iv I would like to thank my supervisor, Michael Parkhouse, not only for all the support and advice given throughout all these years, but also for making us laugh, for the cookies and chocolates and world music to balance my heavy metal tendencies. In summary, thank you for the unique and special working environment.
A special thanks to Rute Nascimento, my “supervisor in the bench”, for everything that she taught me and for the contagious enthusiasm with science. I am very lucky indeed, for the opportunity to work with you.
To everyone from Infection and Immunity group, past and present, specially to Silvia Correia and Sónia Ventura, for sharing with me the good days and the bad years in science.
I would like to acknowledge Instituto Gulbenkian de Ciência, one of the best places not only for post-docs but also for PhD students.
To my friend, Andreia, because after four thesis, and everything in between, the desert is still the same.
I would like to thank all my friends from “Cacém e arredores” not only for their friendship, but also for letting me know how non-scientists live and think, and especially to Pedro Ferreira and Silvia Batista for forming an efficient moral support group. From different research areas, but with the same PhD problem (and some others..) to solve.
v
BAC bacterial artificial chromosome CDK cyclin-dependent kinases
CLT cytomegalovirus latency-expressed transcripts CPE cytopathogenic effect
DAI DNA-dependent activator of IFN‑regulatory factors EBV Epstein-Barr virus
GM-P granulocyte-monocyte precursor cells
GPCR seven-transmembrane G protein-coupled receptor
HA Haemaglutinin peptide
HCMV Human cytomegalovirus HFF Human foreskin fibroblast HSV-1 Herpes simplex virus 1 HSV-2 Herpes simplex virus 2
IE Immediate early
IFN interferon
IL-8 Interleukin-8
KSHV Kaposi’s sarcoma-associated herpesvirus LAT latency-associated transcripts
MCMV Murine cytomegalovirus
MHC major histocompatibility complex MHV-68 Murine herpesvirus strain 68
MIEP major immediate-early promoter region MOI Multiplicity of infection
NEMO NF- B essential modulator NHEJ non-homologous end-joining
NK Natural killer
PAMP pathogen-associated molecular patterns PBS Phosphate buffered saline
PRR pattern-recognition receptors PVDF Polyvinylidene difluoride
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel TLR Toll‑like receptors
vi
O Citomegalovírus Humano (HCMV), incluído na subfamília dos -Herpesvírus, infecta indivíduos saudáveis, geralmente, de forma assintomática, no entanto, em indivíduos imunossuprimidos, como pacientes com SIDA, pode provocar doenças graves ou fatais. Após uma infecção primária, o HCMV estabelece uma infecção latente com reactivações periódicas. De modo a assegurar a sua sobrevivência e propagação, o HCMV desenvolveu vários mecanismos para subverter a resposta da imunidade inata e adaptativa do hospedeiro. Infecção pelo HCMV induz a produção da interleucina-8 (IL-8), uma quimiocina pro-inflamatória quimiotáctica, principalmente, para neutrófilos. Os neutrófilos têm um papel importante na disseminação do HCMV ao transportarem o vírus para diferentes órgãos do corpo e transmitindo-o a outros tipos de células. Por outro lado, a IL-8 aumenta a replicação e produção de viriões do HCMV.
vii
via de sinalização do NF-kB pode ser activada pelo reconhecimento de danos no DNA. De facto, não se observou indução de IL-8 pelo gene UL76 na ausência de ATM (células com mutação no gene ATM) ou após inibição da proteína ATM (inibidor específico para ATM, KU55933). De maior importância clínica, é o facto de infecção de células ATM -/- com o HCMV induzir níveis de IL-8 reduzidos comparando com a infecção de células normais. Por outro lado, um vírus HCMV com uma mutação no gene UL76 é significativamente menos eficiente a induzir a expressão de IL-8.
viii
Human cytomegalovirus (HCMV) is a -herpesvirus that infects healthy individuals, usually asymptomatically, but can cause severe or fatal disease in immunocompromised individuals such as transplant recipients or AIDS patients. Primary HCMV infection, as with other herpesviruses, is followed by establishment of lifelong latency and periodic reactivation. To ensure its survival and propagation within the host, HCMV has evolved many strategies to subvert both innate and adaptive host immunity. It is known that HCMV infection induces production of interleukin-8 (IL-8), a pro-inflammatory chemokine with neutrophil chemotatic activity. Significantly, neutrophils are a major carrier of HCMV during viremia and they are able to transmit infectious virus to other cells, playing a key role in virus dissemination through their contact with endothelial cells. In addition, IL-8 enhances HCMV virion production.
ix
pathway by the DNA damage response, we show the requirement of ATM in IL-8 induction by UL76. Specifically, there was no induction of IL-8 by UL76 using an ATM -/- cell line and a specific ATM inhibitor, KU55933. More importantly, there was a significant reduction of IL-8 secretion when ATM -/- cells were infected with wild type HCMV virus, suggesting that ATM is involved in IL-8 induction by the virus. To further demonstrate the impact of UL76 on HCMV-induced IL-8, we have established that a UL76 deletion mutant HCMV was significantly less efficient in stimulating IL-8 production than the wild type virus.
x
Resumo iii
Summary v
Table of contents
Chapter 1: Introduction
1.1. Virus 1
1.1.1. Herpesviruses 1
1.1.1.1. Virus structure and genome organization 1
1.1.1.2. Life cycle 2
1.1.1.3. Classification 3
1.1.1.3.1. The Alphaherpesvirinaesubfamily 5 1.1.1.3.2. The Gammaherpesvirinae subfamily 6 1.1.1.3.3. The Betaherpesvirinae subfamily 6 1.1.1.3.3.1. Human Cytomegalovirus (HCMV) 7
1.1.1.4. The UL24 gene family 10
1.2. HCMV-host interaction 14
1.2.1. Virus modulation of cell cycle and DNA damage 14 1.2.2. Host immune response to virus infection 20
1.2.3. Virus modulation of immune system 26
1.2.4. Virus modulation of chemokines 29
1.2.4.1. IL-8 30
1.2.4.1.1. Gene regulation 31
1.2.4.1.1.1. NF-KB 32
1.2.4.1.1.2. NF-kB Canonical pathway 34 1.2.4.1.1.3. NF-kB Non-Canonical pathway 34 1.2.4.1.1.4. NF-kB activation by genotoxic stress 35
xi Chapter 2: Human Cytomegalovirus UL76 protein induces
IL-8 expression
2.1. Summary 56
2.2. Introduction 57
2.3. Materials and Methods 59
2.3.1. Cells 59
2.3.2. Plasmids 59
2.3.3. Lentivirus production and titration 60 2.3.4. Lentivirus transduction and RNA isolation 60 2.3.5. Target Synthesis and Hybridization to Affymetrix
GeneChips 61
2.3.6. Microarrays Data Analysis 62
2.3.7. Reverse transcriptase polymerase chain reaction
(RT-PCR) 62
2.3.8. Luciferase assays 63
2.3.9. Enzyme-linked Immunoabsorbent Assay (ELISA) 63
2.3.10. Western blot 64
2.3.11. Statistical Analysis 64
2.4. Results 64
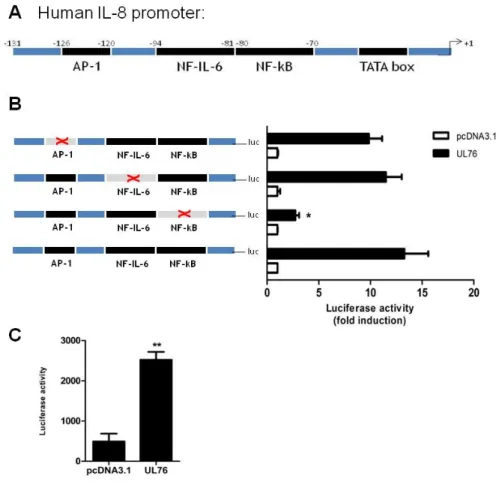
2.4.1. The HCMV UL76 gene induces transcription of IL-8 64 2.4.2. HCMV UL76 gene induces secretion of IL-8 protein 67
2.5. Discussion 68
xii
3.1. Summary 73
3.2. Introduction 74
3.3. Materials and Methods 76
3.3.1. Cells 76
3.3.2. Plasmids 76
3.3.3. Luciferase assays 77
3.3.4. Immunofluorescence 77
3.3.5. Nuclear extracts preparation 78
3.3.6. Western blot 78
3.3.7. Enzyme-Linked Immunoabsorbent Assay (ELISA) 79
3.3.8. Cell cycle analysis 79
3.3.9. Statistical analysis 80
3.4. Results 80
3.4.1. Induction of IL-8 by HCMV UL76 is
NF-kB-dependent 80
3.4.2. Induction of IL-8 by UL76 requires IKK activation
and IkB degradation. 82
3.4.3. UL76 induces translocation of p65 to the nucleus 83 3.4.4. IL-8 induction by HCMV UL76 is ATM-dependent 86 3.4.5. UL76 endonuclease motifs mutation reduced IL-8
induction and has no effect on cell cycle arrest 90
3.5. Discussion 92
3.6. Acknowledgements 95
xiii
4.1. Summary 99
4.2. Introduction 100
4.3. Materials and Methods 101
4.3.1. Cells 101
4.3.2. HCMV stock production 101
4.3.3. Enzyme-linked Immunoabsorbent Assay (ELISA) 102
4.3.4. Western blot 103
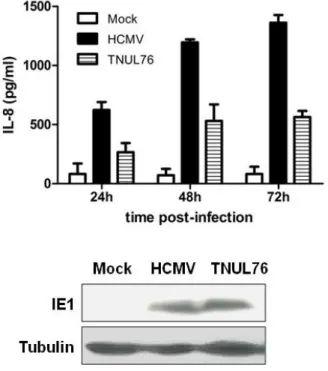
4.4. Results 103
4.4.1. Induction of IL-8 by HCMV is reduced in the
absence of UL76 103
4.4.2. ATM is required for IL-8 induction by HCMV 105
4.5. Discussion 106
4.6. References 109
Chapter 5: Final Considerations 112
xiv
Figure 1.1. Canonical and non-canonical signaling to NF- B 33
Figure 1.2. Activation of NF- B pathway by genotoxic stress 36
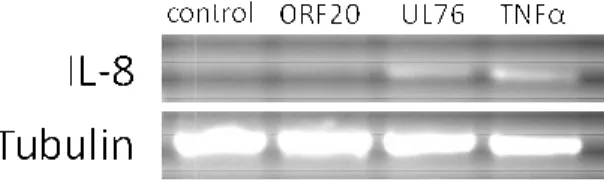
Figure 2.1. UL76 induces IL-8 gene transcription. 65
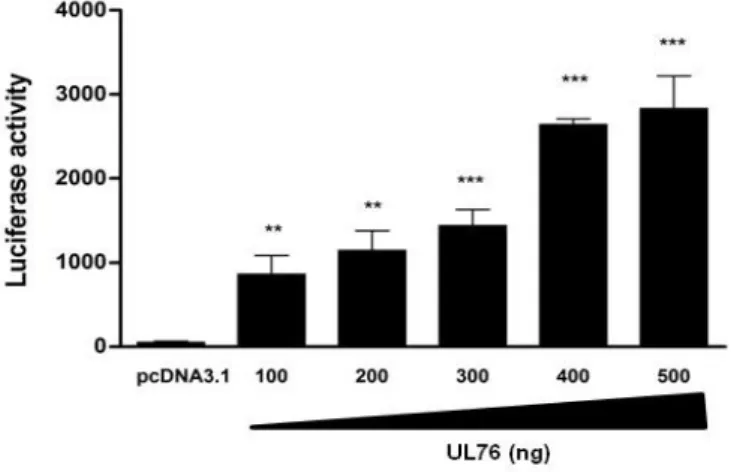
Figure 2.2. UL76 activates IL-8 promoter transcription. 66
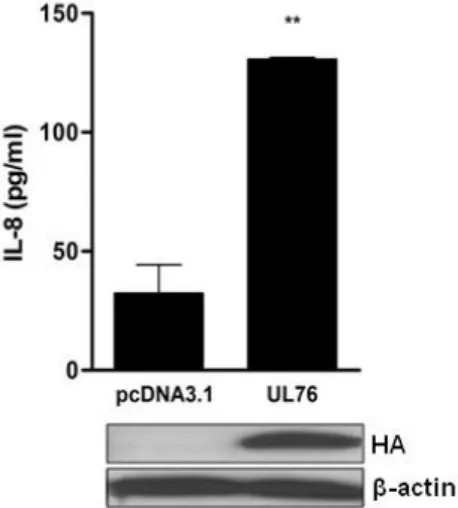
Figure 2.3. UL76 induces IL-8 secretion. 67
Figure 3.1 IL-8 induction by HCMV UL76 is NF-kB-dependent. 81
Figure 3.2 Induction of IL-8 by UL76 requires IKK activation
and IkB degradation. 83
Figure 3.3 UL76 induces p65 translocation to the nucleus. 85
Figure 3.4 UL76 activates NEMO through DNA damage
pathway. 87
Figure 3.5 IL-8 induction by UL76 is ATM-dependent. 89
Figure 3.6. Putative endonuclease activity impact on UL76
functions. 92
Figure 4.1 Requirement of UL76 for optimal IL-8 induction by
HCMV. 104
Figure 4.2 IL-8 induction by HCMV is ATM-dependent. 106
Table 1. Human herpesviruses 5
1 1.1. Virus
Viruses are obligate intracellular parasites that present uniquely close relationships with the organisms that they infect. Viruses have a limited coding genome thus, they rely on the infected cell to supply the energy, chemicals and much of the machinery required for their replication. Their success, like all parasites, is dependent on the balance that they maintain with their hosts, with which they need to coexist. The different components involved in this equilibrium will be reviewed in this introduction, with specific focus on herpesviruses, a classical example of a successful group of viruses.
1.1.1. Herpesviruses
Herpesviruses are a large group of successful, and widely distributed, double stranded DNA viruses of serious medical and veterinary importance. Although herpesviruses are well adapted to their natural host, their infection can result in severe diseases, particularly in children and immunocompromised individuals (Pellett & Roizman, 2007). Herpesviruses are characterized by their distinct virion morphology and the ability to establish latency during their life cycle.
1.1.1.1. Virus structure and genome organization
2
enclosed by the lipid envelope containing several glycoproteins (Davison, 2007b).
The variability of genome organization of herpesviruses provides the basis for their classification into six groups, ranging from simple forms showing a large sequence repeated at both terminus (e.g. HHV-6), or without repetitions at all (e.g. tupaia herpesvirus), to more complex organizations where the terminus sequences are internally repeated in an inverted orientation, dividing the genomes into two segments, each one consisting in unique sequences flanked by inverted repeats (e.g. HSV-1 and HCMV) (Pellett & Roizman, 2007).
1.1.1.2. Life cycle
3
modifications of proteins (e.g. protein kinases), although it can vary between different herpesviruses (Pellett & Roizman, 2007).
Viral replication occurs in the nucleus by circularization, followed by production of concatemers and cleavage of unit-length molecules which are packaged into the capsids (Boehmer & Lehman, 1997). The assembly of the capsid and packaging of the replicated viral genome occurs in the nucleus. The nucleocapsid is translocated to the cytoplasm by budding at the inner nuclear membrane followed by fusion of the primary envelope with the outer nuclear membrane. This process is mediated by viral and cellular proteins and involves reorganization of the nuclear architecture. During the final maturation process in the cytoplasm, tegument proteins associate with the translocated nucleocapsid and with the future envelope containing viral membrane proteins. This complex network of interactions results in the formation of an infectious virion, released by infected cells during the lytic phase of herpesviruses infection (Mettenleiter et al., 2009).
Latency, on the other hand, is characterized by limited gene expression and lack of virion production. During this phase, viral genomes form closed circular molecules that retain the capacity to replicate upon reactivation. Although all herpesviruses known are able to establish a latent infection in a specific type of cells, the mechanisms for establishment, maintenance, or termination of the latent phase differ between the viruses (Pellett & Roizman, 2007).
1.1.1.3. Classification
4
containing herpesviruses that infect mammalian, reptilian and avian hosts; Alloherpesviridae, which infect fish and amphibian hosts; and Malacoherpesviridae, with only one member, targeting the invertebrate bivalve mollusk. Eight different herpesviruses, encompassing all three herpesvirinae subfamilies, are known to infect humans (Davison et al., 2009). Based on phylogenetic studies the estimated date for the divergence from the most recent common ancestor of the three Herpesvirinae subfamilies is around 400 million years ago (McGeoch & Gatherer, 2005). Another study, based on the number of shared functions between the different subfamilies, supported previous observations of an early split, around 220 million years ago, of the - and -herpesviruses from the -herpesviruses (Albà et al., 2001).
5
Table 1. Human herpesviruses (Pellett & Roizman, 2007).
A brief description of each subfamily will be presented, focusing on the human herpesviruses and, with emphasis, on human cytomegalovirus (HCMV).
1.1.1.3.1. The Alphaherpesvirinaesubfamily
This subfamily contains the genera Simplexvirus (1 and HSV-2) and Varicellovirus (VZV) which infect mammalian hosts and, in addition, other genera that infect avian hosts (Davison et al., 2009).
The -herpesviruses are characterized by their relatively short life cycle and variable host range. Alpha-herpesviruses latency is established in sensory ganglia and neurons. During this phase, specific latency-associated transcripts (LATs) are expressed and there is no production of infectious virus. In the case of VZV, however, there is only expression of a restricted number of
Subfamily Virus Disease (ex.) Latency
alpha
Human Herpesvirus 1 (HSV-1) Fever, blisters
Sensory ganglia
neurons Human Herpesvirus 2 (HSV-2) Genital ulceration
Human Herpesvirus 3 (VZV) Chicken pox, shingles
beta
Human Herpesvirus 5 (HCMV)
Mononucleosis
Skin rash Mononuclear cells Human Herpesvirus 6
Human Herpesvirus 7
gamma
Human Herpesvirus 4 (EBV) Burkitt’s lymphoma
6
transcripts and proteins that are present in early phases of normal lytic infection (Hay & Ruyechan, 2007).
1.1.1.3.2. The Gammaherpesvirinae subfamily
The human -herpesviruses are divided in two genera, Lymphocryptovirus which includes human Epstein–Barr virus (EBV) and Rhadinovirus, which includes human Kaposi’s sarcoma-associated herpesvirus (KSHV). Recently, two lineages that are separable from the established genera were formed: Macavirus and Percavirus (Davison et al., 2009).
The members of this subfamily have cellular tropism for lymphocytes and are characterized by their ability to maintain latent infection in quiescent and proliferating cells. In addition, -herpesviruses are able to induce lymphoproliferation and cancers. Tumors associated with EBV and KSHV include mainly lymphoproliferative diseases and lymphomas (Pellett & Roizman, 2007).
1.1.1.3.3. The Betaherpesvirinae subfamily
7 The replication cycle of -herpesviruses is relatively long, with a slow progression in culture, in contrast to -herpesviruses. All members of the subfamily have a restricted host range and establish latency in hematopoietic cells in the myeloid lineage (Mocarski et al., 2007; Sissons et al., 2002). Both variants of HHV-6 and HHV-7 are predominately T-lymphotropic, although HHV-HHV-6 can also infect cells of myeloid lineage (Lusso et al., 1994; Santoro et al., 1999).
1.1.1.3.3.1. Human Cytomegalovirus (HCMV)
Human cytomegalovirus is the largest herpesvirus known, with a genome of approximately 235 kbp encoding more than 200 genes (Mocarski et al., 2007). The importance of HCMV as a human pathogen is emphasized by its prevalence in the population (estimated to be 50-90% in developed countries), and the fact that HCMV is the leading cause of congenital viral infection in humans. Primary infection with HCMV is frequently asymptomatic in immunocompetent individuals. In immunocompromised patients, however, such as transplant recipients or AIDS patients, HCMV infections frequently result in severe, even fatal, disease (Sissons & Carmichael, 2002).
Cells infected with HCMV show a unique cytopathogenic effect (CPE) consisting in rounded and enlarged cells, a characteristic designated cytomegaly which is responsible for the virus name (Albrecht & Weller, 1980).
8
fibroblasts and smooth muscle cells being the major targets for virus replication. Productive replication in such ubiquitous cell types permits HCMV to replicate in nearly every organ in the human host (Sinzger et al., 2008).
Fibroblasts are the standard cell culture system for propagation of HCMV, but several strains have endothelial cell tropism. It has been observed that extended propagation of HCMV in fibroblasts frequently results in loss of endothelial cell tropism, whereas propagation in endothelial cells maintains the broad cell tropism of the respective strain (Waldman et al., 1991). Differences between the cell tropisms of HCMV strains from clinical isolates from different patients may be related to the variable clinical profile of HCMV infections (Sinzger et al., 1999).
9
based on infection of granulocyte-monocyte precursor cells (GM-Ps) permitted the identification of cytomegalovirus latency-expressed transcripts (CLTs) consisting of spliced and un-spliced RNAs mapped to both strands of the MIE region of the HCMV genome (Kondo et al., 1996) and the UL111.5A transcript which encodes a viral homologue of interleukin-10 (vIL-10) (Jenkins et al., 2004). Other transcripts shown to be expressed during latency in CD34+ progenitor cells were encoded by UL138 (Goodrum et al., 2007) and LUNA (Bego et al., 2005).
Virus reactivation is associated with myeloid differentiation as viral lytic gene expression only occurs when these cells differentiate into macrophage or dendritic cell phenotypes (Reeves & Sinclair, 2008).
10 1.1.1.4. The UL24 gene family
The UL24 gene is located in the unique long segment of HSV-1 genome, overlapping the thymidine kinase gene (McGeoch et al., 1988). It is conserved not only in all three subfamilies of human herpesviruses, but also in other mammalian, avian and reptilian herpesviruses, with exception of amphibians or fish (Table 2).
Of the core herpesviruses genes, UL24 is the only one that remains unassigned to any functional category (Davison et al., 2002). Its universal presence in herpesviruses and lack of homology with cellular genes suggests that UL24 gene family has a relevant role in the viral life cycle and/or host evasion mechanisms.
11
Table 2. The human herpesvirus UL24 gene family
Deletion of the UL24 gene in HSV-1 resulted in a virus with significantly reduced plaque size and an associated decreased viral yield, suggesting that the UL24 function, although not essential, is important for virus growth, at least in cell culture (Jacobson et al., 1998; Jacobson et al., 1989). Similar results were obtained for HSV-2 UL24 (Blakeney et al., 2005), VZV ORF35 (Ito et al., 2005) and HCMV UL76 from AD169 strain (Yu et al., 2003). Global functional analysis conducted using HCMV Towne complete genome indicated that UL76 is an essential gene for viral replication of this viral strain (Dunn et al., 2003). This result, however, may be due to the impact of UL76 mutation on the overlapping UL77 gene as a recent study demonstrated that UL76 from Towne strain is not essential for viral growth, although it showed a slower replication cycle at low multiplicities of infection. Interestingly, the same study identified UL76 as a gene regulator of UL77 expression, possibly to control appropriate time and
Subfamily Virus Gene
alpha
Human Herpesvirus 1 (HSV-1) UL24
Human Herpesvirus 2 (HSV-2) UL24
Human Herpesvirus 3 (VZV) ORF35
beta
Human Herpesvirus 5 (HCMV) UL76
Human Herpesvirus 6 U49
Human Herpesvirus 7 U49
gamma
Human Herpesvirus 4 (EBV) BXRF1
12
concentration of this viral gene for efficient viral replication (Isomura et al., 2010).
Further deletion studies revealed that the absence of UL24 from HSV-1, HSV-2 and VZV leads to a syncytial plaque phenotype, similar to that observed for other HSV-1 viral proteins, suggesting that UL24, like UL20 and gK, may have a role in viral egress (Blakeney et al., 2005; Ito et al., 2005; Pearson & Coen, 2002), although the possible involvement of UL24 in assembly and egress of virus particles remains to be explored.
During viral infection, UL24 homologues are detected predominantly in the nucleus, and transiently localize in the nucleoli (Hong-Yan et al., 2001; Nascimento & Parkhouse, 2007; Pearson & Coen, 2002; Wang et al., 2000). Several studies demonstrated that UL24 is responsible for nucleolin and B23 redistribution in the nucleus during HSV-1 infection (Lymberopoulos & Pearson, 2007). Thus, it is possible that UL24 plays a role in nuclear egress through its impact on nucleoli, although how nucleolin affects HSV-1 nuclear egress is still unclear. The fact that deletion of conserved homology domains of UL24, including the putative endonuclease motifs, resulted in loss of nucleolin and B23 dispersal activity, suggests that this function may be shared among all herpesviruses and must be relevant for the viral life cycle (Bertrand et al., 2010; Bertrand & Pearson, 2008; Lymberopoulos et al., 2011).
13
study of herpesviruses biology in vivo. Similar to the human homologues, ORF20 is located in the nucleus and, when transiently expressed in human and murine cell lines, it induces cell cycle arrest at the G2/M phase, followed by apoptosis at later time points. During the G2 phase, the cyclin B/Cdc2 complex is inactive as the Cdc2 protein is in the inhibitory phosphorylated form. Consistent with the observed G2 arrest, cells expressing MHV-68 ORF20 showed an increased phosphoryation of Cdc2 at the inhibitory Tyr15 site and a consequent inactivation of Cdc2-cyclin B complex was demonstrated (Nascimento & Parkhouse, 2007). As observed for MHV-68 ORF20, the UL24 homologues from human herpesviruses representative of each subfamily (HSV-1 UL24, HCMV UL76 and KSHV ORF20) also induced cell cycle arrest followed by apoptosis through the same mechanism (Nascimento et al., 2009). The precise mechanism of cell cycle arrest induced by UL24 homologues remain to be clarified. An interesting and possible explanation is the recent report that HCMV UL76 induces chromosomal aberrations and DNA damage (Siew et al., 2009).
14
would be important to analyze the ORF20 mutant MHV-68 pathogenesis in its natural host, the wood mice (members of the genus Apodemus) (Ehlers et al., 2007) as a recent comparative analysis revealed that MHV-68 infection of BALB/c (M. musculus) and laboratory-bred wood mice are markedly different. The chemokine binding protein M3 of MHV-68 modulates the host response to infection in the natural host, although it has no effect during infection of Mus musculus-derived strains via the respiratory tract (Hughes et al., 2010).
1.2. HCMV-host interaction
1.2.1. Virus modulation of cell cycle and DNA Damage response
15
during cellular metabolism and collapsed replication forks, or from exogenous sources including ionizing radiation or chemicals that directly or indirectly damage DNA and are commonly used in cancer therapy (e.g. etoposide, camptothecin) (Shrivastav et al., 2008). The DNA damage sensor molecules (e.g. RPA, MRN) that activate the checkpoints appear to be shared by all the three pathways, or at least, to play a primary sensor role in one pathway and a secondary role in the others. The same is observed for the signal transducers molecules, such as protein kinases (ATM and ATR) and phosphatases, which are shared by the different checkpoints to varying degrees. The specificity of the checkpoint is due to the effector proteins, which inhibit phase transition (Sancar et al., 2004).
16
include expression and phosphorylation of repair proteins and chromatin modulation of repair factor accessibility (Shrivastav et al., 2008).
17
suggesting that they may have a role in HCMV manipulation of cell cycle progression (Castillo et al., 2005; Lu & Shenk, 1996; Wiebusch & Hagemeier, 1999).
Recent studies with HHV-6A, one of the less studied -herpesviruses, demonstrated that infection of T cells with HHV-6A results in cell cycle arrest at the G2/M phase. In this case, G2 arrest may serve to block the clonal expansion and proliferation of HHV-6A specific T cells to maintain immune suppression and evade the antiviral immune response (Li et al., 2011).
A common strategy of several gamma-herpesviruses is the expression of a viral cyclin (v-cyclin) with homology to the cellular D-type cyclin (Upton et al., 2005; van Dyk et al., 1999; Verschuren et al., 2004). Reminiscent of cellular cyclins, v-cyclin interacts with, and thus activates, the Cdk4 and Cdk6 kinases. Although the v-cyclin is functionally similar to cellular v-cyclin, it is resistant to inhibition by p21 and p16 and it is not regulated by the cyclin-dependent kinases (Cdks) (Direkze & Laman, 2004).
18
viruses defend themselves from the host cell DNA damage response machinery. Paradoxically, recent reports indicate that such cellular responses may have a beneficial role in viral replication (Luo et al., 2011).
19
DNA damage response in an ATM- and DNA-PK-dependent manner in the absence of DNA lesions (Soutoglou & Misteli, 2008). Thus, it is possible that the trigger of herpesvirus-induced DNA damage response is not the recognition of viral DNA as double strand breaks or actual damage to DNA, but it is the recruitment of DNA damage repair factors observed during viral infection.
The beneficial role of activation of DNA damage in viral replication is not restricted to herpesviruses as it has also been observed for several other viruses. Simian virus 40 (SV40) replication is dependent of ATM-mediated phosphorylation of large tumour antigen (LTag), an essential viral protein involved in viral replication (Shi et al., 2005). Human papillomavirus (HPV) infection also induces an ATM response in both undifferentiated and differentiated cells. Importantly, ATM kinase activity is required for viral genome amplification in differentiating cells, but not for episome maintenance in undifferentiated cells. This suggests that activation of the DNA damage signaling response by HPV is tailored to different requirements, depending on the differentiation stage of the host cell (Moody & Laimins, 2009). Adenovirus, however, has evolved mechanisms to inhibit DNA repair during infection, by degradation and mislocalization of the Mre11–Rad50– NBS1 complex, thus preventing activation of DNA damage checkpoints and viral DNA concatemerization. The model proposed is that the DNA damage response results in the masking of the origins of adenovirus DNA replication such that viral replication proteins are unable to gain access (Stracker et al., 2002).
20
viral pathology and persistence, but also new ideas for the development of antiviral and anti-tumour drugs. Moreover, the study of virus host evasion mechanisms can provide new tools to study recognition and repair of damaged DNA by cellular machinery.
1.2.2. Host immune response to virus infection
Host cells have at least three layers of defense mechanisms which include autonomous innate mechanisms, such as RNA interference and cytidine deamination (these will not be discussed further), nonautonomous innate mechanisms induced by exposure to viruses and other stimuli such as interferon and proinflammatory cytokines and adaptive (or acquired) immunity resulting in an antigen-specific response via antibodies and lymphocytes. The aim of these systems is to clear the viruses and virus-infected cells, or at least to limit the pathogenicity and viral spread. To achieve this goal, there is a significant interplay between them, although they present significant differences related with their rapidity, specificity, persistence, evolutionary antiquity and consequences for the infected cell (Biron & Sen, 2007).
21
Innate immune recognition of viruses as foreign, and thus potentially dangerous, is performed by a limited number of germline-encoded pattern-recognition receptors (PRRs). The host cell PRRs immediately distinguish self molecules from viruses through recognition of microbial components, known as pathogen-associated molecular patterns (PAMPs), which are expressed constitutively by pathogens. Toll‑like receptors (TLRs), membrane‑bound receptors that are localized in the plasma membrane and endosomes, are the best characterized PRRs. Of particular interest for virus infection is the endossomal TLR3, which recognizes viral double stranded RNA, and TLR7, TLR8 and TLR9 which recognise viral DNA. Intracellular PRRs detect pathogen nucleic acids in the cytoplasm. RNA is recognized by the RIG‑I‑like receptors (RLRs) retinoic acid‑inducible gene I (RIG‑I) and melanoma differentiation‑associated gene 5 (MDA5). Intracellular DNA sensors include DNA-dependent activator of IFN‑regulatory factors (DAI), absent in melanoma 2 (AIM2), RNA polymerase III, leucine‑rich repeat flightless interacting protein 1 (LRRFIP1) and, most recently, IFN ‑inducible protein 16 (IFI16). Activation of TLRs and intracellular nucleic acid sensors results in the induction of signalling pathways that lead to the expression of proteins with pro‑inflammatory and microbicidal activities, including cytokines and type I IFNs (IFN and IFN ) (Mogensen, 2009; Takeuchi & Akira, 2010).
22
importance of the interferon response against viral infections has been emphasized by the increased susceptibility to virus infection of mice deficient for different components of the IFN system (Arnheiter et al., 1996; Hefti et al., 1999).
Interferons are a large family of related cytokines which include type I IFNs, type II IFNs and type III IFN. The IFNs that are induced directly in response to virus infection are IFN- and – , which bind to a widely distributed receptor. Interferon- , a type II IFN and commonly called “immune IFN”, on the other hand, binds to a different cell-surface receptor and is only secreted by activated T cells and NK cells (Farrar & Schreiber, 1993). The type III IFNs are also secreted after viral infection and following stimulation of key molecules involved in type I IFN induction, such as IRF-3, RIG-I, and NF- B (Onoguchi et al., 2007). Once secreted, Type III IFNs bind to specific cellular receptors eliciting an antiviral response similar to IFN / , with the induction of transcription of hundreds of inferferon stimulated genes (ISGs) (Sommereyns et al., 2008).
23
The innate immunity mechanisms are clearly ineffective in the clearance of the herpesviruses and in preventing the consequent establishment of latency, although it controls the viral replication leading to a generally asymptomatic infection. In the case of HCMV, the innate immune response initiated after the binding and entry of the virus into the cell involves the activation of IRF-3 that will lead to the production of type I interferon via the DNA sensor DAI (DeFilippis et al., 2010a) and pro-inflammatory cytokines by CD14 and TLR2 recognition (Compton et al., 2003). The possible involvement of structural viral proteins in the induction of innate immune responses was confirmed by the identification of the HCMV envelope glycoproteins gB and gH as proteins essential for TLR2 activation (Boehme et al., 2006). Recently, however, it has been demonstrated that glycoprotein B is not essential for the activation of IRF-3-dependent innate immune response. In fact, IRF3 signaling activated by HCMV infection is very similar to the pathway activated by cytoplasmic double-stranded DNA (DeFilippis et al., 2010b).
An important component of the innate immune response to viral infections is the natural killer (NK) cell population. The importance of this subset of cells in cytomegalovirus immunity is evident in the MCMV infection of mice but there is limited evidence in the case of HCMV infection. The fact that HCMV encodes several proteins that prevent NK cell activation by different mechanisms underlines the conclusion that NK cells have an important role in the immune response to HCMV infection as well (Jackson et al., 2011).
24
the adaptive immune system, B lymphocytes and T lymphocytes, are activated by the recognition of viral constituents, through the binding to highly specific cell surface recognition receptors. These receptors are specific to the constituents, designated by antigens, of that particular virus. Importantly, the adaptive immune system adapts to repeated exposure to a particular virus or viral protein, resulting in a more rapid response of higher magnitude by the specific adaptive immune cells during a secondary infection with that virus. This phenomenon is called immunological memory and it is the basis for vaccination (Braciale et al., 2007).
25
response to primary HCMV infection includes the production of specific antibodies for several structural and non-structural proteins, as well as for envelope glycoproteins (Landini & Michelson, 1988) and these play an important role in preventing transmission to the fetus during pregnancy (Fowler et al., 1992). Similarly, T lymphocytes maintain their antigen-specific receptors on the cell surface. Upon activation T cells are important in the recognition and elimination of virus-infected cells. The T lymphocytes antigen receptors recognize processed peptide fragments of intracellular viral proteins presented on the infected cell surface by the major histocompatibility complex (MHC) class I. Alternatively, antigens may be internalized by antigen presenting cells, such as dendritic cells and macrophages. After intracellular degradation, the resulting peptides are incorporated into the MHC class II molecules which are transported to the surface of the antigen presenting cell and are recognized by CD4+ T cells, thus leading to their activation and participation in both serological and CD8+ cytotoxic immune responses.
Activated CD8+ T lymphocytes limit the spread of infection by killing virus-infected cells through direct cell-to-cell contact and by the release of soluble mediators which include cytokines such as interferon gamma and tumor necrosis factor alpha (TNF-α). The activated T lymphocytes, both CD4+ and CD8+, through these and other soluble mediators, recruit and coordinate the response of the innate immune cells which, in turn, act to clear infection.
26
HCMV. Similar studies are limited during primary infection, however, partially because it is frequently asymptomatic (Crough & Khanna, 2009). Decades of research identified several HCMV proteins that are targets of the CD8+ T cell response to HCMV. However, the majority of the studies on the immunobiology of the CD8+ T cell response in primary infection and the generation of long term memory have been using the first identified viral targets, namely pp65 and IE proteins (Borysiewicz et al., 1988; McLaughlin-Taylor et al., 1994). Early studies with MCMV infected mice showed that CD4+ T cell are also an important component of the cellular immunity response to cytomegaloviruses as long term depletion of the whole CD4+ T cell compartment resulted in persistent virus replication at specific sites (Jonji et al., 1989). Furthermore, the maintenance of HCMV specific CD8+ T cell infusions in bone marrow transplant patients was dependent on the presence of HCMV-specific CD4+ cells indicating a critical role of CD4+ helper T cells in the effective response of CD8+ T cells (Einsele et al., 2002; Walter et al., 1995).
Finally, there is evidence that a subset of gamma delta ( ) T cells (the minor V 2 negative subpopulation) is expanded following HCMV reactivation in transplant patients and is able to mediate cytotoxicity of HCMV infected target cells (Knight et al., 2010).
1.2.3. Virus modulation of immune system
27
herpesviruses, in the establishment of persistence during the host’s life time.
The interferon system is a powerful and first line of defence against virus infections, and so it is not surprising that viruses have evolved multiple means of down regulating the induction and impact of IFN responses. These include inhibiting IFN production, inhibiting the mediated signalling pathways, and blocking the action of IFN-induced enzymes with antiviral activity. The exact strategy exploited by a virus will presumably depend on the biology of the host-virus interaction and will be a major factor that will influence the pathogenesis of that virus infection (Randall & Goodbourn, 2008). Herpesviruses induce expression of type I IFN during the primary infection (Ankel et al., 1998; Boehme et al., 2004). Thus, it is not surprising that an effective evasion of these initial type I IFN responses is essential for virus replication and establishment of latency.
28
On the other hand, UL83 has been also shown to inhibit IFN- production by inhibiting IRF-3 phosphorylation and translocation into the nucleus (Abate et al., 2004). HCMV also encode two PKR antagonists, proteins IRS1 and TRS1 (Cassady, 2005).
Natural killer (NK) cells are crucial in controlling cytomegalovirus infections in both the human and the murine hosts. NK cells are inhibited by signals delivered via inhibitory receptors interacting with class I MHC molecules on the surface of target cells (Jackson et al., 2011). Interestingly, cells infected with HCMV are remarkable resistant to NK cell-mediated cytolysis in vitro. Not surprisingly, several genes encoding proteins (UL16, UL18, UL40, UL83, UL141 and UL142) and one encoding a microRNA (miR-UL112) have been identified as capable of suppressing NK cell recognition through presentation of inhibitory signals or suppression of activating signals (Wilkinson et al., 2008). One of the mechanisms of HCMV-mediated inhibitory receptor signalling is the expression of UL18 gene, a MHC class I homologue present at the cell surface where it binds the inhibitory NK cell receptor LILRB1 (LIR-1) inhibiting activation of NK cells (Prod'homme et al., 2007). On the other hand, HCMV infection efficiently activates expression of ligands for the NK cell activating receptor NKG2D. However, at least two proteins, UL16 and UL142, and the miR-UL112 collaborate to suppress presentation of these ligands on the cell surface inhibiting NK cells activation (Wilkinson et al., 2008).
29
HCMV and MCMV interfere with CD4+ T cell recognition of infected cells by inhibition of MHC class II expression (Heise et al., 1998; Scholz et al., 1992; Sedmak et al., 1995). Moreover, it has been demonstrated that HCMV evolved different mechanisms to downregulate MHC class II molecules at early and late times after infection. The early inhibition is probably due to a structural component while the late effect was dependent on virus replication and expression of US1-US9 or US11 (Odeberg & Söderberg-Nauclér, 2001).
1.2.4. Virus modulation of chemokines
Chemokines are structurally related molecules that regulate the trafficking and activation of various types of leukocytes through the interaction with a subset of seven-transmembrane G protein-coupled receptor (GPCR) and thus may be important mediators of host defense mechanisms. Chemokines can be divided in three subfamilies: -chemokines (C-X-C), which act primarily on neutrophils (chemotaxis), -chemokines (C-C), which act on monocytes, lymphocytes, basophils and eosinophils (chemoattraction and degranulation), and -chemokines, which act upon lymphocytes (Zlotnik & Yoshie, 2000).
30
1999) and the vMIP-I (K6 gene) and vMIP-III (K4.1) from KSHV which show angiogenic activity (Boshoff et al., 1997; Kledal et al., 1997; Stine et al., 2000). The chemokine homologues are not shared by closely related herpesviruses, suggesting that they may be related to specific aspects of pathogenesis. Other common evasion mechanism includes the inhibition of chemokine signaling by the secretion of soluble chemokine-binding proteins or by encoding GPCR homologues which mimic the functions of cellular GPCR (Alcami, 2003). A well studied example is the US28 gene from HCMV encoding a vGPCR for β-chemokines (Gao & Murphy, 1994). Constitutively activated, US28 protein can regulate the chemokine environment by sequestering CC chemokines and thus, depleting them from the medium (Bodaghi et al., 1998). In addition, viruses hijack chemokine receptors as co-receptors for viral entry and intracellular signaling. For example, chemokine receptors CCR5 and CXCR4 are major HIV-1 co-receptors which mediate viral entry into susceptible cells (Deng et al., 1996; Feng et al., 1996).
In addition to encoding viral homologues of cellular proteins involved in the signaling of chemokines, generally viruses are able to induce or inhibit expression of several chemokines. Interleukin-8 is an important example as it is induced by several viruses and it will be described in more detail.
1.2.4.1. IL-8
31
angiogenesis (Koch et al., 1992) and interferon- inhibition (Khabar et al., 1997). IL-8 expression is low or absent under normal conditions but highly inducible in vivo, as well as in a wide variety of cells in vitro, by a wide range of extracellular stimuli in addition to viruses, such as the proinflammatory cytokines IL-1 or TNF-α
(Kasahara et al., 1991) or by bacteria (Aihara et al., 1997) and cell-stressing agents.
1.2.4.1.1. Gene regulation
32
repression factor (NRF). The low amount of IL-8 found in unstimulated cells results not only from a repressed transcription but also due to mRNA instability. Expression of IL-8 is regulated post-transcriptionally by the p38 MAPK pathway that contributes to cytokines/stress-induced IL-8 by stabilizing the IL-8 mRNA (Hoffmann et al., 2002).
In conclusion, IL-8 production is actively repressed in the absence of external stimuli. During stimulation, maximal IL-8 amounts can be produced as a result of promoter derepression, activation of NF-kB and JNK pathways to induce transcription and the rapidly stabilization of the resulting mRNA by the p38 MAPK pathway. In this way, cells are able to rapidly increase and at the same time, regulate the amount of IL-8 secreted and thereby control the extent of leukocytes attracted to sites of tissue injury.
1.2.4.1.1.1. NF-KB
Nuclear factor-kB (NF-kB) consists of a family of transcription factors which in mammals include p65 (RelA), RelB, c-Rel, p105/p50 (NF-kB1), and p100/52 (NF-kB2). Each subunit can associate with each other to form distinct transcriptionally active homo- and hetero-dimeric complexes, with p50/p65 heterodimer being the most abundant and present in almost all cell types (Oeckinghaus & Ghosh, 2009).
33
The NF-kB signaling pathway plays critical roles in inflammation, immunity, cell proliferation, differentiation, and survival as specific NF-kB binding sites are present in the promoters/enhancers of a large number of genes. Thus, not surprisingly, deregulation of NF-kB is associated with a considerable number of diseases (Perkins, 2007).
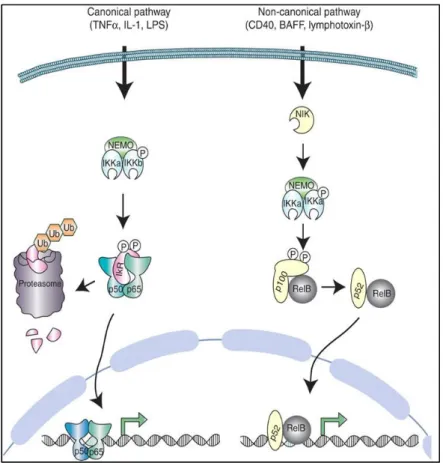
The complexity of the NF-KB signaling is demonstrated by the diverse mechanisms of activation of NF-kB pathway. A simplified view of the two main pathways (canonical and noncanonical) is shown in Figure 1.1 and they will be briefly described.
Figure 1.1. Canonical and non-canonical signaling to NF- B
34 1.2.4.1.1.2. NF-kB Canonical pathway
Diverse inflammatory stimuli such as the cytokines TNF- and IL– 1 activate the ubiquitous p65/p50 dimer through the ‘canonical’ or ‘classical’ NF- B pathway (Fig. 1.1). Stimulation through TNF receptor (TNFR), IL–1 receptor (IL–1 R), or TLRs leads to the phosphorylation of I B kinase (IKK) complex, composed of the two catalytic subunits, IKK and IKK , and the regulatory protein, IKK /NF- B essential modulator (NEMO). The activated IKK complex phosphorylates IkB (inhibitor of NF-kB) proteins leading to their ubiquitination and degradation, thus releasing the NF-kB subunits to translocate to the nucleus and induce transcription of target genes. In most cell types, activation of the canonical pathway results in the nuclear localization of NF-kB dimers within minutes. Regulation of NF-kB signaling involves a series of post-translational modifications of the NF-kB subunits in addition to other proteins of the pathway (Oeckinghaus & Ghosh, 2009; Perkins & Gilmore, 2006).
1.2.4.1.1.3. NF-kB Non-Canonical pathway
35
p52/RelB heterodimers. As different NF-kB dimers combinations have overlapping but distinct DNA-binding specificities, stimulation of both the canonical and noncanonical pathways lead to activation of a different spectrum of genes promoters and enhancers (Perkins & Gilmore, 2006).
1.2.4.1.1.4. NF-kB activation by genotoxic stress
In the canonical and non-canonical pathways, activation results from membrane-associated receptors stimulation. In recent years however, a different pathway activated by genotoxic stimuli, such as ionizing radiation or some chemotherapeutic drugs as etoposide, has been characterized. In this case, the activation signal comes from the nucleus although it converges in IKK complex activation in the cytoplasm as the canonical pathway (Fig. 1.2).
Genotoxic stress induces, in parallel, activation of ATM and translocation of NEMO to the nucleus, where it is sumoylated by a mechanism dependent on PARP1, PIASy and Ubc9. NEMO sumoylation is followed by phosphorylation and mono-ubiquitylation in an ATM-dependent manner, leading to the nuclear export of NEMO as a complex with ATM. The activation of the IKK complex in the cytoplasm by the NEMO–ATM complex requires the associated protein ELK (a protein that is rich in glutamate (E), leucine (L), lysine (K) and serine (S)) (Janssens & Tschopp, 2006; Miyamoto, 2011).
36
Figure 1.2. Activation of NF- B pathway by genotoxic stress.
37 1.2.4.1.2. IL-8 and HCMV
It has been previously described that IL-8 enhances HCMV replication and virus production at 5 and 7 days after the infection, without initiating virus production earlier. An increase in IL-8 receptor was also observed in HCMV-infected cells, suggesting that HCMV infection may alter the responsiveness of cells to IL-8 by induction of IL-8 receptor expression, and in this way, modulate the intracellular environment to be more favorable for viral replication (Murayama et al., 1994). In addition, HCMV infection increased the levels of IL-8 protein in human lung fibroblasts, astrocytoma cells, endothelial cells, peripheral blood mononuclear cells and a human monocytic cell line (THP-1) (Craigen et al., 1997; Grundy et al., 1998; Murayama et al., 1997). Infection with high passage laboratory HCMV strains (AD169, Towne, Davis) or low passage clinical strains (Toledo and ClF isolates) showed that all virus strains significantly increase IL-8 production compared with uninfected cells (Craigen et al., 1997).
38
levels, suggesting the involvement of one or more viral proteins (Cinatl et al., 2000; Cinatl et al., 1999).
The IL-8 induced by HCMV infection is functional since IL-8 in supernatants from HCMV-infected cells significantly enhanced neutrophil transendothelial migration compared to those from uninfected cells. Moreover, infected endothelial cells were able to transmit the virus to neutrophils by both cell co-culture and neutrophil transmigration assays. Thus, a possible model is that HCMV-infected endothelial cells, in addition to other cells in the tissues, such as fibroblasts, recruit neutrophils to sites of infection by the secretion of IL-8. The infected endothelial cells then transmit the virus to the neutrophils, which disseminate the virus throughout the body via the bloodstream (Craigen et al., 1997; Grundy et al., 1998). Moreover, induction of IL-8 can also be considered to be a strategy of immune response evasion through disruption of normal cell migration patterns or the preferential attraction of leukocytes that cannot efficiently clear the virus (Penfold et al., 1999). Interestingly, HCMV encodes two genes, UL146 and UL147 which are homologues to CXC chemokines. The UL146 gene encodes the protein vCXCL1 which functions as a selective agonist for CXCR2 and, although with lower affinity and potency, for CXCR1 (Lüttichau, 2010; Penfold et al., 1999). The gene product of UL147 has not yet been characterized. The presence of a viral gene that mimics the effect of IL-8 in neutrophil chemotaxis reinforces the importance of neutrophils in the pathogenesis of HCMV. Thus, the absence of UL146 in HCMV AD169 strain or sequence divergence observed in several clinical strains may contribute to the observed attenuation of their associated pathogenesis.
39
such as IL-8. A recent example is HCMV UL7, a homologue of the SLAM-family receptor CD229, which impairs the production of IL-8, among other proinflammatory cytokines, in different cell types (Engel et al., 2011). Thus, HCMV may have evolved different mechanisms to induce IL-8 for its benefit, on the other hand, whilst also evolving genes that limit its upregulation, thus preventing excessive inflammation and consequent deleterious effects. Another interesting effect of IL-8 related to viral infections is the reduction of antiviral activity of IFN observed in vitro (Khabar et al., 1997).
In summary, IL-8 induced by HCMV may aggravate HCMV infection by enhancing virus replication, virion production and viral dissemination due to neutrophil chemotaxis, in addition to inhibiting the antiviral activities of IFN. Thus, IL-8 might be a novel target for HCMV infection and associated diseases therapy.
1.3. Aim of the project
40
such as x-ray crystallography. The UL24 gene family is one of the approximately 40 core genes that are conserved in all three herpesviruses subfamilies and the only one which still has no assigned function. Previously, functional assays demonstrated that all homologues of UL24 gene family induce cell cycle arrest (Nascimento et al., 2009; Nascimento & Parkhouse, 2007), confirming that genes conserved in all herpesviruses are likely to manipulate conserved cellular pathways in a similar manner in all herpesviruses. Other strategies, on the other hand, may be restricted to one subfamily or species, with a function related to a more restricted and specific aspect of the virus life cycle. In this work, microarray analysis of cells expressing the HCMV UL24 homologue (UL76) gave new insight into herpesviruses host evasion strategies. Specifically, the aim of this project is to identify and characterize a new function for UL76, the UL24 homologue from HCMV, consisting in the induction of IL-8 and its impact during HCMV infection.
1.4. References
Abate, D. A., Watanabe, S. & Mocarski, E. S. (2004). Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 78, 10995-11006.
Abraham, R. T. (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15, 2177-2196.
Ahn, J. H. & Hayward, G. S. (1997). The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol 71, 4599-4613.
41 Albrecht, T. & Weller, T. H. (1980). Heterogeneous morphologic features of plaques induced by five strains of human cytomegalovirus. Am J Clin Pathol 73, 648-654.
Albà, M. M., Das, R., Orengo, C. A. & Kellam, P. (2001). Genomewide function conservation and phylogeny in the Herpesviridae. Genome Res 11, 43-54.
Alcami, A. (2003). Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3, 36-50.
Ankel, H., Westra, D. F., Welling-Wester, S. & Lebon, P. (1998). Induction of interferon-alpha by glycoprotein D of herpes simplex virus: a possible role of chemokine receptors. Virology 251, 317-326.
Arnheiter, H., Frese, M., Kambadur, R., Meier, E. & Haller, O. (1996). Mx transgenic mice--animal models of health. Curr Top Microbiol Immunol 206, 119-147.
Bego, M., Maciejewski, J., Khaiboullina, S., Pari, G. & St Jeor, S. (2005). Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J Virol 79, 11022-11034.
Bertrand, L., Leiva-Torres, G. A., Hyjazie, H. & Pearson, A. (2010). Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J Virol 84, 109-118.
Bertrand, L. & Pearson, A. (2008). The conserved N-terminal domain of herpes simplex virus 1 UL24 protein is sufficient to induce the spatial redistribution of nucleolin. J Gen Virol 89, 1142-1151. Biron, C. A. & Sen, G. C. (2007). Innate Responses to Viral Infections. In
Fields Virology, 5th Edition. Edited by D. M. Knipe & P. M. Howley: Lippincott Williams & Wilkins.
Blakeney, S., Kowalski, J., Tummolo, D., DeStefano, J., Cooper, D., Guo, M., Gangolli, S., Long, D., Zamb, T., Natuk, R. J. & Visalli, R. J. (2005). Herpes simplex virus type 2 UL24 gene is a virulence determinant in murine and guinea pig disease models. J Virol 79, 10498-10506.
Bodaghi, B., Jones, T. R., Zipeto, D., Vita, C., Sun, L., Laurent, L., Arenzana-Seisdedos, F., Virelizier, J. L. & Michelson, S. (1998). Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J Exp Med 188, 855-866.
42 for TLR2 activation in permissive cells. J Immunol 177, 7094-7102.
Boehme, K. W., Singh, J., Perry, S. T. & Compton, T. (2004). Human cytomegalovirus elicits a coordinated cellular antiviral response via envelope glycoprotein B. J Virol 78, 1202-1211.
Boehmer, P. E. & Lehman, I. R. (1997). Herpes simplex virus DNA replication. Annu Rev Biochem 66, 347-384.
Borysiewicz, L. K., Hickling, J. K., Graham, S., Sinclair, J., Cranage, M. P., Smith, G. L. & Sissons, J. G. (1988). Human cytomegalovirus-specific cytotoxic T cells. Relative frequency of stage-cytomegalovirus-specific CTL recognizing the 72-kD immediate early protein and glycoprotein B expressed by recombinant vaccinia viruses. J Exp Med 168, 919-931.
Boshoff, C., Endo, Y., Collins, P. D., Takeuchi, Y., Reeves, J. D., Schweickart, V. L., Siani, M. A., Sasaki, T., Williams, T. J., Gray, P. W., Moore, P. S., Chang, Y. & Weiss, R. A. (1997). Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278, 290-294.
Braciale, T. J., Hahn, Y. S. & Burton, D. R. (2007). The Adaptive Immune Response to Viruses. In Fields Virology, 5th Edition. Edited by D. M. Knipe & P. M. Howley: Lippincott Williams & Wilkins.
Bresnahan, W. A., Boldogh, I., Thompson, E. A. & Albrecht, T. (1996). Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224, 150-160.
Cann, K. L. & Hicks, G. G. (2007). Regulation of the cellular DNA double-strand break response. Biochem Cell Biol 85, 663-674.
Cassady, K. A. (2005). Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol 79, 8707-8715.
Castillo, J. P., Frame, F. M., Rogoff, H. A., Pickering, M. T., Yurochko, A. D. & Kowalik, T. F. (2005). Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J Virol 79, 11467-11475. Cinatl, J., Kotchetkov, R., Weimer, E., Blaheta, R. A., Scholz, M., Vogel, J.
U., Gümbel, H. O. & Doerr, H. W. (2000). The antisense oligonucleotide ISIS 2922 prevents cytomegalovirus-induced upregulation of IL-8 and ICAM-1 in cultured human fibroblasts. J Med Virol 60, 313-323.
43 inhibition of cytomegalovirus immediate-early expression in combination with antioxidants as a novel treatment strategy? Intervirology 42, 419-424.
Compton, T., Kurt-Jones, E. A., Boehme, K. W., Belko, J., Latz, E., Golenbock, D. T. & Finberg, R. W. (2003). Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 77, 4588-4596.
Craigen, J. L., Yong, K. L., Jordan, N. J., MacCormac, L. P., Westwick, J., Akbar, A. N. & Grundy, J. E. (1997). Human cytomegalovirus infection up-regulates interleukin-8 gene expression and stimulates neutrophil transendothelial migration. Immunology 92, 138-145.
Crough, T. & Khanna, R. (2009). Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 22, 76-98, Table of Contents.
Davison, A. J. (2007a). Comparative analysis of the genomes. In Human Herpesviruses
Biology, Therapy, and Immunoprophylaxis: Cambridge University Press. Davison, A. J. (2007b). Overview of classification. In Human
Herpesviruses
Biology, Therapy, and Immunoprophylaxis: Cambridge University Press. Davison, A. J., Dargan, D. J. & Stow, N. D. (2002). Fundamental and
accessory systems in herpesviruses. Antiviral Res 56, 1-11. Davison, A. J., Eberle, R., Ehlers, B., Hayward, G. S., McGeoch, D. J.,
Minson, A. C., Pellett, P. E., Roizman, B., Studdert, M. J. & Thiry, E. (2009). The order Herpesvirales. Arch Virol 154, 171-177. DeFilippis, V. R., Alvarado, D., Sali, T., Rothenburg, S. & Früh, K. (2010a).
Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol 84, 585-598.
DeFilippis, V. R., Sali, T., Alvarado, D., White, L., Bresnahan, W. & Früh, K. J. (2010b). Activation of the interferon response by human cytomegalovirus occurs via cytoplasmic double-stranded DNA but not glycoprotein B. J Virol 84, 8913-8925.
44 Direkze, S. & Laman, H. (2004). Regulation of growth signalling and cell cycle by Kaposi's sarcoma-associated herpesvirus genes. Int J Exp Pathol 85, 305-319.
Dittmer, D. & Mocarski, E. S. (1997). Human cytomegalovirus infection inhibits G1/S transition. J Virol 71, 1629-1634.
Dunn, W., Chou, C., Li, H., Hai, R., Patterson, D., Stolc, V., Zhu, H. & Liu, F. (2003). Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100, 14223-14228.
E, X., Pickering, M. T., Debatis, M., Castillo, J., Lagadinos, A., Wang, S., Lu, S. & Kowalik, T. F. (2011). An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog 7, e1001342.
Ehlers, B., Küchler, J., Yasmum, N., Dural, G., Voigt, S., Schmidt-Chanasit, J., Jäkel, T., Matuschka, F. R., Richter, D., Essbauer, S., Hughes, D. J., Summers, C., Bennett, M., Stewart, J. P. & Ulrich, R. G. (2007). Identification of novel rodent herpesviruses, including the first gammaherpesvirus of Mus musculus. J Virol 81, 8091-8100.
Ehmann, G. L., McLean, T. I. & Bachenheimer, S. L. (2000). Herpes simplex virus type 1 infection imposes a G(1)/S block in asynchronously growing cells and prevents G(1) entry in quiescent cells. Virology 267, 335-349.
Einsele, H., Roosnek, E., Rufer, N., Sinzger, C., Riegler, S., Löffler, J., Grigoleit, U., Moris, A., Rammensee, H. G., Kanz, L., Kleihauer, A., Frank, F., Jahn, G. & Hebart, H. (2002). Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99, 3916-3922.
Engel, P., Pérez-Carmona, N., Albà, M. M., Robertson, K., Ghazal, P. & Angulo, A. (2011). Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunol Cell Biol 89, 753-766.
Farrar, M. A. & Schreiber, R. D. (1993). The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol 11, 571-611.
Feng, Y., Broder, C. C., Kennedy, P. E. & Berger, E. A. (1996). HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872-877.
45 Fortunato, E. A., Dell'Aquila, M. L. & Spector, D. H. (2000). Specific chromosome 1 breaks induced by human cytomegalovirus. Proc Natl Acad Sci U S A 97, 853-858.
Fowler, K. B., Stagno, S., Pass, R. F., Britt, W. J., Boll, T. J. & Alford, C. A. (1992). The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med 326, 663-667. Gao, J. L. & Murphy, P. M. (1994). Human cytomegalovirus open reading
frame US28 encodes a functional beta chemokine receptor. J Biol Chem 269, 28539-28542.
García, M. A., Gil, J., Ventoso, I., Guerra, S., Domingo, E., Rivas, C. & Esteban, M. (2006). Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70, 1032-1060.
Gaspar, M. & Shenk, T. (2006). Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A 103, 2821-2826.
Goodrum, F., Reeves, M., Sinclair, J., High, K. & Shenk, T. (2007). Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110, 937-945.
Grundy, J. E., Lawson, K. M., MacCormac, L. P., Fletcher, J. M. & Yong, K. L. (1998). Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis 177, 1465-1474.
Haller, O., Stertz, S. & Kochs, G. (2007). The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect 9, 1636-1643.
Hay, J. & Ruyechan, W. (2007). Alphaherpesvirus DNA replication. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis: Cambridge University Press.
Hefti, H. P., Frese, M., Landis, H., Di Paolo, C., Aguzzi, A., Haller, O. & Pavlovic, J. (1999). Human MxA protein protects mice lacking a functional alpha/beta interferon system against La crosse virus and other lethal viral infections. J Virol 73, 6984-6991.