNA
genômico humano. Para isso, utilizou-se os seguintes primers
5’-GACTCGAGACTTCAAGCCCTATTC-3’ sense e 5’-CTAAGCTTCTCGCCCAGTCGTG -3’
antisense. Após a purificação do produto do PCR (GFX™ PCR DNA and Gel Band
Purification Kit – Amersham Biosciences) e digestão com Xho I e Hind III overnight
a 37°C (10 U de Xho I e 10 U de Hind III), o inserto foi clonado no vetor pGL3
básico (Promega – Fig. 8 A), gentilmente cedido pela Dr. Andrea S. Heimann. Para
isso, 1 µg de pGL3 foi também digerido com as mesmas enzimas,
overnight
, sendoMRE MRE MRE CRE LUCIFERASE
MRE MRE MRE CRE LUCIFERASE
MRE
Materiais e Métodos
posteriormente semeadas em placas de LB agar com ampicilina. Foram escolhidas
10 colônias crescidas na placa de LB. Estas 10 colônias foram crescidas
separadamente em 5 mL de LB líquido+ampicilina
overnight
, isolando-se após estetempo o DNA plasmidial destas bactérias com auxílio de kit de miniPrep
(Promega). A presença do inserto (CRE) assim como sua correta seqüência de
DNA foram checadas através de seqüenciamento automático de DNA (MegaBACE
100 DNA Analysis System – Amersham Bioscience, Sunnyvale, CA, USA).
3.10.1.2 Clonagem dos MREs no pGL3 + CRE: oligos contendo 3 cópias
in
tandem
da região promotora do gene humano da IL6, dos nucleotídeos 173 a-142 (que contem o elemento de múltipla resposta-MRE) foram adquiridos
comercialmente (IDT). Os oligos foram desenhados de forma a possuirem nas
extremidades sítios para as enzimas de restrição Mlu I e Xho I (nas porções 5´e
3´, respectivamente) de acordo com a seqüência:
MRE sense 5’ – 3’:
CGCGTATGCTAAAGGACGGTCACATTGCACAATCTTAAATGCTAAAGGACGTCACATTGC
ACAATCTTAAATGCTAAAGGACGTCACATTGCACAATCTTAAC
MRE antisense 5’ – 3’:
TCGAGTTAAGATTGTGCAATGTGACGTCCTTTAGCATTTAAGATTGTGCAATGTGACGTC
CTTTAGCATTTAAGATTGTGCAATGTGACCGTCCTTTAGCATA
As porções sense e antisense do MRE foram aneladas em termociclador e
inseridas no pGL3 + CRE previamente aberto com Mlu I e Xho I. Para a reação de
Materiais e Métodos
10 ηg de MREs anelados, e 15 unidades de DNA ligase. A reação ocorreu a 14°C
por 16 em termociclador (MJ Research, USA), purificada e bactérias competentes
XL1 blue foram transformadas por eletroporação com 6 µL do produto de ligação
purificado.
Novamente foram escolhidas 10 colônias crescidas na placa de LB. Estas 10
colônias foram cultivadas separadamente em 5 mL de LB líquido+ampicilina
overnight
, isolando-se após este tempo o DNA plasmidial destas bactérias comauxílio de kit de miniPrep (Promega). A presença do inserto (MREs+CRE) assim
como sua correta seqüência de DNA foram checadas através de seqüenciamento
automático de DNA (Megabace – Amersham Bioscience).
Uma vez confirmada a seqüência correta de DNA, grandes quantidades de
um só clone de bactérias foram crescidas, sendo o DNA plasmidial isolado através
do uso de kit de MaxiPrep (Promega) conforme as instruções do fabricante. Da
mesma forma, o plasmídeo pRL (que codifica um outro tipo de luciferase; a ser
usado para normalização dos resultados; Promega; Fig. 8 B), gentilmente cedido
pela Dr. Nancy Rebouças, foi preparado em grande escala para transfecção de
Materiais e Métodos
Figura 8. Vetores usados no ensaio da luciferase. A região promotora CRE-MREs encontra-se inserida na região de múltipla clonagem do pGL3 básico (A) entre os sítios de XhoI e HindIII e
controla a transcrição da luciferase de Photinus sp, o vagalume. O vetor pRL (B) codifica a
luciferase de Renilla reniformis, um organismo marinho, cuja transcrição é promovida pela região promotora de citomegalovírus (CMV), e foi usado para normalização dos resultados.
3.10.2 Transfecção celular e ensaio da luciferase
Para a transfecção, células HEK293- H e CHO-S foram passadas para placas
de 6 poços na densidade de 1.0 x 106 células/poço e deixadas por 16 h em
incubadora de células. No dia da transfecção, o meio de cultura foi substituído por
DMEM sem soro nem antibióticos e uma mistura de co-transfecção composta por
10 µg de lipossomos (Lipofectamina 2000®) e DNA plasmidial (1µg de pEGFP-AT1,
1 µg de pLuc MRE-CRE e 15 ηg de RLuc) foi acrescentada às células e deixada por
6 h a 37ºC, tempo após o qual, o meio de transfecção foi substituído por DMEM
com antibióticos e 10% de SFB, deixando-se as placas em incubadora até o outro
dia. Após este período, as células foram tripsinizadas e passadas para uma placa
de 96 poços na densidade de 0,1 x 106 células/poço, esperando-se 16 h para sua
adesão e estabilização antes do início do ensaio. Imediatamente antes do
experimento, o meio foi substituído por DMEM sem soro nem antibióticos.
A
B
Materiais e Métodos
Seguindo o esquema temporal ilustrado pela figura 9, os peptídeos foram
pré-incubados 15 min antes e concomitantemente à estimulação com o agonista
dos receptores AT1, a ang II (1 µM) ou com o agonista dos receptores β
-adrenérgicos, o isoproterenol (10 µM). Doses de reforço de peptídeo foram
adicionadas 30, 60, 120, 180 e 240 min após a estimulação. Após um período total
de 5 h, a quantificação da atividade da luciferase foi realizada com o kit
Dual-Luciferase® Reporter Assay System (Promega), seguindo as instruções do
fabricante, em luminômetro (SpectraMax L, Molecular Devices).
Figura 9. Esquema temporal do ensaio da luciferase. As células receberam um pré-tratamento com
os peptídeos 15 min antes e concomitantemente à estimulação das células com and II (1 µM) ou
isoproterenol (10 µM). Reforços subseqüentes de peptídeos foram adicionados às células
periodicamente. Após um tempo total de 5 h, as células foram lisadas e a atividade da luciferase foi avaliada no sobrenadante. P=peptídeo.
0 1 2 3 4 5
P P P P P
Lise celular
-0.25 0.5
P
P + Agonista
Tempo (h)
0 1 2 3 4 5
P P P P P
Lise celular
-0.25 0.5
P
P + Agonista
Tempo (h)
0 1 2 3 4 5
P P P P P
Lise celular
-0.25 0.5
P
P + Agonista
Materiais e Métodos
HealthCare, Uppsala, Sweden) com um gradiente isocrático (fase móvel: 2% de
acetonitrila em água e 0,1 % de ácido fórmico) e um fluxo de 200 µL/min. A
adição do grupamento fosfato aos peptídeos foi analisada por espectrometria de
massas utilizando a ionização por eletro spray no modo positivo (ESI+), voltagem
da fonte de 3,5 kV e voltagem do cone de 40 V (Finnegan, USA).
3.12 Aumento da expressão da EP24.15 em células CHO-S e
HEK293 e ensaio da luciferase – Para investigar o efeito do aumento da
expressão da EP24.15 sobre a transdução de sinal dos receptores AT1 e β
-adrenérgico, células CHO-S e HEK293 foram transfectadas com os vetores da
luciferase anteriormente mencionados mais 1 µg do vetor de expressão da
EP24.15 selvagem ou 1 µg do vetor vazio, usado como controle (CARREÑO
et al.
,2005). Após 48 h, a expressão da luciferase foi estimulada pelo tratamento das
células CHO-S com ang II e das células HEK293 com isoproterenol por 5 h. Para
avaliar o aumento da expressão da EP24.15, uma alíquota das células utilizadas no
ensaio da luciferase foi separada e a atividade da EP24.15 foi quantificada no
lisado celular, utilizando o substrato fluorogênico
QFS
conforme descrito no tópico3.5 (SHRIMPTON
et al.
, 1997; TULLAIet al.
, 2000).3.13 Acoplamento dos peptídeos a colunas de purificação por
afinidade e captura de ligantes - Para construção das colunas de afinidade foi
Materiais e Métodos
Uppsala, Sweden). Uma solução 10 mg/mL de cada peptídeo foi prepara e
incubada por 1 h 30 com a coluna, à temperatura ambiente, para ligação do
N-terminal dos peptídeos à resina. O excesso de grupos reativos da resina foram
inativados e a coluna foi lavada com os tampões apropriados segundo as
instruções do fabricante.
Os cérebros de cinco ratos foram homogeneizados em tampão fosfato (PBS:
cloreto de sódio 137 mM, cloreto de potássio 2 mM, tampão fosfato 10 mM, pH
7.4) com inibidores de proteases na proporção de tampão:tecido 5:1 (v/m). O
homogenato foi incubado em gelo por 20 min e centrifugado por 10 min a 4°C,
1000 x g. A fração citosólica foi separada do pellet e ultracentrifugada por 1 h a
4°C, 100000 x g. A concentração de proteínas foi determinada pelo método de
Bradford (BRADFORD, 1976) e 12 mg de proteínas foram incubadas com as
colunas por 1 h 30 a 4°C. Após lavar as colunas com 10 mL (10 x o volume da
coluna) de PBS, as proteínas ligadas foram eluídas com 10 mL de glicina 0.1 M pH
2.7. O primeiro mL da eluição foi descartado uma vez que contém a maior parte
das proteínas que interagiram com baixa afinidade. Os nove mL restantes foram
coletados em um tubo de centrífuga e submetidos à troca de tampão (Tris-Cl 10
Materiais e Métodos
mercaptoetanol e 0.002% de azul de bromofenol), aquecido a 100°C por 5 min e
mantido em freezer -80°C.
As proteínas contidas no eluato foram separadas por eletroforese em gel de
poliacrilamida 10%. As bandas protéicas foram visualizadas após a coloração do
gel com azul de Comassie, sendo a seguir recortadas e acondicionadas
separadamente em tubos
eppendorf
. As bandas de gel foram enviadas para afacility
de espectrometria de massa na universidade TUFTS (The Tufts UniversityCore Facility, Tufts Medical School, Dept. of Physiology Boston, MA, USA), onde
foram submetidas à digestão por tripsina sendo os peptídeos resultantes extraídos
do gel e analisados em um espectrômetro de massa (Thermo LTQ ion trap,
Thermo Corp., San Jose, CA). Os espectros de MS/MS foram comparados ao banco
de dados de proteínas não redundante do NCBI usando o algorítmo SEQUEST
(YATES
et al.
, 1995).3.14 Análise estatística – Os resultados foram expressos como média ±
EPM. As comparações estatísticas foram realizadas utilizando-se o teste t ou
ANOVA de uma via seguida pelo teste de Tukey. Valores de
P
< 0.05 foramResultados
4 RESULTADOS
4.1 I dentificação de novos peptídeos de cérebro de ratos - Nove
novos peptídeos, ligantes da oligopeptidase EP24.15, foram identificados em
cérebro de ratos e encontram-se listados na tabela 1. As análises das seqüências
em bancos de dados revelou que diversos dos peptídeos apresentavam sítios de
modificação pós-traducional.
Tabela 1. Peptídeos extraídos de cérebro de ratos com a metodologia de captura pela EP24.15 inativa e identificados por espectrometria de massa.
SEQÜÊNCI A PEPTÍ DI CA SÍ TI O PROTEÍ NA PRECURSORA
HRDTGI LDSLGR Nenhum proteína básica da mielina
SPQLEDEAKELQ Nenhum Pré-próencefalina
PGANAAAAKI QASFR miristoilação
PKC neurogranina
GSAKVAFSAI RSTNH PKC cerebelina
SFAKAPHLDL Nenhum cromogranina B
SSGAHGEEGSARI WKA PKC citocromo C oxidase
FHNPHMNPLPTGYEDE CKI I citocromo C oxidase
EETVRKAMEAVAAQGKVKK PKC Fosfoglicerato mutase
DVRPQVHPNYRI TV Nenhum ATPase lisossomal
Resultados
encomendados para síntese em duas formas. Uma forma simples, isto é, apenas a
seqüência originalmente identificada e uma forma fluorescente e permeável à
célula, ligada à uma seqüência derivada do peptídeo TAT do HI V por ponte
dissulfeto (Tabela 2), de forma que, uma vez no ambiente redutor dentro das
células (SAITO et al., 2003), a ponte dissulfeto se rompesse, liberando o peptídeo do TAT. Além deles, foi também escolhida uma seqüência aleatória (RD), que não
é ligante de EP24.15 e não possui sítios de modificação-pós traducional, sendo
usada como controle (RI OLI et al., 2003).
Tabela 2. Peptídeos escolhidos para síntese e análise.
Nome Seqüência Sítio
5 A* 5-FAM-LTLRTKLC(SS)CRKKRRQRRR PKC
FE3# 5-FAM-SSGAHGEEGSARI WKAC(SS)CRKKRRQRRR PKC
FE2# 5-FAM-PGANAAAAKI QASFRC(SS)CRKKRRQRRR PKC
FE4# 5-FAM-GSAKVAFSAI RSTNHC(SS)CRKKRRQRRR PKC
RD# 5-FAM-VNMVPVGWASRC(SS)CRKKRRQRRR -
Peptídeos extraídos de * tecido adiposo de camundongo e #cérebro de rato. Em itálico, peptídeo
derivado da proteína TAT do HIV. Em negrito sublinhado, sítio de modificação pós-traducional previsto pelo PROSITE. 5-FAM corresponde ao marcador fluorescente fluoresceína. O peptídeo 5 A
Resultados
4.2 Ensaios cinéticos com a EP24.15 recombinante ativa e
determinação dos sítios de hidrólise - Os resultados do ensaio cinético
representados na figura 10 (A, B, C e D) mostram que os quatro peptídeos
escolhidos para estudo (5A, FE2, FE3 e FE4) foram capazes de inibir de forma
dependente da concentração a quebra do QFS, indicando que eles interagem com
o sítio catalítico da EP24.15. Como esperado, o peptídeo RD não foi capaz de
alterar a quebra do QFS de forma dependente da concentração (Fig. 10 E). As constantes de inibição aparente calculadas a partir deste ensaio estão listadas na
tabela 3 e encontram-se dentro da faixa calculada para outros substratos da
enzima (RI OLI et al., 2003).
Tabela 3. Constantes de inibição da EP24.15 calculadas para os peptídeos estudados.
Peptídeo Ki aparente (µM)
5A 7.97± 1.59 FE2 1.30± 0.12 FE3 2.28± 0.59 FE4 3.09± 0.48
RD > 100
Resultados
Figura 10.Cinética enzimática da EP 24.15 na presença dos peptídeos estudados. Os peptídeos
foram incubados em diferentes concentrações (1-50 µM) com 50 ηg de EP24.15 ativa recombinante
e 20 µM de seu substrato QFS.
As análises cromatográficas do produto de incubação dos peptídeos com a
EP24.15 (Fig. 11 A, B, C e D) mostram que embora os quatro peptídeos interajam
com o sítio catalítico da enzima, apenas os peptídeos 5 A e FE2 são clivados,
enquanto que os peptídeos FE3 e FE4 permanecem intactos. Desta forma, é
possível sugerir que os dois primeiros peptídeos são substratos enquanto os outros
dois são inibidores competitivos da EP24.15. O produto de incubação dos
substratos também foi analisado por MALDI para determinação do padrão de
clivagem dos peptídeos pela EP24.15. As análises mostraram que o peptídeo 5A é
0 5500 11000
5A (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE2 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE3 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE4 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5
0 1 2 5 10 50
0 5500 11000
RD (µM)
η M Q FS /m in /m g d e E P 2 4 .1 5 A B C D E 0 5500 11000
5A (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE2 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE3 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5 0 5500 11000
FE4 (µM) 0 1 2 5 10 50
η M Q FS /m in /m g d e E P 2 4 .1 5
0 1 2 5 10 50
0 5500 11000
RD (µM)