Bioassay-guided isolation of cyanobacterial metabolites with anticancer activity
93
0
0
Texto
Imagem
+7


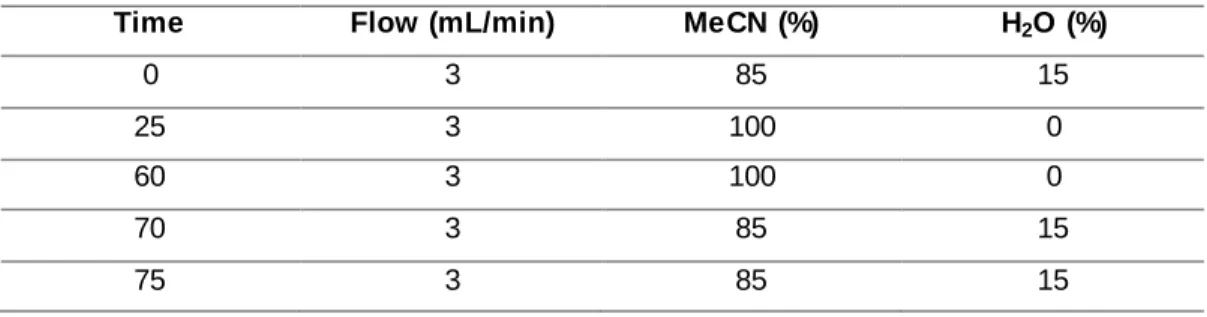

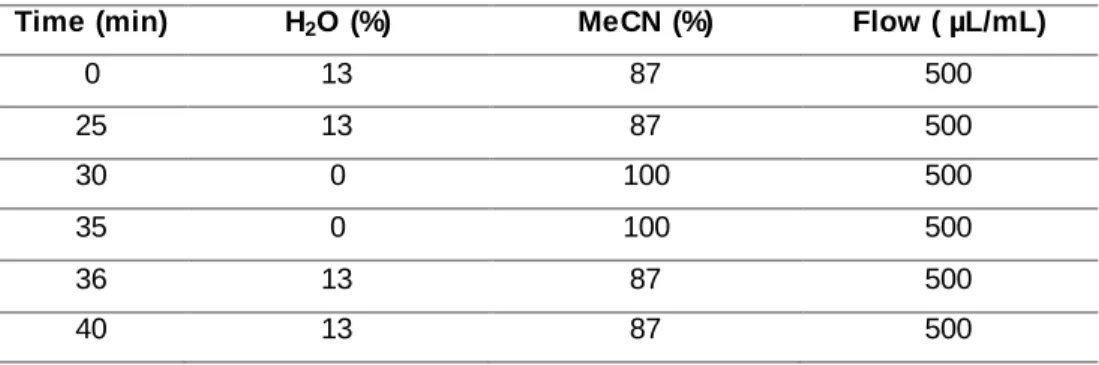
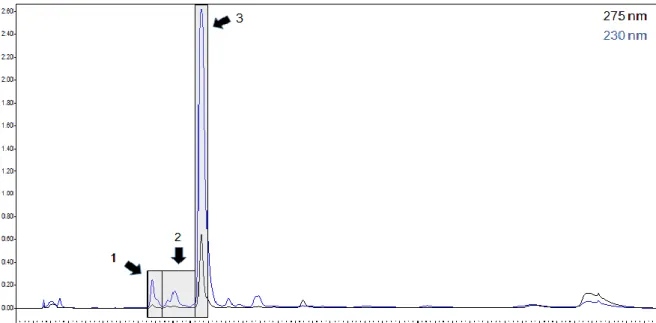
Documentos relacionados