Artigo
*e-mail: [email protected]
DESENVOLVIMENTO DE UM MÉTODO ANALÍTICO BASEADO EM MICROEXTRAÇÃO LÍQUIDO-LÍQUIDO PARA A DETERMINAÇÃO DE CROMO (VI) EM AMOSTRAS AQUOSAS COM DETECÇÃO POR ESPECTROMETRIA DE ABSORÇÃO ATÔMICA EM CHAMA
Renata Mior, Joyce Nunes Bianchin Dutra e Eduardo Carasek*
Departamento de Química, Universidade Federal de Santa Catarina, 88040-900 Florianópolis – SC, Brasil Edmar Martendal
Departamento de Química, Universidade do Estado de Santa Catarina, 89219-710 Joinville – SC, Brasil
Recebido em 13/7/12; aceito em 18/2/13; publicado na web em 15/4/13
DEVELOPMENT OF ANANALYTICAL METHOD FOR THE DETERMINATION OF HEXAVALENT CHROMIUM IN WATER SAMPLES BASED ON LIQUID-LIQUID MICROEXTRACTION WITH DETECTION BY FLAME ATOMIC ABSORPTION SPECTROMETRY. In this study, a method for determination of hexavalent chromium in aqueous samples using liquid-liquid microextraction (LLME) and detection by Flame Atomic Absorption Spectrometry (F AAS) was developed. The LLME procedure was based on the extraction of Cr (VI) by acetone at a sample pH of 1.2. The use of saturated ammonium sulphate solution allowed effective separation of the aqueous and organic phases and acetone extracted chromium. The sample pH, acetone volume and stirring time were optimized by a full factorial design.
Keywords: hexavalent chromium; liquid-liquid microextraction; F AAS.
INTRODUÇÃO
Entre as inúmeras atividades industriais as quais utilizam cromo destacam-se a galvanoplastia, siderurgia, preservação da madeira e curtume.1,2 Como resultado do crescimento dessas atividades
industriais devido a sua ampla utilização, o metalcromo tem sido dispersado cada vez mais no meio ambiente. E os resíduos gerados em consequência de tais atividades são as principais fontes de con-taminação de cromo em corpos d’água.1
Segundo a legislação brasileira, para as águas de classe II e III, o teor máximo de Cr é de 50 µg L-1.3 Em águas naturais as principais
espécies do cromo trivalente - Cr (III) - são dependentes do pH: Cr3+, Cr(OH)2+, Cr(OH)
2+2 e Cr(OH)3. As principais espécies de Cr
(VI) presentes em soluções aquosas são os íons hidrogenoicromato (HCrO4-), cromato (CrO
42-) e dicromato (Cr2O7-2), além de ácido
crômico (H2CrO4), que existe em meio ácido concentrado.
O Cr (III) é considerado não essencial para plantas, mas um elemento traço essencial para animais. Por outro lado, as espécies de Cr (VI) são consideradas carcinogênicas, e, devido à sua natureza fortemente oxidante, e à fácil permeabilidade na membrana biológica, o Cr (VI) danifica as macromoléculas, proteínas e o DNA.4-6
As técnicas instrumentais frequentemente utilizadas para deter-minação elementar incluem a espectrometria de absorção atômica em chama (F AAS), espectrometria de emissão óptica com plasma indutivamente acoplado (ICP OES),7 espectrometria de absorção
atômica com atomização eletrotérmica (ET AAS),8 espectrometria
de massas com plasma indutivamente acoplado (ICP MS)9 e a
espec-trometria de absorção atômica com geração química de vapor (CVG AAS).10 Dentre as técnicas mencionadas, umas das mais utilizadas
para a determinação de vários elementos em diferentes amostras é a F AAS, a qual apresenta uma excelente precisão instrumental e, comparativamente aos outros instrumentos citados, apresenta um menor custo de aquisição e manutenção.
Entretanto, a F AAS possui algumas limitações, tal como o fato desta conseguir detectar concentrações na faixa de mg L-1, o que a
torna inadequada para análise de traços (na região de µg L-1 ou mais
baixo). Além disto, tal técnica pode apresentar dificuldades quanto à determinação de analitos em certas amostras complexas devido à baixa tolerância a interferentes, dificultando a quantificação.11 Desta
maneira, algum método de preparo de amostra deve ser empregado para utilização da técnica de F AAS para determinação de analitos presentes em amostras complexas e em concentrações abaixo do limite de quantificação do instrumento.
De maneira a superar as limitações de detectabilidade da F AAS, e, consequentemente, conseguir atingir baixos limites de detecção, é necessário promover uma etapa de extração e pré-concentração dos analitos de interesse. Isto permite a aplicação de uma instrumentação simples e de menor custo de aquisição e manutenção para a medida final da quantidade do analito. Técnicas como a extração com sol-ventes (sólido-líquido, líquido-líquido),12 extração em fase sólida
(SPE),13 extração no ponto nuvem,14 entre outras, têm sido utilizadas
para pré-concentração de metais traço. Dentre as técnicas de extração comumente utilizadas para concentrar analitos destaca-se a extração em fase sólida (SPE) e suas variações em escala micro,15 como, por
exemplo, a microextração em fase sólida (SPME).16
A técnica de extração com solvente é uma das técnicas mais clássicas utilizadas, sendo denominada de extração líquido-líquido (LLE) quando aplicada a amostras líquidas. Ela consiste na sepa-ração dos analitos da matriz pela distribuição do(s) analito(s) entre dois líquidos imiscíveis ou fases. Porém, há algumas desvantagens neste clássico método de extração. Por exemplo, o uso relativamente grande de solventes orgânicos tóxicos que não são miscíveis em amostras aquosas, o que pode acarretar em problemas ambientais e de saúde, o custo elevado desses solventes que devem ser de alta pureza e ainda morosidade do processo. Por estes motivos, a LLE tem sido modificada para novas configurações, tais como, a micro-extração líquido-líquido (LLME),17,18 microextração com gota única
(SDME)19 e microextração líquido-líquido dispersiva (DLLME).20,21
Y. Cai e colaboradores23 usaram a LLE para determinação de
ésteres de ftalatos em meio aquoso com solventes orgânicos miscíveis, adicionando sais inorgânicos (efeito “salting-out”) para promover a separação de fases. Como vantagem do método desenvolvido os autores apontam o fato de que solventes orgânicos miscíveis em água são geralmente bem menos tóxicos que os solventes apolares convencionalmente utilizados. Para LLE, a adição de sais inorgânicos reforça a partição do analito favoravelmente ao solvente de extração, pois diminui sua solubilidade em fase aquosa, mostrando uma efi-ciência notável na extração.24 Vários parâmetros importantes afetam
a eficiência de extração. Entre eles está o tipo e a quantidade de sal, a razão de fases (volume de amostra/volume de solvente), tempo de extração, grau de agitação e pH da amostra, entre outros.
Neste trabalho, o planejamento fatorial completo foi usado para otimização simultânea das variáveis de um sistema de microextra-ção líquido-líquido baseado no efeito “salting-out” para promover a separação das fases aquosa e de um solvente miscível em água e de baixa toxicidade – acetona. Este sistema foi utilizado para a pré--concentração e a determinação de Cr (VI) em matrizes aquosas com detecção por F AAS.
PARTE EXPERIMENTAL
Materiais e métodos
Instrumentação
Um espectrômetro de absorção atômica Varian SpectrAA 50 (Varian, Victoria, Austrália), equipado com atomizador em chama e lâmpada de cátodo oco de cromo (Hitachi, HLA-4S) foi utilizado. As leituras de absorvância obtidas em área de pico foram realizadas conforme as condições operacionais descritas na Tabela 1.
Reagentes e soluções
As soluções foram preparadas empregando-se água deioniza-da proveniente de um sistema de purificação de água Milli-Q® da
Millipore® (Bedford, MA, EUA). Todos os reagentes usados foram
de grau analítico e utilizados sem purificação prévia. Soluções de trabalhoforam preparadas diariamente a partir de diluições adequadas de uma solução estoque (grau absorção atômica) de Cr (VI) 100 mg L-1 (SPEX, EUA).
Os solventes orgânicos usados no processo de extração foram acetona, n-propanol, metanol e etanol (Merck, Brasil).
As soluções ácidas, utilizadas na substituição do solvente acetona pelo meio aquoso, foram preparadas com HNO3 0,1 mol L-1 (Merck,
Brasil). Ácido clorídrico (Merck, Brasil) foi utilizado para ajustar o pH das amostras. O efeito da força iônica do meio (efeito salting-out) foi investigado com a utilização do sal sulfato de amônio (Nuclear, Brasil). Brancos foram realizados regularmente para verificação da presença do analito nos reagentes utilizados.
Sistema de pré-concentração
O sistema de microextração líquido-líquido empregado neste
trabalho (Figura 1) tem como princípio a extração do Cr (VI) pela acetona em pH em torno de 1. Nesse valor de pH, o Cr (VI) está majoritariamente na sua forma eletricamente neutra, H2CrO4, e é
extraído com solvente orgânico de alta ou média polaridade. Na primeira etapa, 10 mL de amostra aquosa de Cr (VI) 100 µg L-1
foi saturada com sulfato de amônio e o pH foi ajustado com HCl concentrado. A seguir, o volume de solvente extrator requerido pelo planejamento foi adicionado. O frasco foi agitado objetivando uma eficiente extração, e em seguida levado para a centrífuga (3000 rpm, 1 min), sendo esse procedimento necessário para acelerar a separação de fases. Logo após, com as fases claramente separadas, a fase orgâ-nica foi coletada com auxílio de uma pipeta de Pasteur e armazenada em um pequeno frasco plástico (Eppendorf®) de 2 mL. Na segunda
etapa do procedimento foram adicionados 200 µL de solução aquosa ácida (HNO3 0,1 mol L-1) ao extrato orgânico. A seguir esses extratos
acidificados foram levados para a estufa a 80 ºC por cerca de 20 min com a finalidade de evaporar a acetona. O volume final de extrato aquoso permaneceu praticamente inalterado em 200 µL. A fase aquosa contendo o metal foi analisada diretamente por F AAS, sendo o sinal analítico registrado como absorvância integrada. O procedimento descrito foi realizado em todas as etapas de otimização do sistema de microextração líquido-líquido proposto.
RESULTADOS E DISCUSÃO
Para iniciar a otimização deste método foi necessária a realiza-ção de alguns testes preliminares para estudar o comportamento da extração do Cr (VI) com o solvente extrator. Em todas as etapas de otimização do sistema foram realizadas, em paralelo, extrações do branco (amostra sem a adição do metal).
Seleção de sais inorgânicos - efeito “salting out”
Para a escolha do sal foram consideradas a força iônica e a capacidade de separação de fases. Soluções saturadas dos seguintes sais foram estudadas: cloreto de alumínio, cloreto de cálcio, cloreto de sódio e sulfato de amônio e os solventes testados foram acetona, metanol, etanol e n-propanol. A solução saturada de sulfato de amô-nio foi escolhida, pois foi a única em que houve separação de fases usando n-propanol e acetona como solventes, provavelmente devido ao fato de que a solubilidade desse sal é superior a dos outros sais estudados. Além disso, o sulfato de amônio apresenta a dupla carga do íon sulfato que leva a uma maior força iônica. Esperava-se que o uso de uma solução saturada com cloreto de alumínio levasse a uma separação de fases devido à tripla carga do íon alumínio. Entretanto, Tabela 1. Parâmetros operacionais do espectrômetro de absorção atômica
em chama Parâmetro
Comprimento de onda (nm) 357,9 Corrente da lâmpada (mA) 7,0 Largura da fenda (nm) 1,0 Altura do queimador (mm) 20,0 Vazão de acetileno (L min-1) 2,0
devido ao alto grau de hidrólise desse cátion, a fração em que o íon Al3+ se encontra em solução é baixa, não contribuindo tanto como
esperado para a força iônica na solução. O mesmo não acontece com os íons do sal sulfato de amônio, pois ambas são espécies pouco se dissociam/hidrolisam. A solubilidade dos sais avaliados pode ser observada na Tabela 2.
Seleção de solventes orgânicos como fase extratora
A escolha do solvente extrator foi realizada a partir de extrações com sulfato de amônio saturado em pH em torno de 1. O solvente ideal tem que ser miscível em água (por isso, em princípio, de menor toxicidade), extrair o Cr(VI) da solução saturada de sulfato de amônio e facilmente separar as fases aquosa e orgânica. Para isto, os seguintes solventes foram testados: n-propanol, metanol, acetona e etanol. Com o uso de propanol houve separação de fases, mas o analito não foi extraído. Os solventes orgânicos metanol e etanol foram totalmente miscíveis na solução saturada de sulfato de amônio, provocando precipitação desse sal, e, com isso, não houve separação das fases líquidas. Acetona como solvente extrator mostrou-se viável, uma vez que houve uma clara separação de fases e esta extraiu o Cr (VI) da amostra saturada com sulfato de amônio. Dessa maneira, escolheu-se acetona como solvente extrator.
De maneira a desenvolver um método com menor uso de rea-gentes, nenhum agente complexante para o Cr (VI) foi utilizado. No sistema Cr (VI), em pH baixo, a espécie H2CrO4 prevalece, e por estar em meio saturado com sulfato de amônio, essa espécie apresenta maior afinidade pela fase orgânica. Sugere-se que a acetona tenha a capacidade de extrair o ácido crômico devido à interação entre essas duas espécies eletricamente neutras.
Planejamento multivariado para otimização do sistema de pré- concentração
Usando planejamento fatorial completo (N = 2k + C, com k = 3,
onde: N = número de experimentos e k = fatores) em duplicata e com ponto central (C), três fatores foram simultaneamente otimizados. Os três fatores foram pH da amostra, volume de acetona e tempo de agitação para a extração. As condições fixadas do sistema de microextração líquido-líquido foram volume de amostra de 10 mL saturada com sulfato de amônio e concentração de cromo de 100 µg L-1. Na Tabela 3 estão dispostos os valores de nível mínimo, máximo
e central de cada variável estudada.
A partir dos resultados obtidos e usando análise de variância (ANOVA) e probabilidade estatística (p = 0,05) foi construído o gráfi-co de Pareto para determinar a influência dos fatores e suas interações
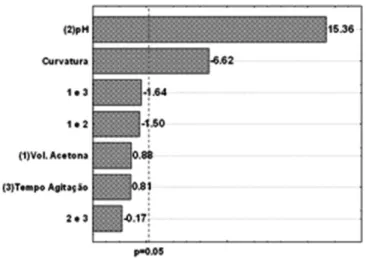
no sistema. O sinal negativo obtido para o pH (Figura 2) indica que a redução de tal variável leva ao aumento na resposta analítica. Isso acontece, pois a espécie neutra H2CrO4 é mais abundante em pH 1,2
comparativamente a pH 2,2, como pode ser observado no diagrama de distribuição de espécies de Cr (VI) em função do pH (Figura 3).
Um sinal positivo do efeito (4,36) obtido para o volume de acetona indica que um aumento no volume do solvente orgânico (nesse caso, de 500 para 700 µL) acarreta em um aumento na resposta analítica. Como todo o volume de solvente extrator que se separa da amostra aquosa foi coletado para obtenção da resposta analítica, é esperada uma melhora na resposta analítica com o aumento do volume extrator. Com relação ao tempo de agitação, observa-se um fato interessante. Embora o efeito principal da variável aponte para melhores respos-tas em direção a menores tempos de extração, o efeito de interação entre as variáveis volume de solvente e tempo de extração possui maior valor (em módulo) e sinal positivo. Dessa forma, esse efeito de interação foi levado em conta para a escolha dos níveis de tempo de extração para o planejamento fatorial seguinte.
De acordo com os resultados do primeiro planejamento fatorial, esse estudo foi repetido, porém desta vez com os níveis de cada variável modificados, com o objetivo de se aproximar de condições mais robustas. As variáveis e níveis escolhidos para a continuação da otimização encontram-se na Tabela 4 e o gráfico de Pareto resultante é apresentado na Figura 4.
Tabela 2. Solubilidade dos sais utilizados no método proposto Sal Solubilidade em g/100 g de H2O (20° C)
(NH4)2SO4 75,4
AlCl3 45,8
CaCl2 74,5
NaCl 35,9
Tabela 3. Fatores e níveis usados no planejamento fatorial 1, para a pré--concentração e extração do Cr (VI) por LLME e determinação por F AAS Fator Mínimo (-1) Central (0) Máximo(1)
pH da amostra 1,2 1,7 2,2
Volume de acetona (µL) 500 600 700 Tempo de agitação (min) 1,00 2,00 3,00
Figura 2. Primeirográfico de Pareto obtido através de planejamento fatorial
completo para pré-concentração e extração do Cr (VI) e determinação por F AAS
Figura 3. Diagrama de distribuição das espécies Cr (VI) em baixa
De acordo com a Figura 4, observa-se um efeito positivo do pH na eficiência de extração, ou seja, as respostas em pH 1,2 são, na média, maiores que em pH 0,7. Embora a espécie extraível de cromo seja mais abundante em pH cada vez mais baixo, o solvente extrator (acetona, pKa = -2,85) é parcialmente protonado pelo forte meio ácido. Isso foi de fato observado, pois em pH 0,7 um volume menor de acetona foi coletado comparativamente a pH 1,2, sugerindo uma solubilização parcial da acetona devido ao pH mais reduzido. Assim, o pH ótimo foi atribuído como 1,2. As outras variáveis não foram significativas, indicando que os intervalos estudados correspondem ao intervalo ótimo de resposta, ou seja, correspondem ao intervalo de robustez do método para essas variáveis. Dessa forma, o volume de acetona e o tempo de agitação foram fixados em 600 µL e 4 min, respectivamente.
A partir das variáveis otimizadas foram obtidos os parâmetros analíticos de mérito, mostrados na Tabela 5, sendo y = 0,12164 + 0,00194 x a equação de reta obtida. Os limites de detecção e quan-tificação foram estimados como 3 e 10 vezes a relação sinal/ruído, respectivamente. O método desenvolvido alcançou limite de quantifi-cação satisfatório comparativamente ao que estabelece a resolução do CONAMA 357/05 para águas classe II e III, que é de 50 µg L-1. A faixa
linear do método corresponde ao método que utiliza o procedimento de pré-concentração. O fator de pré-concentração foi estimado pela relação entre as inclinações entre as curvas de calibração com e sem pré-concentração. O método proposto foi aplicado na determinação de Cr (VI) em amostras de água de captação do Rio Carreiro (Serafina Corrêa, RS) e água da rede de abastecimento de Florianópolis (SC). Devido ao fato da concentração do Cr (VI) ter sido inferior ao limite de detecção do método, as amostras foram fortificadas em diversos níveis de concentração para a verificação da exatidão do método. As inclinações das curvas de calibração das amostras adicionadas de Cr (VI) foram comparadas com a curva de calibração externa com água deionizada. Por essa forma de avalição da exatidão, o método é considerado satisfatório se as inclinações entre as curvas
obtidas por adição de analito e a curva de calibração externa em água deionizada, ambas obtidas pelo método de pré-concentração, não diferirem significativamente. A recuperação (exatidão) foi expressa como a relação entre a inclinação da curva padrão em água com as inclinações das curvas das amostras. (Tabela 6). Excelentes recupe-rações na faixa de 96-106% foram obtidas, indicando que o método sofre pouca influência dos componentes da matriz. Desta forma, a quantificação pode ser realizada com a curva de calibração externa obtida com água deionizada.
CONCLUSÃO
O sistema de LLME proposto nesse trabalho apresenta como vantagens o baixo consumo de solventes orgânicos, fatores de enri-quecimento satisfatórios, dispensa o uso de agentes complexantes e de reagentes de alta toxicidade, simplificando assim a etapa de pre-paro de amostra. O método apresentou limites de detecção bastante satisfatórios, especialmente se for levada em consideração a técnica instrumental relativamente simples utilizada. Menores limites de detecção poderiam ser obtidos pelo uso de uma técnica de detecção mais sensível. Sugere-se que outros elementos que formem espécies não carregadas em um determinado pH também possam ser extraídos por essa técnica. Sugere-se também que reações químicas específicas entre o solvente e o analito, formando espécies solúveis no solvente extrator, possam ser utilizadas como força motriz no processo de extração, aumentando a seletividade/especificidade do método. REFERÊNCIAS
1. Martendal, E.; Maltez, H.F.; Carasek, E.; J. Hazard. Mater.2009, 161, 450.
2. Abreu, M.A.; Toffoli, S.M.; Ceram. Int.2009, 35, 2225.
3. http://www.mma.gov.br/port/conama/res/res05/res35705.pdf, acessada em Janeiro 2010.
4. Sena, M.M.; Collins, C.H.; Collins, K.E.; Quím. Nova 2011, 24, 331. 5. Ferreira, A.D.Q.; Quim. Nova2002, 25, 572.
6. Béni, Á.; Karosi, R.; Posta, J.;Microchem. J. 2007, 85, 103.
7. Boevski, I.; Daskalovau, N.; Havezov, I. Spectrochim. Acta, Part B 2000, 55, 1643.
8. Oliveira, E.P.; Santelli, R.E.; Cassella, R.J.; Microchem. J. 2008, 89, 116. 9. Bravoa, I.D.B.; Castroa, R.S.; Riquelme, N.L.; Diaz, C.T.; Goyenagaa,
D.A.; J. Trace Elem. Med. Biol. 2007, 21, 14. Tabela 4. Fatores e níveis usados no planejamento fatorial 2, para a
pré--concentração e extração do Cr (VI) por LLME e determinação por F AAS Fator Mínimo (-1) Central (0) Máximo(1) pH da solução 0,70 0,95 1,20 Volume de acetona (µL) 550 600 650 Tempo de agitação (min) 3,00 4,50 6,00
Tabela 5. Parâmetros analíticos de mérito para determinação de Cr (VI) em amostras aquosas e determinação por F AAS
Faixa linear, µg L-1 10 – 100
Coeficiente de correlação (R) 0,998 Desvio padrão relativo (DPR%) (45 µg L-1, n = 5) 4,7
Limite de detecção, µg L-1 3
Limite de quantificação, µg L-1 10
Fator de enriquecimento (FE) 38 vezes Tabela 6. Valores de exatidão do método proposto para determinação de Cr (VI) obtidos pela fortificação das amostras submetidas ao procedimento de pré-concentração
Amostras Faixa adição ( µg L-1)
Exatidão (%) Água do Rio Carreiro (RS) 0 – 100 95,6 – 102,2 Água da rede de abastecimento de
Florianópolis(SC)
0 – 100 97,8 – 105,7
Figura 4. Segundo gráfico de Pareto obtido através de planejamento
10. Sola-Larrañaga, C.; Navarro-Blasco, I.;Food Chem. 2009, 115,1048. 11. Budziak, D.; Silva, E.L.; Carasek, E.; Campos, S.D.; Microchim. Acta
2003, 141,169.
12. Shahawi, M.S.E.; Bashammakh, A.S.; Bahaffi, S.O.; Talanta 2007, 72, 1494.
13. Capelo, J.L.; Filgueiras, A.V.; Lalilla, I.; Bendicho, C.; Talanta 1999, 50,905.
14. Sussulini, A.; Arruda, M.A.Z.; Eclet. Quím.2006, 31, 73.
15. Budziak, D.; Martendal, E.; Carasek, E.; J. Chromatogr., A,2008, 1198, 54.
16. Kaur, V.; Malik, A.K.; Talanta 2007, 73, 425.
17. Xu, L.; Basheer, C.; Lee, H.K.; J. Chromatogr., A 2009, 1216, 701. 18. Nazari S.; J. Hazard. Mater.2009, 165,200.
19. Pena, F.; Lavilla, I.; Bendicho, C.;Spectrochim. Acta, Part B 2008, 63, 498.
20. Rivas, R.E.; Garcia, I.L.; Córdoba,M.H.;Spectrochim. Acta, Part B 2009, 64, 329.
21. Anthemidis, A.N.; Ioannou,K.I.G.; Talanta 2009, 79, 86. 22. Carasek, E.; Tonjes, J.W.; Scharf, M.; Talanta 2002, 56, 185. 23. Cai, Y.; Shi, Y.; Liu, J; Mou, S.; Lu, Y.; Microchim. Acta2007, 157, 73.