FACULDADE DE CI ˆENCIAS FARMAC ˆEUTICAS DE RIBEIR ˜AO PRETO
An´alises de propriedades eletrost´aticas e estruturais
de complexos de prote´ınas para o desenvolvimento de preditores de
complexac¸˜ao em larga escala
Tulio Marcus Ribeiro Calixto
FACULDADE DE CI ˆENCIAS FARMAC ˆEUTICAS DE RIBEIR ˜AO PRETO
An´alises de propriedades eletrost´aticas e estruturais
de complexos de prote´ınas para o desenvolvimento de preditores de
complexac¸˜ao em larga escala
Dissertac¸˜ao de Mestrado apresentada ao Programa de P´os-Graduac¸˜ao em Ciˆencias Farmacˆeuticas para obtenc¸˜ao do T´ıtulo de Mestre em Ciˆencias
´
Area de Concentrac¸˜ao: F´ısica Biol´ogica
Orientado: Tulio Marcus Ribeiro Calixto
Orientador: Fernando Lu´ıs Barroso da Silva
AUTORIZO A REPRODUC¸ ˜AO E DIVULGAC¸ ˜AO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETR ˆONICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE.
Calixto, Tulio Marcus Ribeiro
An´alises de propriedades eletrost´aticas e estruturais de complexos de prote´ınas para o desenvolvimento de preditores de complexac¸˜ao em larga escala. Riber˜ao Preto, 2010.
228p.; 30cm.
Dissertac¸˜ao de Mestrado, apresentada `a Faculdade de Ciˆencias Farmacˆeuticas de Ribeir˜ao Preto/USP - ´Area de concentrac¸˜ao: F´ısica Biol´ogica.
Orientador: da Silva, Fernando Lu´ıs Barroso.
Tulio Marcus Ribeiro Calixto
An´alises de propriedades eletrost´aticas e estruturais de complexos de prote´ınas para o desenvolvimento de preditores de complexac¸˜ao em larga escala
Dissertac¸˜ao de Mestrado apresentada ao Programa de P´os-Graduac¸˜ao em Ciˆencias Farmacˆeuticas para obtenc¸˜ao do T´ıtulo de Mestre em Ciˆencias
´
Area de Concentrac¸˜ao: F´ısica Biol´ogica
Orientador: Fernando Lu´ıs Barroso da Silva
Aprovado em:
Banca Examinadora
Prof. Dr.
Instituic¸˜ao: Assinatura:
Prof. Dr.
Instituic¸˜ao: Assinatura:
Prof. Dr.
Agradecimentos
Inicio agradecendo a Deus, pela minha vida e pela forc¸a nos momentos de desˆanimo e cansac¸o.
A minha fam´ılia pelo amor, carinho e compreens˜ao durante todo o tempo de realizac¸˜ao da minha p´os-graduac¸˜ao, especialmente ao meu irm˜ao Jos´e Sim˜ao Calixto J´unior, pela boa convivˆencia e paciˆencia para comigo.
Ao amigo Rodrigo Faccioli, pela hospedagem em S˜ao Carlos, estudos, parceria no desenvolvimento desoftwares, desabafos e pelas longas conversas e discuss˜oes acadˆemicas e alheias.
Ao Centro de Inform´atica de Ribeir˜ao Preto, especialmente a minha chefe Cl´elia Cardoso Camargo, que sempre me apoiou durante a p´os-graduc¸˜ao e ao amigo Ali Faiez Taha, pelas discuss˜oes, cr´ıticas e incentivo aos estudos.
Agradec¸o ao meu orientador, Prof. Dr. Fernando Lu´ıs Barroso da Silva, que me iniciou na ´area de F´ısica Biol´ogica e forneceu o conhecimento necess´ario para o desenvolvimento deste trabalho. Reconhec¸o e agradec¸o a oportunidade, apoio, dedicac¸˜ao, paciˆencia, confianc¸a e amizade.
A todos os amigos que passaram pela rep´ublica, Andr´e Lara, Lucas At´ılio, F´abio Marcondez, Leandro Nassif, Marco Antˆonio, L´ıvio Leite, Ivan Farjala, Rodrigo Takeuchi, Fl´avio Neto, Eduardo, Jos´e Regis, Guilherme e em especial Fl´avio Henrique Alves, por todo apoio nesta nova fase da minha vida que se iniciou no ano 2000, pelas conversas e reflex˜oes noturnas, projetos, sonhos, festas, caronas pra Ita´u de Minas, enfim todos os momentos alegres e outros nem tanto.
Aos colegas do laborat´orio de F´ısica Biol´ogica, Jo˜ao Dalmolin, Ricardo, Lariani, Eliamar, Andr´e, pela amizade e companheirismo.
Aos membros da banca do exame geral de qualificac¸˜ao Prof. Dr. Antˆonio Caliri e Prof. Dr. Renato Tin´os pela disponibilidade de ler, criticar e fazer valiosas su-gest˜oes para a melhoria deste trabalho.
A todos os funcion´arios da sec¸˜ao de p´os-graduac¸˜ao e a Faculdade de Ciˆencias Far-macˆeuticas de Ribeir˜ao Preto pelo oportunidade de cursar o mestrado.
Resumo
CALIXTO, T. M. R.An´alises de propriedades eletrost´aticas e estruturais de complexos de
prote´ınas para o desenvolvimento de preditores de complexac¸˜ao em larga escala. 2010.
228f. Dissertac¸˜ao (Mestrado) - Faculdade de Ciˆencias Farmacˆeuticas de Ribeir˜ao Preto,
Uni-versidade de S˜ao Paulo, Ribeir˜ao Preto, 2010.
Estudos te´oricos dos mecanismos moleculares respons´aveis pela formac¸˜ao e estabilidade de complexos moleculares vˆem ganhando relevˆancia pelas possibilidades pr´aticas que oferecem, por exemplo, na compreens˜ao de diversas doenc¸as e no desenho racional de f´armacos. Neste projeto, nossa ˆenfase est´a no estudo de complexos de prote´ınas, extra´ıdos do banco de dados de prote´ınas (PDB), onde desenvolvemos ferramentas computacionais as quais permitem efetuar an´alises em duas direc¸˜oes: 1) efetuar previs˜oes b´asicas, atrav´es do emprego de propriedades ele-trost´aticas de prote´ınas, em diferentes condic¸˜oes e n´ıveis preditivos e 2) realizac¸˜ao de um con-junto de an´alises estat´ısticas, como freq¨uˆencia de contato, em busca de preditores de complexos de prote´ınas e identificar padr˜oes de interac¸˜ao entre seus amino´acidos em func¸˜ao da distˆancia de separac¸˜ao. Com base nos resultados obtidos por ambos os estudos, objetivamos quantificar as forc¸as f´ısicas envolvidas na formac¸˜ao dos complexos prot´eicos. O foco do projeto, a longo prazo, ´e prever o fenˆomeno da complexac¸˜ao atrav´es da fus˜ao dessas duas linhas de estudos: pre-ditor b´asico de complexos prot´eicos e an´alise do potencial estat´ıstico entre os amino´acidos que formam o complexo. O presente projeto ´e conclu´ıdo com a construc¸˜ao de portaiswebque dis-ponibilizar˜ao os resultados obtidos por nossos trabalhos bem como a possibilidade de qualquer usu´ario, efetuar consultas por propriedades de prote´ınas e/ou grupo de prote´ınas.
Abstract
CALIXTO, T. M. R.Analysis of electrostatics and structural properties of protein
comple-xes to the development of complexation predictors in high-throughput computing. 2010.
228f. Dissertation (Master) - Faculdade de Ciˆencias Farmacˆeuticas de Ribeir˜ao Preto,
Univer-sidade de S˜ao Paulo, Ribeir˜ao Preto, 2010.
Theoretical studies of the molecular mechanisms responsible for the formation and stability of molecular complexes are gaining relevance for the practical possibilities that they offer, for example, in the understanding of diverse diseases and the rational drug design. In this project, our emphasis is on the study of protein complexes, extracted from the protein data bank (PDB). We have developed computational tools which allow to perform analyses in two directions: 1) to make basic complexation forecasts, through the use of electrostatic properties of proteins, in different conditions and predictive levels, and 2) to carry out a set of statistical analyses, as contacts frequency, in order to build up predictors of protein complexes and to identify patters of interactions between the amino acids as a function of their separation distance. Based on the results obtained on both studies, we aim quantify the physical forces involved in the formation of protein complexes. The focus of the project, in the long run, is to foresee the phenomenon of the protein complexes through the fusing of these two study lines: a coarse-grained predictor of protein complexes and analysis of the statistical potentials between the amino acids that form the complex. The present project is concluded with the construction of web services where we make available the results obtained on our works. This server also has the possibility to be used by any computer user, that wishes to perform search on protein and/or protein group properties.
Lista de Figuras
1 Diagrama esquem´atico dos portais desenvolvidos. Elementos em cinza indicam as ferramentas que ser˜ao implementadas no futuro. . . 15
2 Ilustrac¸˜ao esquem´atica de funcionamento dos portais web propostos neste tra-balho. . . 16
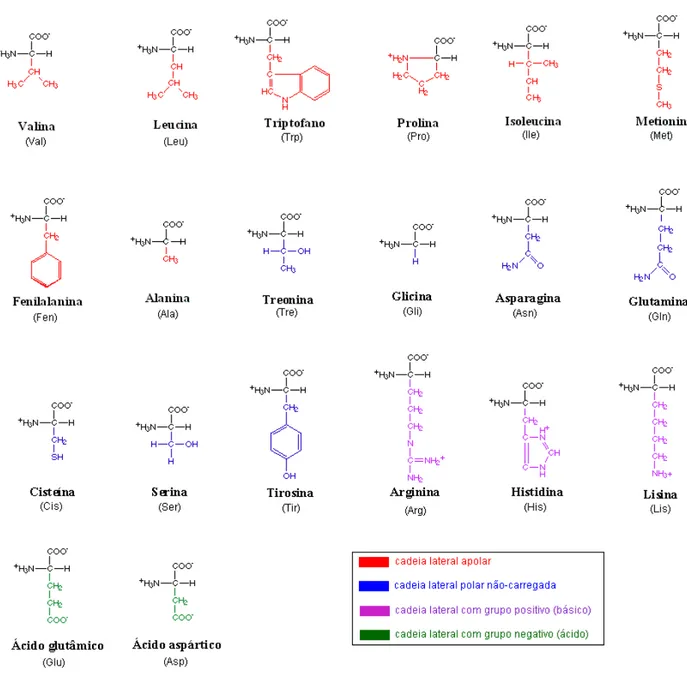
3 Estrutura geral de um amino´acido. . . 21
4 Relac¸˜ao dos 20 amino´acidos existentes na natureza, adaptado da referˆencia (1). 22
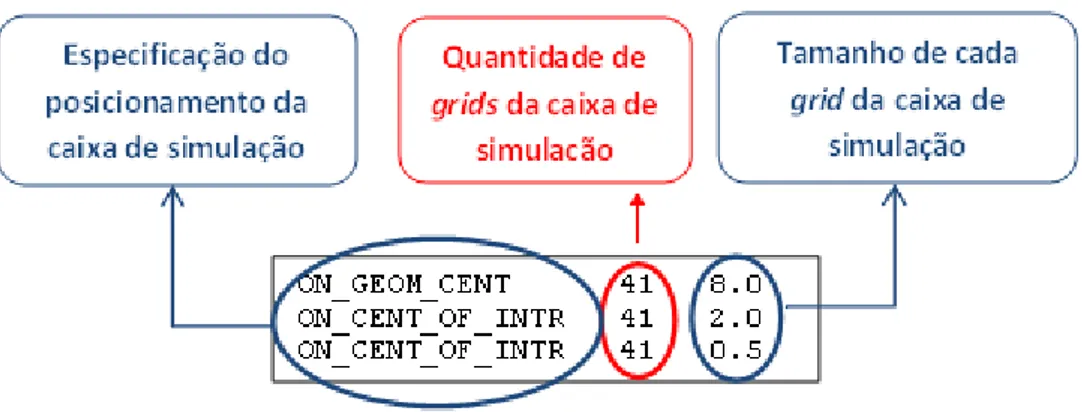
5 Exemplo de um arquivo no formato ogm, utilizado como entrada pelo pacote MEAD v.2.2.7. . . 30
6 Ilustrac¸˜ao esquem´atica de uma prote´ına inserida em uma rede para execuc¸˜ao do m´etodo de diferenc¸as finitas para a soluc¸˜ao da EPBL. . . 30
7 Modelo relacional do banco de dados. . . 47
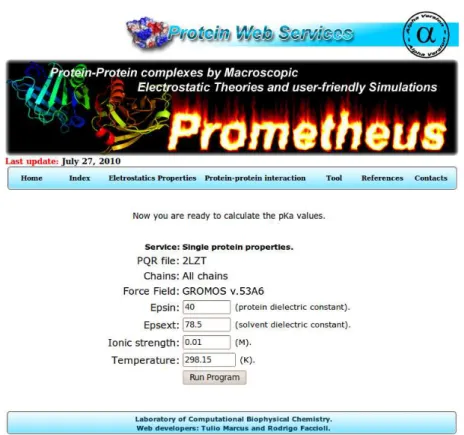
8 P´agina inicial do portalwebPROMETHEUS. Dispon´ıvel em: http://glu. fcfrp.usp.br/services.htm. . . 50
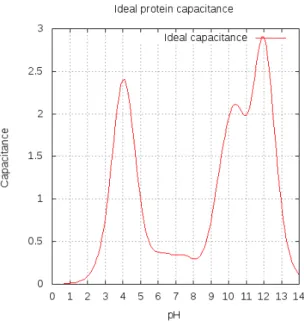
9 Ferramenta ”Single amino acid properties”. . . 51 10 Curvas de titulac¸˜ao e capacitˆancia ideais em func¸˜ao do pH do amino´acido ´acido
glutˆamico (GLU), obtidas pela ferramenta ”Single Amino acid Properties”. . . . 52 11 Curva de titulac¸˜ao ideal do amino´acido ´acido glutˆamico (GLU). pKa= 4,4 (2). . 52
12 Curva da capacitˆancia ideal em func¸˜ao do pH, do amino´acido ´acido glutˆamico (GLU). . . 53
13 Tela de aquisic¸˜ao de parˆametros para utilizac¸˜ao da ferramenta “Single protein properties”. . . 54 14 Tela para apresentac¸˜ao dos resultados obtidos pela ferramenta “Single protein
16 Curva da capacitˆancia, em func¸˜ao do pH, da prote´ına lisozima (PDB: 2LZT). . 56
17 Tela para configurac¸˜ao dos parˆametros iniciais do portal PROMETHEUS. . . . 57
18 Tela para a especificac¸˜ao dos parˆametros f´ısico-qu´ımicos para entrada no pro-gramamultiflex. . . 58 19 Tela para apresentac¸˜ao das curvas de titulac¸˜ao e capacitˆancia em func¸˜ao do pH,
geradas pela ferramenta “Single protein properties”. . . 58 20 Curva de titulac¸˜ao da prote´ına lisozima (PDB: 2LZT), no n´ıvel de predic¸˜ao
Poisson-Boltzmann. . . 59 21 Curva da capacitˆancia, em func¸˜ao do pH, da prote´ına lisozima (PDB: 2LZT),
no n´ıvel de predic¸˜aoPoisson-Boltzmann. . . 59 22 Tela para entrada dos parˆametros iniciais para a realizac¸˜ao dos c´alculos da
predic¸˜ao de complexac¸˜ao entre prote´ınas. . . 60
23 Tela para entrada dos parˆametros f´ısico-qu´ımicos para predic¸˜ao de complexos prot´eicos, no n´ıvel de predic¸˜ao ideal (anal´ıtico). . . 62
24 Tela para apresentac¸˜ao dos c´alculos anal´ıticos de ∆Gele (em unidades de kBT)
em func¸˜ao da distˆancia de separac¸˜ao (em ˚Angstr¨om) no pH 10,4 e doB23 em func¸˜ao do pH, em forc¸a iˆonica nula, para a complexac¸˜ao entre as prote´ınas liso-zima (PDB: 2LZT) e tirosina kinase (PDB: 1LCJ). . . 63
25 ∆Gele (anal´ıtico), no pH 10,4 e forc¸a iˆonica nula, para a complexac¸˜ao entre as prote´ınas lisozima (PDB: 2LZT) e tirosina kinase (PDB: 1LCJ). . . 64
26 B23(anal´ıtico) em func¸˜ao do pH, em forc¸a iˆonica nula, para a complexac¸˜ao entre as prote´ınas lisozima (PDB: 2LZT) e tirosina kinase (PDB: 1LCJ). . . 64
27 Tela para entrada dos parˆametros que ser˜ao utilizados para a construc¸˜ao dos arquivos de configurac¸˜ao utilizados pelo pacote MEAD para o c´alculo dospKa’s
dos amino´acidos ioniz´aveis. . . 65
28 Tela para definic¸˜ao das condic¸˜oes experimentais das simulac¸˜oes com as estru-turas tridimensionais. . . 66
30 Tela para apresentac¸˜ao do∆Gele, no pH 11,1 em forc¸a iˆonica igual a 0,01M, e do
B23, para a complexac¸˜ao entre as prote´ınas lisozima (PDB: 2LZT) e calbindina (PDB: 3ICB). . . 68
31 ∆Gele entre as prote´ınas calbindina (PDB: 3ICB) e lisozima (PDB: 2LZT), em forc¸a iˆonica igual a 0,01M. . . 69
32 B23 entre as prote´ınas calbindina (PDB: 3ICB) e lisozima (PDB: 2LZT). . . 69 33 Tela da ferramenta que permite criar um arquivo no formato PQR a partir de um
arquivo PDB. . . 71
34 Ferramenta “Create MEAD files”. . . 72 35 Tela para aquisic¸˜ao dos parˆametros experimentais, utilizados pela ferramenta
“Split proteins”. . . 73 36 Complexos de prote´ınas separados em prote´ınas independentes pela ferramenta
“Split proteins”. . . 74 37 Curvas de titulac¸˜ao ideal de cada prote´ına individual, presente no complexo
proteinase-inibidor (PDB: 2PTC). . . 75
38 Curvas de capacitˆancia ideal em func¸˜ao do pH de cada prote´ına individual, pre-sente no complexo proteinase-inibidor (PDB: 2PTC). . . 75
39 Tela de aquisic¸˜ao dos parˆametros utilizados pela ferramenta “Find best case”. . 76 40 Tela para apresentac¸˜ao do resultado obtido pela ferramenta “Find best case”,
para um conjunto de prote´ınas, no pH 7,5. . . 77
41 Ferramenta que efetua a preparac¸˜ao inicial de um arquivo no formato PDB. . . 79
42 Exemplo de um arquivo no formato PDB ap´os ser processado pela ferramenta “Clean PDB”. . . 80 43 Tela inicial para ordenac¸˜ao de prote´ınas e/ou complexos de prote´ınas pelo pI. . 81 44 Tela para entrada dos parˆametros f´ısico-qu´ımicos utilizados para a predic¸˜ao da
complexac¸˜ao entre duas prote´ınas. . . 82
48 Tela para inserc¸˜ao dos c´odigos PDB’s dos complexos de prote´ınas que ser˜ao analisados pela ferramenta “Statistical potential” do portal MOLESA. . . 85 49 Tela para configurac¸˜ao dos parˆametros que ser˜ao utilizados pela ferramenta
“Statistical potential”. . . 87 50 Ilustrac¸˜ao de um complexo prot´eico esquem´atico formado por duas prote´ınas
(representadas pelas cadeiasAeB) para a realizac¸˜ao do c´alculo da distˆancia de separac¸˜ao entre os res´ıduos presentes em cada prote´ına. . . 87
51 Ilustrac¸˜ao do contador de freq¨uˆencias entre os amino´acidos i e j de um complexo prot´eico esquem´atico. . . 88
52 Freq¨uˆencia de contatos e potencial de forc¸a m´edia entre os res´ıduosie j. . . 89 53 Freq¨uˆencia de contatos (com e sem normalizac¸˜ao) entre os res´ıduosie j. . . 90 54 Potencial de forc¸a m´edia entre os res´ıduosie j, normalizados de forma
proba-bil´ıstica e com base na FDR. . . 90
55 Curva de titulac¸˜ao ideal do amino´acido alanina (ALA). . . 94
56 Curva da capacitˆancia ideal em func¸˜ao do pH, do amino´acido alanina (ALA). . 95
57 Curva de titulac¸˜ao ideal do amino´acido arginina (ARG). pKa= 12,0 (2). . . 96
58 Curva da capacitˆancia ideal em func¸˜ao do pH, do amino´acido arginina (ARG). . 96
59 Comparac¸˜ao entre as curvas de titulac¸˜ao te´orica e experimental da prote´ına li-sozima (PDB: 2LZT) em forc¸a iˆonica igual a 0,1M. . . 97
60 Comparac¸˜ao entre as curvas de titulac¸˜ao te´oricas obtidas analiticamente e por simulac¸˜ao Monte Carlo da prote´ına calbindina (PDB: 3ICB) em forc¸a iˆonica nula. 98
61 Comparac¸˜ao entre a titulac¸˜ao ideal (curva vermelha), obtida pelo portal PROMETHEUS, e titulac¸˜ao MA, obtida por simulac¸˜ao Monte Carlo (curva verde - concentrac¸˜ao da prote´ına: 150µM; concentrac¸˜ao de sal: 0,15M (3)) da prote´ınaβ-lactoglobulina (PDB: 1BEB). pIexperimental: 5,18 (4). . . 99 62 Comparac¸˜ao entre a capacitˆancia ideal em func¸˜ao do pH, da prote´ına lisozima
(PDB: 2LZT) provida pelo portal PROMETHEUS e a obtida da referˆencia (5). . 100
64 RMSD dos pKa’s da lisozima (PDB: 2LZT) em func¸˜ao de diferentes valores de
EPSIN para o campo de forc¸a AMBER99. A concentrac¸˜ao de sal foi variada de 0,01M a 0,15 M. A temperatura e a constante diel´etrica do solvente foram fixadas em 298 K e 80, respectivamente. Os dados experimentais das referˆencias (6–8) foram usados para o c´alculo do RMSD. . . 115
65 RMSD dos pKa’s da BPTI (PDB: 4PTI) em func¸˜ao de diferentes valores de
EPSIN para o campo de forc¸a GROMOS96. A concentrac¸˜ao de sal foi variada de 0,01M a 0,15 M. A temperatura e a constante diel´etrica do solvente foram fixadas em 298 K e 80, respectivamente. Os dados experimentais das referˆencias (9–12) foram usados para o c´alculo do RMSD. . . 116
66 RMSD dos pKa’s da lisozima (PDB: 2LZT) em forc¸a iˆonica igual a 0,1M em
func¸˜ao de diferentes valores de EPSIN para os campos de forc¸a GROMOS96 e AMBER99. A temperatura e a constante diel´etrica do solvente foram fixadas em 298 K e 80, respectivamente. Os dados experimentais das referˆencias (6–8) foram usados para o c´alculo do RMSD. . . 117
67 RMSD dos pKa’s da BPTI (PDB: 4PTI) em forc¸a iˆonica igual a 0,1M em
func¸˜ao de diferentes valores de EPSIN para os campos de forc¸a GROMOS96 e AMBER99. A temperatura e a constante diel´etrica do solvente foram fixadas em 298 K e 80, respectivamente. Os dados experimentais das referˆencias (9–12) foram usados para o c´alculo do RMSD. . . 117
68 Comparac¸˜ao entre a titulac¸˜ao ideal e a titulac¸˜ao baseada na estrutura 3D (PB e MC), para a cabindina (PDB: 3ICB). pI experimental: 4,5 (14). Os dados de MC foram retirados da referˆencia (15). . . 119
69 Comparac¸˜ao entre a capacitˆancia ideal e a capacitˆancia baseada na estrutura 3D (PB e MC) em func¸˜ao do pH, para a cabindina (PDB: 3ICB). Os dados de MC foram retirados da referˆencia (15). . . 119
71 Curva de titulac¸˜ao da prote´ına lisozima (PDB: 2LZT), em v´arias concentrac¸˜oes de sal. A temperatura, a constante diel´etrica da prote´ına e a constante diel´etrica do solvente foram fixadas em 298 K, 40 e 80, respectivamente. Campo de forc¸a: GROMOS96. . . 122
72 Curva de titulac¸˜ao da prote´ına calbindina (PDB: 3ICB), em v´arias concentrac¸˜oes de sal. A temperatura, a constante diel´etrica da prote´ına e a cons-tante diel´etrica do solvente foram fixadas em 298 K, 40 e 80, respectivamente. Campo de forc¸a: GROMOS96. . . 122
73 Comparac¸˜ao do∆Gele do complexo hirundina–trombina (PDB: 4HTC), com e sem o mecanismo de regulac¸˜ao de cargas em forc¸a iˆonica igual a 0,01M. . . 123
74 Comparac¸˜ao do B23, do complexo hirundina–trombina (PDB: 4HTC), com e sem o mecanismo de regulac¸˜ao de cargas em forc¸a iˆonica igual a 0,01M. . . 124
75 Curva de titulac¸˜ao do complexo hirundina–trombina (PDB: 4HTC), separado em duas prote´ına, em forc¸a iˆonica nula. . . 125
76 Curva da capacitˆancia em func¸˜ao do pH, do complexo hirundina–trombina (PDB: 4HTC), separado em duas prote´ına, em forc¸a iˆonica nula. . . 125
77 Curvas de titulac¸˜ao de cada prote´ına que forma o complexo prot´etico tripsina– inibidor (PDB: 2PTC). . . 126
78 Curvas da capacitˆancia de cada prote´ına que forma o complexo prot´etico tripsina–inibidor (PDB: 2PTC). . . 127
79 ∆Gele formando o complexo prot´eico tripsina–inibidor (PDB: 2PTC). O pH foi fixado em 10 e forc¸a iˆonica nula. . . 127
80 B23 formando o complexo prot´eico tripsina–inibidor (PDB: 2PTC). pH experimental:10 (16, 17). . . 128
81 Comparac¸˜ao do ∆Gele do complexo HyHEL-10 Fab–lisozima (PDB: 3HFM), com e sem o mecanismo de regulac¸˜ao de cargas. O pH e a forc¸a iˆonica foram fixados 10,6 e 0,01M, respectivamente. . . 129
82 Comparac¸˜ao do B23, do complexo HyHEL-10 Fab–lisozima (PDB: 3HFM), com e sem o mecanismo de regulac¸˜ao de cargas em forc¸a iˆonica igual a 0,01M. 129
84 ∆Gele do complexo formado por duas lisozimas (PDB: 2LZT) com e sem o mecanismo de regulac¸˜ao de cargas, em forc¸a iˆonica nula e 0,01M. O pH, a temperatura e a constante diel´etrica do solvente foram fixados em 10,6, 298,15 K e 78,5, respectivamente. . . 131
85 ∆Gele, nos n´ıveis de predic¸˜ao anal´ıtico ePoisson-Boltzmann, do complexo for-mado por duas lisozimas (PDB: 2LZT) com e sem o mecanismo de regulac¸˜ao de cargas em forc¸a iˆonica igual a 0,01M. A temperatura, a constante diel´etrica da prote´ına e a constante diel´etrica do solvente foram fixadas em 298 K, 40 e 80, respectivamente. O pH foi mantido constante em 10,6 para os c´alculos anal´ıticos e 11,2 para os c´alculos utilizando PB. Campo de forc¸a: GROMOS96. 132
86 B2 de complexac¸˜ao entre duas lisozimas (PDB: 2LZT), com o mecanismo de regulac¸˜ao de cargas, em v´arios regimes de forc¸a iˆonica. . . 133
87 Comparac¸˜ao do B2 de complexac¸˜ao entre duas lisozimas (PDB: 2LZT), nos n´ıveis de predic¸˜ao anal´ıtico e PB, com o mecanismo de regulac¸˜ao de cargas, com medidas experimentais e outras previs˜oes te´oricas. A forc¸a iˆonica foi fixada em 0,005M. Nos c´alculos por PB, εpfoi definido como igual a 40. Os campos
de forc¸a est˜ao citados nas legendas das curvas do pr´oprio gr´afico. Os dados experimentais foram obtidos da referˆencia (18). . . 133
88 Comparac¸˜ao do B2 de complexac¸˜ao entre duas lisozimas (PDB: 2LZT), nos n´ıveis de predic¸˜ao anal´ıtico e PB, com o mecanismo de regulac¸˜ao de cargas, com medidas experimentais e outras previs˜oes te´oricas. A forc¸a iˆonica foi fixada em 0,1M. Nos c´alculos por PB, εp foi definido como igual a 40. Os campos
de forc¸a est˜ao citados nas legendas das curvas do pr´oprio gr´afico. Os dados experimentais foram obtidos da referˆencia (18). . . 134
89 Comparac¸˜ao do B2 de complexac¸˜ao entre dois quimotripsinogˆenios (PDB: 1CHG), nos n´ıveis de predic¸˜ao anal´ıtico e PB, com o mecanismo de regulac¸˜ao de cargas, com medidas experimentais e outras previs˜oes te´oricas. A forc¸a iˆonica foi fixada em 0,005M. Nos c´alculos por PB,εpfoi definido como igual a
90 Comparac¸˜ao do B2 de complexac¸˜ao entre dois quimotripsinogˆenios (PDB: 1CHG), nos n´ıveis de predic¸˜ao anal´ıtico e PB, com o mecanismo de regulac¸˜ao de cargas, com medidas experimentais e outras previs˜oes te´oricas. A forc¸a iˆonica foi fixada em 0,01M e 0,005. Nos c´alculos por PB,εpfoi definido como
igual a 40. As cargas foram definidas de acordo com o campo forc¸a AMBER99. Os dados experimentais foram obtidos da referˆencia (18). . . 136
91 Comparac¸˜ao do B2, com e sem o potencial de dispers˜ao de Hamaker, de complexac¸˜ao entre duas lisozimas (PDB: 2LZT), com o mecanismo de regulac¸˜ao de cargas e forc¸a iˆonica nula. . . 137
92 Comparac¸˜ao do∆Gele com e sem o mecanismo de regulac¸˜ao de cargas. O pH foi fixado em 4,5 e forc¸a iˆonica nula. . . 138
93 Freq¨uˆencia de contatos entre os res´ıduos ALA–ALA para as prote´ınas dos conjuntos 3, 4, 5 e controle. A normalizac¸˜ao das curvas seguiu o crit´erio 1. Os conjuntos est˜ao especificados nas legendas no interior do gr´afico. . . 141
94 Freq¨uˆencia de contatos entre os res´ıduos GLU–GLU para as prote´ınas dos conjuntos 3, 4, 5 e controle. A normalizac¸˜ao das curvas seguiu o crit´erio 1. Os conjuntos est˜ao especificados nas legendas no interior do gr´afico. . . 142
95 Freq¨uˆencia de contatos entre os res´ıduos ILE–VAL para as prote´ınas dos conjuntos 3, 4, 5 e controle. A normalizac¸˜ao das curvas seguiu o crit´erio 1. Os conjuntos est˜ao especificados nas legendas no interior do gr´afico. . . 142
96 PFM obtido a partir da freq¨uˆencia de contatos entre os res´ıduos ALA-ALA presentes nas prote´ınas pertencentes aos conjunto 3, 4, 5 e controle. A normalizac¸˜ao foi feita pelo crit´erio 1. . . 143
97 PFM obtido a partir da freq¨uˆencia de contatos entre os res´ıduos GLU-GLU presentes nas prote´ınas pertencentes aos conjunto 3, 4, 5 e controle. A normalizac¸˜ao foi feita pelo crit´erio 1. . . 144
98 PFM obtido a partir da freq¨uˆencia de contatos entre os res´ıduos ILE-VAL presentes nas prote´ınas pertencentes aos conjunto 3, 4, 5 e controle. A normalizac¸˜ao foi feita pelo crit´erio 1. . . 144
100 PFM obtido a partir da freq¨uˆencia de contatos entre os res´ıduos GLU-GLU presentes nas prote´ınas pertencentes aos conjunto 3, 4, 5 e controle. A normalizac¸˜ao foi feita pelo crit´erio 2. . . 145
101 PFM obtido a partir da freq¨uˆencia de contatos entre os res´ıduos ILE-VAL presentes nas prote´ınas pertencentes aos conjunto 3, 4, 5 e controle. A normalizac¸˜ao foi feita pelo crit´erio 2. . . 146
102 Comparac¸˜ao entre os crit´erios de normalizac¸˜ao 1 e 2 no c´alculo do PFM entre os res´ıduos ALA–ALA presentes nas prote´ınas pertencentes ao conjunto controle. 147
103 Modelo do arquivo de informac¸˜oes criado ap´os a conclus˜ao do processamento da ferramenta “Single protein properties”, para o c´alculo da titulac¸˜ao ideal da prote´ınaβ-lactoglobulina bovina (PDB: 1BEB). . . 151
104 Ilustrac¸˜ao do modelo MVC (Model View Control) utilizado no desenvolvimento dos portaisweb. . . 153 105 Organizac¸˜ao do sistema em relac¸˜ao `a origem da fonte de dados que ser˜ao
pro-cessados. . . 154
106 Exemplo de um arquivo no formato PQR, mostrando o primeiro amino´acido de uma prote´ına. . . 188
107 Exemplo de um arquivo de configurac¸˜ao no formatosites. . . 189 108 Exemplo de um arquivo de configurac¸˜ao no formato st do amino´acido ´acido
glutˆamico (GLU). Neste exemplo utilizamos o campo de forc¸a AMBER99 para prover as cargas e os raios de cada ´atomo. . . 189
109 Exemplo de um arquivo de configurac¸˜ao no formatomgm. . . 190 110 Estrutura gerada pela classePDBParser, obtida do tutorial do Biopython v.1.52. 193 111 Arquivos no formato st utilizando o campo de forc¸a GROMOS96, conforme
proposto pela referˆencia (19). . . 197
112 Arquivos no formatostutilizando o campo de forc¸a AMBER99, conforme pro-posto pela referˆencia (20). . . 198
Lista de Tabelas
1 Valores depKa’s dos amino´acidos “isolados” obtidos experimentalmente a
tem-peratura de 25◦C. . . 38
2 Valores do raio, volume e peso molecular de cada amino´acido. . . 41
3 Comparac¸˜ao entre os pI’s experimentais e os calculados pelo PROMETHEUS no n´ıvel anal´ıtico. Os dados experimentais foram obtidos da referˆencia (21). . . 95
4 Comparac¸˜ao entre os pontos isoel´etricos experimentais e te´oricos, com os cal-culados pelo PROMETHEUS no n´ıvel de predic¸˜ao anal´ıtico. . . 101
5 Comparac¸˜ao dos valores de pKa’s da prote´ına lisozima em diversas
concentrac¸˜oes de sal, alterando-se a constante diel´etrica da prote´ına (εp). A
´ultima coluna apresenta os pKa’s obtidos pelo servic¸o H++ e a pen´ultima os
pKa’s experimentais. A temperatura e a constante diel´etrica do solvente foram
fixadas em 298,0 K e 80,0, respectivamente. Dados obtidos utilizando o campo de forc¸a GROMOS96. . . 103
6 Comparac¸˜ao dos valores de pKa’s da prote´ına lisozima em diversas
concentrac¸˜oes de sal, alterando-se a constante diel´etrica da prote´ına (εp). A
´ultima coluna apresenta os pKa’s obtidos pelo servic¸o H++ e a pen´ultima os
pKa’s experimentais. A temperatura e a constante diel´etrica do solvente foram
fixadas em 298,0 K e 80,0, respectivamente. Dados obtidos utilizando o campo de forc¸a AMBER99. . . 104
7 Comparac¸˜ao dos valores de pKa’s da prote´ına BPTI em diversas concentrac¸˜oes
de sal, alterando-se a constante diel´etrica da prote´ına (εp). A ´ultima coluna
apre-senta os pKa’s obtidos pelo servic¸o H++ e a pen´ultima os pKa’s experimentais.
8 Comparac¸˜ao dos valores de pKa’s da prote´ına BPTI em diversas concentrac¸˜oes
de sal, alterando-se a constante diel´etrica da prote´ına (εp). A ´ultima coluna
apre-senta os pKa’s obtidos pelo servic¸o H++ e a pen´ultima os pKa’s experimentais.
A temperatura e a constante diel´etrica do solvente foram fixadas em 298,0 K e 80,0, respectivamente. Dados obtidos utilizando o campo de forc¸a AMBER99. . 106
9 Comparac¸˜ao dos valores de pKa’s dos res´ıduos lisina presentes na prote´ına
cal-bindina, alterando-se a constante diel´etrica da prote´ına (εp). A ´ultima coluna
apresenta os pKa’s medidos experimentalmente. A forc¸a iˆonica, a temperatura
e a constante diel´etrica do solvente foram fixadas em 0,1M, 298,0K e 78,5, res-pectivamente. Dados obtidos utilizando o campo de forc¸a GROMOS96. . . 108
10 Comparac¸˜ao dos valores de pKa’s dos res´ıduos ´acido glutˆamico presentes na
prote´ına calbindina, alterando-se a constante diel´etrica da prote´ına (εp). A
´ultima coluna apresenta os pKa’s medidos experimentalmente. A forc¸a iˆonica,
a temperatura e a constante diel´etrica do solvente foram fixadas em 1M, 298,0K e 77,8, respectivamente. Dados obtidos utilizando o campo de forc¸a GROMOS96.109
11 Comparac¸˜ao dos valores de pKa’s dos res´ıduos presentes na prote´ına
ribonucle-ase A (PDB: 3RN3), em v´arias concentrac¸˜oes de sal, alterando-se a constante diel´etrica da prote´ına (εp). A pen´ultima coluna apresenta os pKa’s medidos
experimentalmente e a ´ultima, os pKa’s preditos pelo servic¸owebPCE. A
tem-peratura e a constante diel´etrica do solvente foram fixadas em 298,0K e 80, respectivamente. Dados obtidos utilizando o campo de forc¸a GROMOS96. . . . 110
12 Comparac¸˜ao dos valores de pKa’s da prote´ına lisozima obtidos pelo servic¸o
H++ e PROMETHEUS alterando-se a constante diel´etrica da prote´ına (εp). A
´ultima coluna apresenta os pKa’s medidos experimentalmente. Dados obtidos
utilizando o campo de forc¸a AMBER99. . . 113
13 Comparac¸˜ao dos valores de pKa’s da prote´ına BPTI obtidos pelo servic¸o H++ e
PROMETHEUS alterando-se a constante diel´etrica da prote´ına (εp). A ´ultima
coluna apresenta os pKa’s medidos experimentalmente. Dados obtidos
utili-zando o campo de forc¸a AMBER99. . . 114
15 C´odigos PDBs dos complexos prot´eicos utilizados no c´alculo da freq¨uˆencia de contato em func¸˜ao da distˆancia de separac¸˜ao entre os res´ıduos de amino´acidos presentes em cadeias distintas da prote´ına. . . 140
16 Relac¸˜ao dos conjuntos de prote´ınas e os respectivos erros encontrados em relac¸˜ao ao conjunto controle, exibidos na Tabela 15. . . 148
17 Quantidade de cada res´ıduo presente nos conjuntos de prote´ınas exibidos na Tabela 15. . . 148
18 Comparativo entre o uso de banco de dados e arquivos texto a respeito da organizac¸˜ao, armazenamento e recuperac¸˜ao de dados. . . 150
19 Comparac¸˜ao dos valores de pKa’s da prote´ına lisozima (distribu´ıdo junto
com o pacote MEAD) e os providos pelo PROMETHEUS com o parˆametro
epsave oldway. . . 163 20 Comparac¸˜ao dos valores de pKa’s da prote´ına lisozima, distribu´ıdo junto
com o pacote MEAD e os providos pelo PROMETHEUS sem o parˆametro
Lista de Algoritmos
1 Pseudoc´odigo utilizado para realizar a normalizac¸˜ao dos dados, pelo crit´erio 1. p. 92
2 Pseudoc´odigo do algoritmo utilizado para realizar o c´alculo do grau de
dissociac¸˜ao de um pr´oton (αi) de um amino´acidoi. . . p. 157
Lista de abreviaturas e siglas
ATOM Atomo.´ Campo pertencente ao arquivo no formato PDB que cont´em as coordenadas atˆomicas dos ´atomos presentes nos grupos de amino´acidos padr˜oes.
BPTI Basic pancreatic trypsin inhibitor (inibidor da tripsina pancre´atica b´asica).
Cr Creighton.
DM Dinˆamica molecular.
EPB Equac¸˜ao dePoisson-Boltzmann. EPBL Equac¸˜ao dePoisson-Boltzmannlinear. EPSIN Constante diel´etrica do soluto.
EPSSOL Constante diel´etrica do solvente. FDR Func¸˜ao de distribuic¸˜ao radial.
GNU General public license(licenc¸a p´ublica geral).
GRASP Graphical representation and analysis of surface-properties
(representac¸˜ao gr´afica e an´alises das propriedades de superf´ıcie). GROMACS Groningen machine for chemical simulations(m´aquina de
Gronin-gen para simulac¸˜oes qu´ımicas).
GROMOS Groningen molecular simulation(simulac¸˜ao molecular de Gronin-gen).
HTML HyperText Markup Language(Linguagem de marcac¸˜ao de texto).
MA Modelo atom´ıstico.
MC Monte Carlo.
MDC Mapas de contato.
MEAD Macroscopic electrostatics with atomic detail (eletrost´atica ma-crosc´opica com detalhes atom´ısticos).
MI M´etodos inversos.
MJ Miyazawa e Jernigan.
MOLESA Molecular structures analysis(an´alise de estruturas molecular). MVC Model view control(modelo vis˜ao controle).
NBO N´ıvel deBorn-Oppenheimer. NMM N´ıvel deMcMillan-Mayer.
NMR Nuclear magnetic resonance(ressonˆancia magn´etica nuclear). NS N´ıvel deSchr¨odinger.
NT Nozaki e Tanford.
PDB Protein data bank(banco de dados de prote´ınas).
PDBid C´odigo de identificac¸˜ao de uma prote´ına utilizado pelo PDB. PFM Potencial de forc¸a m´edia.
PROMETHEUS Protein-Protein complexes by macroscopic electrostatic theories and user-friendly simulations(complexos prote´ına-prote´ına por te-oria eletrost´atica macrosc´opica e simulac¸˜oes amig´aveis).
Lista de s´ımbolos
∆Gele Variac¸˜ao da energia livre eletrost´atica de complexac¸˜ao (dada em unidades de kBT).
αi Grau de dissociac¸˜ao de um amino´acidoi.
λ Parˆametro de carregamento.
φ(r) Potencial eletrost´atico em uma determinada posic¸˜aor.
ρ(r) Densidade de carga m´edia na posic¸˜aor.
σ Distˆancia m´ınima de separac¸˜ao entre duas prote´ınas.
ε0 Constante diel´etrica do v´acuo (ε0=8,85.10−12C2/Nm2).
εp Constante diel´etrica do interior da prote´ına.
εs Constante diel´etrica do solvente (para H2O,εs=77,8, em T = 298
K).
◦C Grau Celsius.
a Coeficiente de atividade qu´ımica.
B23 Segundo coeficiente cruzado de virial.
B23(ele) Termo eletrost´atico do segundo coeficiente cruzado de virial.
B23(er) Termo repulsivo do segundo coeficiente cruzado de virial.
B2 Segundo coeficiente de virial.
Cideal Capacitˆancia ideal de uma prote´ına.
Caideal Capacitˆancia ideal de um amino´acidoa.
CMpx Coordenada no eixo X do centro geom´etrico de uma prote´ına.
CMpy Coordenada no eixo Y do centro geom´etrico de uma prote´ına.
CMpz Coordenada no eixo Z do centro geom´etrico de uma prote´ına.
d Distˆancia do ´atomo mais distante do centro geom´etrico da prote´ına (em ˚Angstr¨om).
dr Variac¸˜ao da distˆancia de separac¸˜ao entre duas prote´ınas.
dx Distˆancia no eixo X em relac¸˜ao ao centro geom´etrico da prote´ına.
dy Distˆancia no eixo Y em relac¸˜ao ao centro geom´etrico da prote´ına.
e Carga elementar (e=1,6.10−19C).
gi j(r) Func¸˜ao de distribuic¸˜ao radial dos res´ıduosie j.
gi j(r)∗ Freq¨uˆencia de contatos entre os res´ıduosi e j na distˆancia r (em
˚
Angstr¨om).
I Forc¸a iˆonica do meio.
K Constante de equil´ıbrio termodinˆamica.
k Inverso do comprimento deDebye.
Ka Constante de equil´ıbrio termodinˆamica de um amino´acidoa.
kB Constante deBoltzmann(kB = 1,381.10−23JK−1).
L Tamanho do lado da caixa de simulac¸˜ao utilizada para a resoluc¸˜ao da EPBL, utilizando o m´etodo das diferenc¸as finitas.
lBox Tamanho do lado de cada elemento c´ubico da rede de simulac¸˜ao
utilizada para resoluc¸˜ao da EPBL, utilizando o m´etodo das diferenc¸as finitas.
lB Comprimento deBjerrum.
Nσ Taxa empregada para o aumento da distˆancia de separac¸˜ao entre duas prote´ınas.
Na N´umero de Avogadro (Na=6,02.1023mol−1).
NBox Quantidade de elementos c´ubicos presentes na caixa de simulac¸˜ao
utilizada para resoluc¸˜ao da EPBL, utilizando o m´etodo das diferenc¸as finitas.
ni Densidade de ´ıons do tipoipor unidade de volume em uma dada
regi˜ao do espac¸o.
n0i Densidade de ´ıons do tipoipor unidade de volume para o seio da soluc¸˜ao.
pH Potencial hidrogeniˆonico.
pI Ponto isoel´etrico de uma mol´ecula.
pK Forma logar´ıtma da constante de equil´ıbrio termodinˆamica.
pKa Forma logar´ıtma da constante de equil´ıbrio termodinˆamica de um
amino´acido da esp´eciea.
qi Carga correspondente ao ´ıoni.
r Distˆancia de separac¸˜ao entre duas prote´ınas (em ˚Angstr¨om).
r′ Pr´oxima distˆancia de separac¸˜ao entre duas prote´ınas.
RF Distˆancia m´axima de separac¸˜ao entre duas prote´ınas.
T Temperatura absoluta em Kelvin (K).
wi j(r) Potencial de forc¸a m´edia em func¸˜ao da distˆancia de separac¸˜ao r
entre os res´ıduosie j.
z Valˆencia de um amino´acido.
zi Valˆencia de um amino´acidoi.
Sum´ario
Resumo i
Abstract ii
Lista de figuras iii
Lista de tabelas xi
Lista de algoritmos xiv
Lista de abreviaturas e siglas xv
Lista de s´ımbolos xvii
1 INTRODUC¸ ˜AO E REVIS ˜AO DA LITERATURA 1
1.1 Biocomputac¸˜ao . . . 1
1.2 Ferramentas para Biologia Estrutural . . . 3
1.2.1 Banco de dados de prote´ınas . . . 3
1.2.2 Validac¸˜ao de estruturas de prote´ınas . . . 3
1.2.3 Servic¸os dispon´ıveis naweb . . . 4 1.3 A importˆancia dos complexos prot´eicos . . . 7
1.4 Abordagem do problema . . . 9
1.4.1 Estrat´egia 1: Propriedades eletrost´aticas de prote´ınas . . . 12
1.4.2 Estrat´egia 2: An´alise de propriedades estruturais para a construc¸˜ao de potenciais estat´ısticos . . . 13
1.5 Sum´ario de nossas contribuic¸˜oes . . . 16
2 OBJETIVOS 18
3 ASPECTOS IMPORTANTES SOBRE PROTE´INAS 20
3.1 Amino´acidos . . . 21
3.2 Ligac¸˜oes pept´ıdicas . . . 21
3.3 Estrutura prim´aria de prote´ınas . . . 22
3.4 Estrutura secund´aria de prote´ınas . . . 23
3.5 Estrutura terci´aria de prote´ınas . . . 23
3.6 Estrutura quatern´aria de prote´ınas . . . 23
3.7 M´etodos para determinac¸˜ao da estrutura tridimensional das prote´ınas . . . 23
4 TRABALHANDO COM INFORMAC¸ ˜OES BIOL ´OGICASIN SIL´ICIO 25
4.1 N´ıveis de detalhamento do modelo . . . 25
4.2 Modelagem do sistema . . . 26
4.3 Soluc¸˜ao do modelo . . . 26
4.4 Equac¸˜ao dePoisson-Boltzmann . . . 27 4.4.1 M´etodo das diferenc¸as finitas . . . 29
4.5 Func¸˜ao de distribuic¸˜ao radial . . . 31
4.6 Potencial de forc¸a m´edia . . . 31
4.7 Campos de forc¸a . . . 32
5 MATERIAL E M ´ETODOS 34
5.1 Teoria . . . 34
5.1.1 Equil´ıbrio ´acido-base . . . 34
5.1.2 C´alculo de pKa’s em prote´ınas . . . 35
5.1.4 N´ıvel de predic¸˜ao ideal (anal´ıtico) – Predic¸˜ao a partir da seq¨uˆencia prim´aria da prote´ına . . . 37
5.1.5 N´ıvel de predic¸˜ao baseado na estrutura 3D da prote´ına – atrav´es da utilizac¸˜ao da EPBL . . . 43
5.1.6 An´alise da freq¨uˆencia de contatos entre os amino´acidos de complexos prot´eicos do PDB e potencial estat´ıstico . . . 44
5.2 Infra-estrutura computacional . . . 45
6 FERRAMENTAS COMPUTACIONAIS DESENVOLVIDAS 49
6.1 Estrat´egia 1: Desenvolvimento de um portal web que permite o estudo de propriedades eletrost´aticas em prote´ınas . . . 49
6.1.1 Cadastramento . . . 50
6.1.2 Propriedades dos amino´acidos isolados (Single amino acid properties) . . . 51 6.1.3 Propriedades de prote´ınas isoladas (Single protein properties) . . . 53 6.1.4 Interac¸˜ao Prote´ına-Prote´ına (Protein-protein interaction) . . . 60 6.1.5 Ferramentas auxiliares desenvolvidas (Tools) . . . 70 6.2 Estrat´egia 2: Desenvolvimento de um portal web que permite a an´alise da
freq¨uˆencia de contatos entre os amino´acidos que formam um complexo prot´eico . 85
6.2.1 Funcionamento do portal MOLESA . . . 91
7 RESULTADOS 93
7.1 O portal PROMETHEUS - predic¸˜ao com base nas propriedades eletrost´aticas das prote´ınas . . . 93
7.1.1 Validac¸˜ao das propriedades eletrost´aticas dos amino´acidos . . . 94
7.1.2 Validac¸˜ao das propriedades eletrost´aticas de prote´ınas . . . 97
7.2 Interac¸˜ao prote´ına–prote´ına . . . 123
7.2.1 Mecanismo de regulac¸˜ao de cargas . . . 137
7.3 O portal MOLESA - an´alise estrutural de complexos de prote´ınas . . . 139
8.1 Enfoque computacional . . . 149
8.1.1 An´alise da complexidade de algoritmos . . . 157
8.2 Enfoque f´ısico . . . 159
8.2.1 Crit´erios para a predic¸˜ao dos pKa’s dos amino´acidos ioniz´aveis . . . 161
9 CONCLUS ˜AO E TRABALHOS FUTUROS 166
9.1 Perspectivas de Trabalho Futuro . . . 167
REFER ˆENCIAS 169
Apˆendice A -- Avaliac¸˜ao dos servic¸os dispon´ıveis naweb 182
Apˆendice B -- Descric¸˜ao das principais classes desenvolvidas 184
Apˆendice C -- Descric¸˜ao dos programas auxiliares utilizados 187
C.1 MEAD . . . 187
C.2 Biopython . . . 192
C.3 GROMACS . . . 194
C.4 PDB2PQR . . . 195
Apˆendice D -- Criac¸˜ao dos arquivos no formatostem func¸˜ao do campo de forc¸a 196
1
INTRODUC
¸ ˜
AO E REVIS ˜
AO DA
LITERATURA
1.1
Biocomputac¸˜ao
A enorme profus˜ao de dados de seq¨uˆencia e estruturais, gerados nas ´ultimas d´ecadas, levaram a criac¸˜ao de um novo campo de investigac¸˜ao, o da Bioinform´atica, o qual ´e definido genericamente como estando na intersec¸˜ao entre a Biotecnologia e a Ciˆencia da Computac¸˜ao (22).
Segundo a revista ERCIM News No. 43, de Outubro de 2000 (23), Biologia Com-putacional e Bioinform´atica s˜ao termos utilizados em um campo interdisciplinar unindo a informac¸˜ao tecnol´ogica com a biol´ogica, que por sua vez tem crescido rapidamente durante os ´ultimos anos. Este campo ´e localizado entre duas ´areas: cient´ıfica e tecnol´ogica, no qual a Biologia Computacional refere-se `a parte mais cient´ıfica desde campo, empregando as t´ecnicas computacionais para a Biologia Molecular, enquanto que a Bioinform´atica ´e mais voltada `a parte de infra-estrutura computacional e an´alises estat´ısticas dos dados, embora na pr´atica h´a uma grande sobreposic¸˜ao entre suas atividades.
Combinando caracter´ısticas dessas duas ´areas afins, temos o que n´os chamamos de Biocomputac¸˜ao1, que compreende ao desenvolvimento de aplicac¸˜oes computacionais (softwares) aplicadas para o entendimento de sistemas biol´ogicos, e que oferec¸am facilidade de uso de forma a dispensar o conhecimento profundo na ´area das ciˆencias da computac¸˜ao para a utilizac¸˜ao das mesmas por um p´ublico externo, provendo ao mesmo tempo a infra-estrutura necess´aria para que as operac¸˜oes realizadas em dados biol´ogicos sejam executadas emlarga escala.
1Entendemos que o termo “Biocomputac¸˜ao” (24, 25) ´e mais abrangente do que “Bioinform´atica” [esse
Dentro deste contexto de conceituac¸˜ao da Biocomputac¸˜ao, ´e not´avel que a utilizac¸˜ao de ferramentas computacionais nas diversas ´areas do saber vem rapidamente crescendo, permitindo um amplo espectro de tarefas, desde as mais corriqueiras, como a otimizac¸˜ao de processos, a acelerac¸˜ao de c´alculos e a armazenagem de um antes inimagin´ario conjunto de dados biol´ogicos, a outras mais amplas, como novas possibilidades e perspectivas de abordagens cient´ıficas e o tratamento em larga escala de sistemas biol´ogicos tradicionalmente estudados em pequena escala. O desenvolvimento da computac¸˜ao e sua inserc¸˜ao na soluc¸˜ao de problemas f´ısicos e qu´ımicos biol´ogicos exemplificam tal tendˆencia (29–32), quer para um maior entendimento dos mecanismos moleculares, quer auxiliando no planejamento de poss´ıveis aplicac¸˜oes industriais, ou ainda, procurando compreender e controlar a fronteira entre a sa´ude e a doenc¸a (26, 33–36).
Em um cen´ario p´os-Genoma, onde um conjunto consider´avel de informac¸˜oes j´a se en-contra dispon´ıvel, a Biocomputac¸˜ao se torna ainda mais relevante, contribuindo com o aux´ılio de diversas ferramentas, como o Sistema Gerenciador de Banco de Dados (SGBD) (37, 38), linguagens de programac¸˜ao multi-plataformas (38–40), poder de processamento relativamente alto a custos reduzidos, os quais possibilitam o armazenamento da informac¸˜ao biol´ogica e, prin-cipalmente, um conjunto de diferentes e complementares formas de extrac¸˜ao e manipulac¸˜ao destes dados (41). Por exemplo, a partir de estruturas tridimensionais de macromol´eculas depo-sitadas no Banco de Dados de Prote´ınas (RCSB Protein Data Bank– PDB) (42), v´arios estudos podem ser realizados (43–47), procurando-se melhor caracterizar um dos grandes paradigmas da Biologia Molecular: correlacionar aestruturacom afunc¸˜aobiol´ogica (48).
Al´em de bancos de dados contendo informac¸˜oes sobre estruturas biol´ogicas, sejam eles de prote´ınas (49), DNA (50), nucleot´ıdeos (51), f´armacos (52), e outros (53), h´a tamb´em diver-sas ferramentas dispon´ıveis gratuitamente `a comunidade (54–57) que permitem a manipulac¸˜ao de estruturas biol´ogicas, dentre as quais destacamos o Biopython (58). Biopython2 ´e um con-junto de programas desenvolvidos em linguagem Python, para a Biologia Molecular, onde ´e poss´ıvel, por exemplo, criar estruturas de dados em Python a partir de informac¸˜oes biol´ogicas contidas em arquivos textos, como os disponibilizados pelo PDB e outros, e, ent˜ao, manipu-lar tais informac¸˜oes de maneira bastante simplificada, atrav´es do uso de m´etodos ou func¸˜oes implementadas pela ferramenta, tornando poss´ıvel dessa forma a manipulac¸˜ao de estruturas biol´ogicas, tanto por profissionais n˜ao familiarizados com m´etodos e estruturas computacionais tradicionalmente empregados para este fim, quanto pelos mais especialistas nesta ´area (enge-nheiros da computac¸˜ao/hardware, “bio-informatas”, analistas de sistemas e outras ´areas afins), o que torna poss´ıvel expandir as funcionalidades providas por esta ferramenta conforme as ne-cessidades das an´alises que ser˜ao feitas.
O trabalho aqui apresentado contempla tanto o uso de banco de dados para extrac¸˜ao da informac¸˜ao, como a gerac¸˜ao de novos bancos de dados, al´em do desenvolvimento de di-versas ferramentas computacionais as quais permitem efetuar diferentes an´alises em complexos prot´eicos. As pr´oximas sec¸˜oes apresentam uma descric¸˜ao de algumas das ferramentas existentes para o uso em Biologia Estrutural.
1.2
Ferramentas para Biologia Estrutural
1.2.1
Banco de dados de prote´ınas
Dentre os diversos bancos de dados de estruturas e informac¸˜oes biol´ogicas dispon´ıveis naweb, como SCOP (59), STING (60) e outros (61), focamos no banco de dados de prote´ınas deBrookhaven (PDB), por ser este uma grande fonte de informac¸˜oes a respeito de estruturas de prote´ınas bem como o ponto de partida inicial escolhido para a realizac¸˜ao das an´alises que ser˜ao desenvolvidas neste trabalho as quais estar˜ao dispon´ıveis gratuitamente `a comunidade.
Criado em 1971 pelo Laborat´orio Nacional deBrookhaven, o PDB, hoje mantido pelo
Research Collaboratory for Structural Bioinformatics(RCSB), armazena estruturas de macro-mol´eculas biol´ogicas (42), obtidas por t´ecnicas experimentais (NMR e Cristalografia de raios X). Al´em das coordenadas espaciais de cada ´atomo da prote´ına, a´ı est˜ao dispon´ıveis, para extrac¸˜ao, dados como a identificac¸˜ao dos res´ıduos de contato, caracterizac¸˜ao da ´area superficial, o n´umero de pontes de hidrogˆenio e de contatos devan der Waals, a magnitude de mudanc¸as conformacionais associadas com a formac¸˜ao de complexos, etc (62). Entretanto, continua sendo dif´ıcil de se prever experimentalmente as conseq¨uˆencias estruturais e funcionais da substituic¸˜ao de amino´acidos espec´ıficos assim como as pr´oprias condic¸˜oes necess´arias para a obtenc¸˜ao dos cristais (63) utilizados no processo de obtenc¸˜ao da estrutura tridimensional de uma prote´ına.
1.2.2
Validac¸˜ao de estruturas de prote´ınas
Apesar do conjunto de testes que s˜ao realizados nos dados antes dos mesmos serem depositados no PDB (42, 62), estes ainda n˜ao est˜ao livres de incertezas e problemas (64). A mai-oria das estruturas tridimensionais de prote´ınas atualmente conhecidas s˜ao obtidas por t´ecnicas de cristalografia de raios X ou ressonˆancia nuclear magn´etica, e, como todo experimento, est˜ao sujeitos a erros (65).
prote´ınas que tiveram sua estrutura resolvida por cristalografia de raios X e verificaram que os resultados podem conter alguns erros os quais s˜ao dif´ıceis de identificar. ´E tarefa do cris-tal´ografo se certificar que estruturas de prote´ınas incorretas n˜ao sejam disponibilizadas na li-teratura (67). Al´em dos erros intr´ınsecos, arquivos PDB podem conter ´atomos faltantes e/ou duplicados, amino´acidos desconhecidos, etc. Por esta raz˜ao ´e necess´ario o tratamento (testes de qualidade e consistˆencia, como os realizados peloWHATIF(64, 67, 68) e outros) dos arqui-vos advindos do PDB, garantindo assim que as an´alises posteriores, por exemplo, os potenciais estat´ısticos e outros resultados gerados, como, por exemplo, o estudo das propriedades ele-trost´aticas de prote´ınas, sejam confi´aveis.
1.2.3
Servic¸os dispon´ıveis na
web
Devido ao alto grau de complexidade do desenvolvimento de programas computacio-nais, principalmente envolvendo sistemas biol´ogicos onde a generalizac¸˜ao de um caso muitas vezes n˜ao atende todos os requisitos de outro sistema, e a grande heterogeneidade de profissio-nais (f´ısicos, qu´ımicos, bi´ologos, engenheiros, analistas de sistemas, farmacˆeuticos, etc.) inte-ressados em estudar sistemas biol´ogicosin sil´ıcio, muitos grupos tˆem disponibilizado, atrav´es de servidoresweb(web sites), ferramentas e/ou servic¸os que proporcionam, ainda que de ma-neira limitada, o estudo de biomol´eculas. Neste cen´ario, o usu´ario pode escolher o sistema de interesse e a metodologia de estudo (n´ıvel de detalhamento do modelo, quais fatores – tempe-ratura do sistema, concentrac¸˜ao do soluto, concentrac¸˜ao do solvente, ´ıons livres na soluc¸˜ao, etc. – ser˜ao considerados, entre outras configurac¸˜oes e/ou possibilidades) e simplesmente aguardar uma resposta (arquivo texto, texto no formato HTML3, e-mail, etc.) do programa (portal,web site) a qual dever´a ser cuidadosamente analisada.
A maioria dos servic¸oswebe programas de computador atualmente dispon´ıveis ´e de-senvolvida valendo-se dos conhecimentos advindos da F´ısica, como o c´alculo de distˆancia, velocidade, massa, carga, pH, etc. A escolha do modelo normalmente est´a relacionada com os recursos computacionais dispon´ıveis, o que se deseja medir e em qual tempo. Por exemplo, po-demos desenvolver programas que computam o sistema no seu n´ıvel m´aximo de detalhamento (quˆantico), onde as posic¸˜oes de todas as part´ıculas (pr´otons, el´etrons, etc.) s˜ao calculadas, ou seja, tem-se um sistema que opera em n´ıvel quˆantico ou, podemos desenvolver ferramentas que trabalham com um modelo mais simplificado, por exemplo, considerando apenas a seq¨uˆencia prim´aria de uma prote´ına, sem se importar com sua estrutura 3D. A escolha do modelo tamb´em est´a intimamente relacionada com um compromisso entre o poder computacional dispon´ıvel, o
tempo de resposta esperado e o tamanho do problema (sistema de interesse).
Com o intuito de exemplificar e ao mesmo tempo familiarizar o leitor com as soluc¸˜oes j´a existentes, exploraremos algumas ferramentas atualmente dispon´ıveis na web. Tais ferra-mentas s˜ao respons´aveis por: validac¸˜ao de estrutura, adic¸˜ao de ´atomos ausentes, c´alculos de propriedades eletrost´aticas em prote´ınas e outros.
1. PDB2PQR (69, 70): Dispon´ıvel em: http://pdb2pqr-1.wustl.edu/
pdb2pqr/ou emhttp://nbcr.net/pdb2pqr/, ´e um programa desenvolvido em linguagem Python que converte um arquivo originalmente no formato PDB para um ar-quivo no formato PQR4. A ferramenta realiza as seguintes tarefas:
• Adic¸˜ao de um n´umero limitado de ´atomos que n˜ao est˜ao presentes no modelo;
• Determinac¸˜ao dos pKa’s5 dos res´ıduos de amino´acidos, utilizando o programa
PROPKA (71, 72);
• Adic¸˜ao de ´atomos de hidrogˆenio6;
• Otimizac¸˜ao das ligac¸˜oes de hidrogˆenio favor´aveis7;
• Provˆe as cargas e os raios para os ´atomos a partir do campo de forc¸a8escolhido.
2. H++ (20): Dispon´ıvel em http://biophysics.cs.vt.edu/H++/ hppdetails.php, ´e um programa que calcula os valores de pKa’s de grupos de
amino´acidos ioniz´aveis presentes em macromol´eculas e acrescenta ´atomos de hidrogˆenio que n˜ao est˜ao presentes na estrutura. O H++ recebe como entrada um arquivo no formato PDB e retorna como sa´ıda arquivos nos formatos PDB, PQR e AMBER (73, 74). Ao submeter uma estrutura no formato PDB, o H++ realiza as seguintes tarefas:
• Remoc¸˜ao de todos os campos HETATM9presentes no arquivo no formato PDB;
• Remoc¸˜ao de todas as mol´eculas de ´agua e contra-´ıons;
4Arquivo contendo as coordenadas, raio e carga para cada ´atomo presente no arquivo PDB. ´E largamente
empregado em pacotes de simulac¸˜ao de propriedades eletrost´aticas de prote´ınas.
5pK: constante de ligac¸˜ao/equil´ıbrio termodinˆamica. Propriedade que pode ser utilizada para analisar v´arios
comportamentos em sistemas biomoleculares.
6Atomos de hidrogˆenio, pertencentes aos amino´acidos, geralmente n˜ao s˜ao identificados nas estruturas das´
prote´ınas resolvidas por cristalografia de raios X devido a limitac¸˜oes da t´ecnica. Uma das necessidades de se obter os ´atomos de hidrogˆenio ´e determinar valores, mais pr´oximos aos dados experimentais, para o volume e o raio de prote´ına, visto que quase 50% dos ´atomos de uma prote´ına s˜ao ´atomos de hidrogˆenio.
7A otimizac¸˜ao da ligac¸˜ao ´e feita buscando um m´ınimo de energia para o sistema. 8Apresentamos a definic¸˜ao e o uso de campo de forc¸a na Sec¸˜ao 4.7 -Campos de forc¸a.
9Campo contido no arquivo no formato PDB que representa as coordenadas atˆomicas de ´atomos que n˜ao
• Verificac¸˜ao da seq¨uˆencia de ´atomos e configurac¸˜ao do nome dos ´atomos para o padr˜ao utilizado pelo pacote AMBER99 (73, 75);
• Adic¸˜ao de ´atomos de hidrogˆenio e otimizac¸˜ao de suas ligac¸˜oes;
• Padronizac¸˜ao dos raios dos ´atomos;
• C´alculos eletrost´aticos atrav´es da utilizac¸˜ao do pacote MEAD (76);
• C´alculo da curva de titulac¸˜ao de cada amino´acido ioniz´avel;
• Adic¸˜ao ou remoc¸˜ao de pr´otons, na estrutura da prote´ına, de acordo com os pKa’s
calculados.
• Minimizac¸˜ao de energia da estrutura da prote´ına em um dado pH utilizando o campo de forc¸a AMBER99.
3. WHATIF (68): Dispon´ıvel em: http://swift.cmbi.ru.nl/servers/html/
index.html, ´e um pacote de programas desenvolvido em linguagem FORTRAN 77 para modelagem molecular especializado para trabalhar com prote´ınas; seu desenvolvi-mento teve in´ıcio em 1987 e prossegue at´e a presente data. WHATIFprovˆe um ambiente flex´ıvel para visualizar, manipular e analisar pequenas mol´eculas tamb´em. Neste contexto ´e poss´ıvel efetuar: comparac¸˜ao de mol´eculas de prote´ınas com base em sua estrutura 3D, visualizar mapas de densidade eletrˆonica de estruturas de prote´ınas, efetuar mutac¸˜oes na seq¨uˆencia de amino´acidos da prote´ına, an´alise e predic¸˜ao de ´atomos de hidrogˆenio, etc.
4. RosettaDock(77): Dispon´ıvel em http://rosettadock.graylab.jhu.edu/, ´e um pacote de programas desenvolvido em linguagem C++ para predic¸˜ao e modelagem (design) de estruturas de prote´ınas, mecanismos de enovelamento (folding) de prote´ına e interac¸˜oes prote´ına-prote´ına.
5. PCE (78): Dispon´ıvel em: http://bioserv.rpbs.jussieu.fr/Help/PCE. html, ´e uma interfacewebpara o programa MEAD. Os servic¸os dispon´ıveis s˜ao: PCE-pot e PCE-pKa, os quais, respectivamente realizam as seguintes tarefas:
• C´alculo do potencial eletrost´atico de uma prote´ına em func¸˜ao de condic¸˜oes expe-rimentais, resolvendo numericamente a equac¸˜ao de Poisson-Boltzmann10. O pro-grama recebe como entrada um arquivo no formato PDB, para o qual ´e gerado um arquivo no formato PQR utilizando o campo de forc¸a PARSE (82, 83), ou um ar-quivo no formato PQR. O programa retorna como resultado imagens que represen-tam graficamente o potencial eletrost´atico na superf´ıcie da mol´ecula em estudo.
• C´alculo dos valores de pKa’s dos amino´acidos ioniz´aveis da prote´ına. O programa
recebe como entrada um arquivo no formato PDB e fornece a resposta em uma p´agina HTML.
Outros servic¸oswebpodem ser encontrados na literatura (52, 60, 84–87).
Nossa primeira contribuic¸˜ao ser´a no sentido de complementar estas ferramentas, atrav´es da construc¸˜ao de portaisweb que auxiliem na predic¸˜ao da complexac¸˜ao de prote´ınas. Propomos, al´em do fornecimento de propriedades dispon´ıveis em outros servic¸os, a possibili-dade de efetuar um estudo das propriepossibili-dades eletrost´aticas em prote´ınas, atrav´es de um preditor inicial de complexos prot´eicos, chamado PROMETHEUS (esta ser´a a primeira frente do traba-lho), em diversas condic¸˜oes e n´ıveis preditivos. No futuro, a ferramenta tamb´em permitir´a:
• Gerac¸˜ao de pept´ıdeos derivados de uma dada seq¨uˆencia, de forma combinat´oria, para classificac¸˜ao dos mais adequados `a complexac¸˜ao com prote´ınas-alvo.
• Acoplamento a outros servic¸os, como o MHOLline (88, 89), utilizado para predic¸˜ao de estrutura de prote´ınas.
Al´em disso, os dados presentes em nossa base de dados ser˜ao disponibilizados gratui-tamente `a comunidade onde, a partir dos mesmos, v´arios estudos podem ser realizados, e com o benef´ıcio que as estruturas de prote´ınas presentes na nossa base de dados j´a estarem validadas.
1.3
A importˆancia dos complexos prot´eicos
Interac¸˜oes prote´ına-prote´ına s˜ao de grande interesse na ind´ustria farmacˆeutica, alimen-tos, biotecnologia, processos de biosseparac¸˜ao, purificac¸˜ao de prote´ınas, micro-encapsulac¸˜ao, biomateriais, etc. (90–92). Al´em disso, complexos prot´eicos est˜ao envolvidos na maioria dos processos biol´ogicos, como por exemplo, cat´alise enzim´atica, transporte de substˆancias (93) e doenc¸as como Alzheimer, Parkinson, Diabetes tipo II, Anemia Falciforme e outros (91, 92, 94), o que torna o entendimento racional dessas interac¸˜oes um tema de grande interesse, sendo ex-plorado pelas mais diversas ´areas do conhecimento, como a F´ısica, Bioqu´ımica, Biologia, etc.
Atrav´es da an´alise das propriedades termodinˆamicas das prote´ınas11 como, por exem-plo, ponto isoel´etrico (pI), energia livre, segundo coeficiente cruzado de virial (B23)12e outros, visamos contribuir para o progresso nas seguintes ´areas e suas correlac¸˜oes:
• Determinac¸˜ao da estrutura prot´eica: A cristalizac¸˜ao de uma prote´ına em soluc¸˜ao ´e o passo inicial para a determinac¸˜ao de sua estrutura utilizando difrac¸˜ao por raios-X13 (95). Entretando a obtenc¸˜ao de um cristal de alta qualidade ´e um dos passos mais dif´ıceis e que consome o maior tempo no processo de determinac¸˜ao da estrutura da prote´ına, uma vez que a condic¸˜ao de cristalizac¸˜ao depende de um grande n´umero de parˆametros e condic¸˜oes experimentais (96).
Estudos demonstram que o segundo coeficiente de virial B2 (e conseq¨uˆentemente o segundo coeficiente cruzado de virial - B23) est´a intimamente relacionado com a cristalizac¸˜ao prot´eica, uma vez que valores negativos deste indicam atrac¸˜ao entre as prote´ınas, o que ´e pr´e-condic¸˜ao para a cristalizac¸˜ao (18, 97, 98).
A utilizac¸˜ao de um preditor b´asico de complexac¸˜ao a partir da seq¨uˆencia linear dos amino´acidos, como dispon´ıvel no PROMETHEUS, auxilia na determinac¸˜ao das poss´ıveis condic¸˜oes f´ısico-qu´ımicas, nas quais poderiam ocorrer a cristalizac¸˜ao das prote´ınas (ja-nelas de cristalizac¸˜ao). Dependendo da qualidade dos resultados que o usu´ario deseja obter, esses testes podem ser realizados em larga escala para um grande conjunto de prote´ınas em diversas condic¸˜oes experimentais, proporcionando dessa forma a obtenc¸˜ao das pr´e-condic¸˜oes f´ısico-qu´ımicas iniciais para a determinac¸˜ao experimental da estrutura prot´eica.
• Ind ´ustrias de alimentos: Produtos aliment´ıcios s˜ao compostos por uma grande diversi-dade de ingredientes como prote´ınas e polissacar´ıdeos (90, 99). As interac¸˜oes entre estas macromol´eculas desempenham um papel importante na estrutura e estabilidades destes produtos. Controlar ou manipular estas interac¸˜oes macromoleculares ´e o fator chave para o desenvolvimento de novos processos e produtos na ind´ustria de alimentos (90). Predizer estas interac¸˜oes facilita tal tarefa.
• Ind ´ustrias farmacˆeucias: A habilidade das prote´ınas se dissolverem em soluc¸˜ao aquosa ´e uma importante propriedade. Esta habilidade ´e medida pela solubilidade da prote´ına, 11Esta abordagem ´e realizada pelas ferramentas computacionais desenvolvidas e acopladas ao portal
PROMETHEUS, o qual permite o estudo e a predic¸˜ao de complexos prot´eicos com base nas propriedades ele-trost´aticas das prote´ınas, em diversos n´ıveis preditivos.
12Esta express˜ao ´e derivada do coeficiente de virial para medir a press˜ao osm´otica de uma soluc¸˜ao.B
23mede a
energia de interac¸˜ao de dois corpos. Uma abordagem mais detalhada sobre oB23´e exibida na Sec¸˜ao 5 -Material e m´etodos.
a qual possui grande importˆancia no processo de purificac¸˜ao de prote´ınas e s´erias implicac¸˜oes em muitas doenc¸as associadas com a agregac¸˜ao prot´eica (91, 92). A so-lubilidade da prote´ına ´e m´ınima ao redor do pI(100), e seu estudo ´e de grande interesse na ind´ustria farmacˆeutica na busca por novos f´armacos e prote´ınas recombinantes para o tratamento destas doenc¸as (Alzheimer, Parkinson, etc.). A solubilidade de uma prote´ına depende de sua carga14 e dos estados de ionizac¸˜ao15 (101) dos res´ıduos ioniz´aveis que a constituem. O portal PROMETHEUS permite o estudo das propriedades eletrost´aticas das prote´ınas em diversas condic¸˜oes experimentais (informadas pelo usu´ario) e n´ıveis de predic¸˜ao, possibilitando que diversas an´alises computacionais sejam realizadas com o objetivo de se obter os melhores ligantes para uma prote´ına e/ou complexo prot´eico.
O processo de purificac¸˜ao de prote´ınas, por precipitac¸˜ao induzida por sal, tem sido lar-gamente empregada na ind´ustria para separar prote´ınas como, por exemplo, prote´ınas do plasma do sangue (102), prote´ınas de extratos vegetais (103) e de bact´erias (104). En-tretanto a obtenc¸˜ao da condic¸˜ao de solubilidade n˜ao ´e trivial, pois depende de uma s´erie de fatores como, pH, distribuic¸˜ao de cargas na prote´ına, concentrac¸˜ao de sal e tempera-tura. Testar cada uma destas configurac¸˜oes experimentalmente ´e uma tarefa que consome muito tempo e possui alto custo. A gama de possibilidades pode ser reduzida atrav´es da utilizac¸˜ao de um preditor, que seja capaz de identificar (em tempo h´abil) quais s˜ao as melhores condic¸˜oes f´ısico-qu´ımicas para a precipitac¸˜ao prot´eica.
1.4
Abordagem do problema
Apesar do conhecimento bem estabelecido que se disp˜oe das principais forc¸as f´ısicas da natureza (105), a quantificac¸˜ao da contribuic¸˜ao de cada uma destas interac¸˜oes envolvidas nos mecanismos moleculares respons´aveis pela formac¸˜ao de um complexo prote´ına-prote´ına ainda ´e um problema n˜ao resolvido (27). A situac¸˜ao ´e equivalente a se conhecer os “ingredientes” (eletrost´atica,van der Waals, hidrofobicidade, etc.) para o preparo de um delicioso bolo, por´em, n˜ao se tem a quantidade necess´aria de cada um deles (falta a “receita”!). De forma an´aloga, n˜ao se conseguiu ainda quantificar a exata contribuic¸˜ao destas interac¸˜oes no processo conhecido comofoldingde prote´ınas (ou “enovelamento” prot´eico), onde se procura obter (e entender os mecanismos) uma estrutura espacial (tridimensional) de uma prote´ına (forma nativa) a partir da seq¨uˆencia prim´aria (sem muitas aplicac¸˜oes pr´aticas) dos elementos que a constituem (seus
14A abordagem te´orica sobre a origem das cargas em prote´ınas ´e exibida na Sec¸˜ao 5.1.1 -Equil´ıbrio ´acido-base. 15Os estados de ionizac¸˜ao s˜ao determinados atrav´es do estudo da diferenc¸a dos valores de pK
a’s, dos res´ıduos
ioniz´aveis, entre a estrutura terci´aria e prim´aria da prote´ına. Detalhes sobre a determinac¸˜ao dos valores depKa’s
amino´acidos). Em ambos os problemas (foldinge complexac¸˜ao), as mesmas interac¸˜oes f´ısicas est˜ao presentes, e h´a a necessidade de se quantificar a participac¸˜ao de cada uma delas para o completo entendimento dos mecanismos moleculares (106). Em outras palavras: procura-se tamb´em pela “receita” (27).
Duas principais tendˆencias de investigac¸˜ao te´orica normalmente s˜ao empregadas com o intuito de se elucidar estes processos: (a) tendˆencia mais f´ısica, e (b) tendˆencia mais com-putacional. Na primeira, assume-se um modelo para o sistema (onde se define as interac¸˜oes f´ısicas que se acredita serem as mais relevantes para o processo e estas s˜ao posteriormente analisadas) e resolve-se este modelo atrav´es de simulac¸˜oes computacionais, calculando-se as propriedades estruturais, dinˆamicas e termodinˆamicas de interesse. O modelo ´e aferido atrav´es de comparac¸˜oes com observac¸˜oes experimentais e/ou previs˜oes por outras teorias16. Na outra abordagem, mais computacional, procura-se valer de informac¸˜oes experimentais dispon´ıveis para descrever, analisar estatisticamente e eventualmente at´e mesmo prever o comportamento do sistema, mesmo que n˜ao se desvende a “f´ısica” do problema. O folding de prote´ınas e a predic¸˜ao de estruturas podem ser citados como exemplos cl´assicos, respectivamente, de cada uma destas duas abordagens, assim como o entendimento do fenˆomeno da complexac¸˜ao e a mera predic¸˜ao da complexac¸˜ao (aqui, sem se importar com as causas).
Por outro lado, os chamados m´etodos inversos (MI) (107, 108) permitem combinar ambas as tendˆencias, possibilitando inclusive a obtenc¸˜ao deHamiltonianas efetivas(modelos) (109, 110) a partir de dados experimentais. Para os sistemas com prote´ınas, o PDB ´e a principal fonte de informac¸˜oes “experimentais” estruturais. Entretanto, al´em das dificuldades intr´ınsecas dos MI, encontramos outras adicionais, por exemplo, a determinac¸˜ao de quais informac¸˜oes ex-tra´ıdas das estruturas prot´eicas s˜ao relevantes e precisam ser empregadas, visto que, mol´eculas de solvente (´agua) e at´e mesmo a posic¸˜ao do amino´acido na cadeia podem interferir diretamente no processo, pois podem alterar as propriedades f´ısico-qu´ımicas do sistema (p.ex. a carga total de uma prote´ına ´e uma func¸˜ao do meio e de sua pr´opria conformac¸˜ao).
Miyazawa e Jernigan (MJ) (111) estabeleceram a possibilidade de se extrair potenciais efetivos (chamados de “estat´ısticos” ou “baseados em conhecimento”) de interac¸˜ao a partir de an´alises de estruturas de prote´ınas dispon´ıveis no PDB. Desde ent˜ao, diversos trabalhos, princi-palmente direcionados para o problema de enovelamento (folding) de prote´ınas, seguiram esta linha (43, 44, 46, 112–114). Apesar de algumas cr´ıticas serem tamb´em reportadas (115, 116), acreditamos que “pistas” podem ser encontradas na an´alise destes potenciais estat´ısticos, especi-almente quando associadas a comparac¸˜oes com resultados obtidos por simulac¸˜oes moleculares
16Esta ´e a linha que adotamos na primeira parte do trabalho (construc¸˜ao de um portal web, chamado