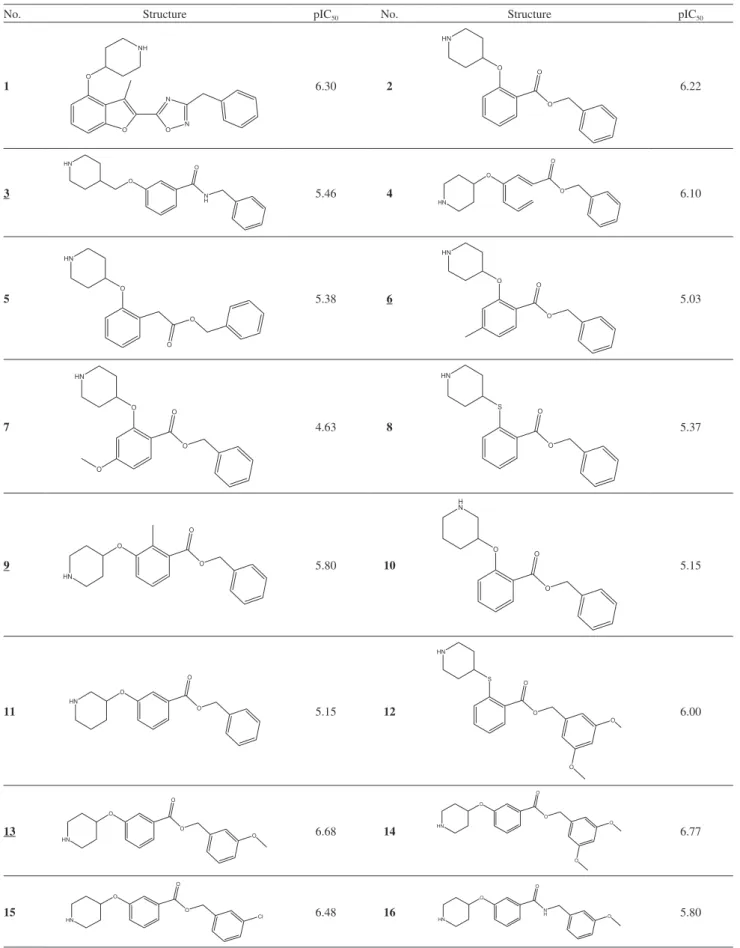
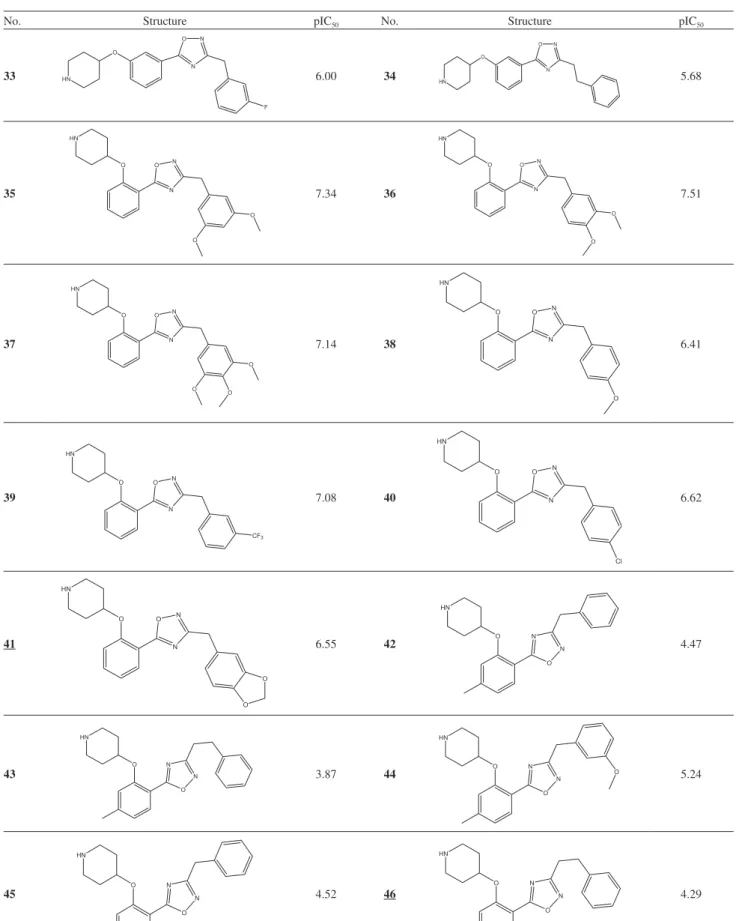
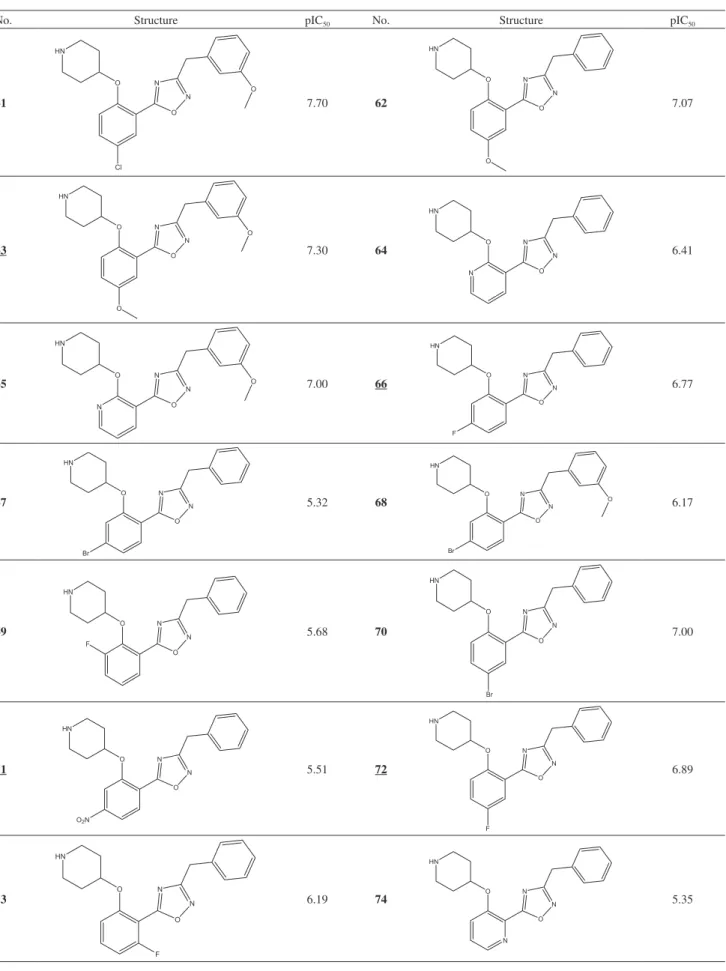
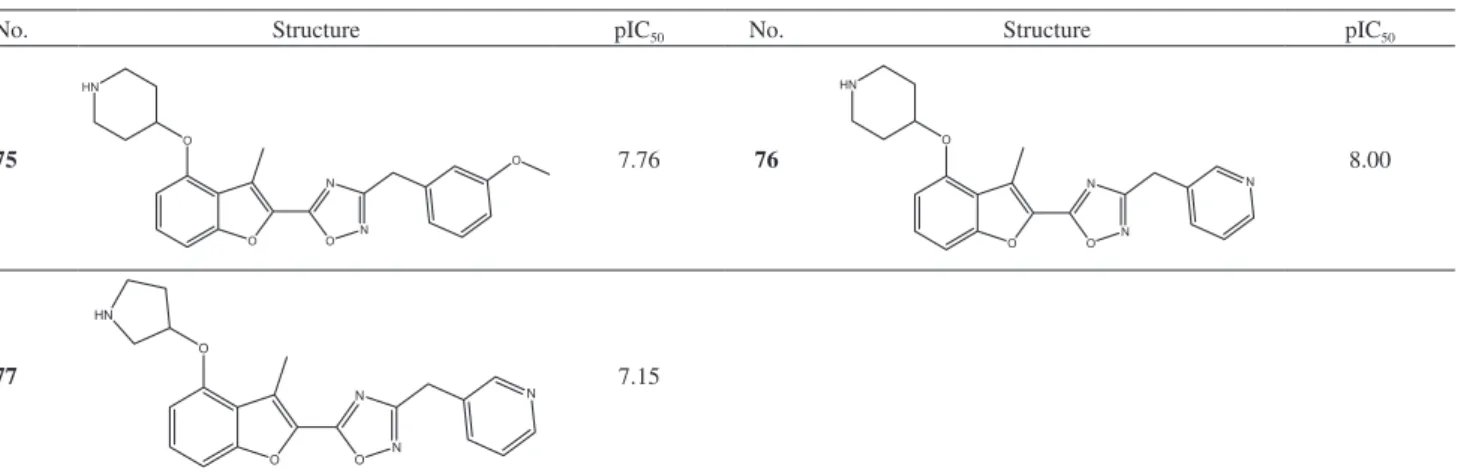
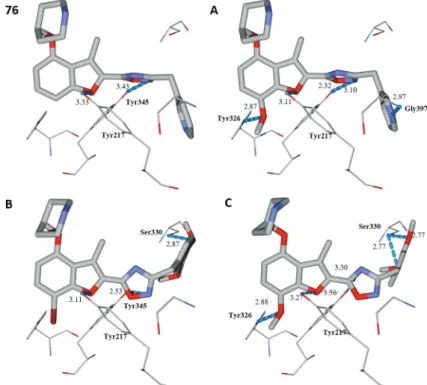
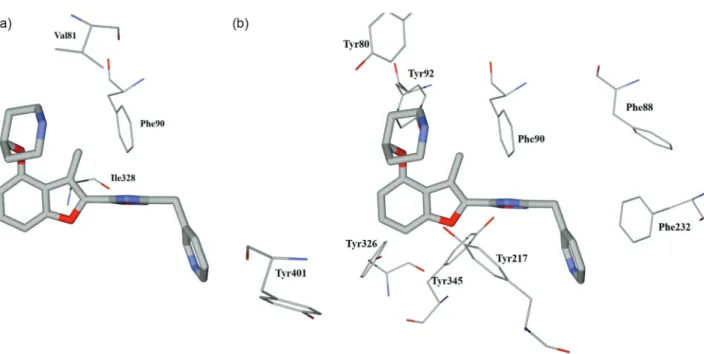
Design of Novel N-Myristoyltransferase Inhibitors of Leishmania donovani Using Four-Dimensional Quantitative Structure-Activity Relationship Analysis
Texto
Imagem
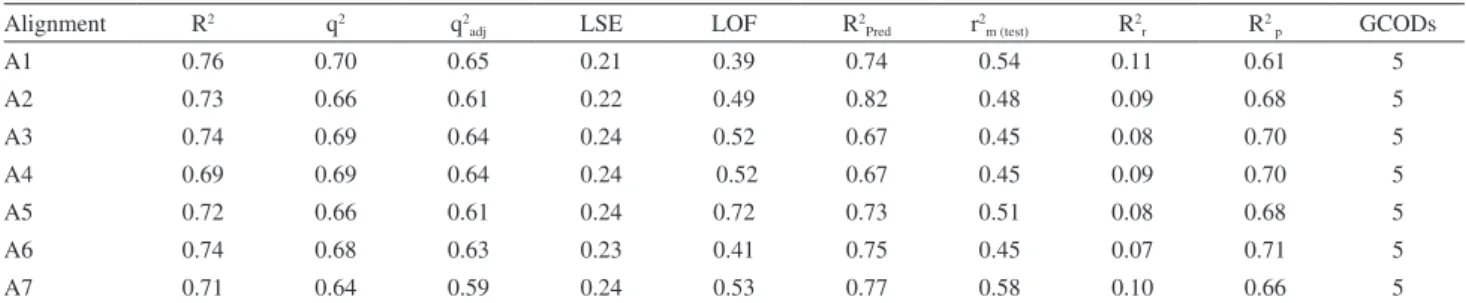
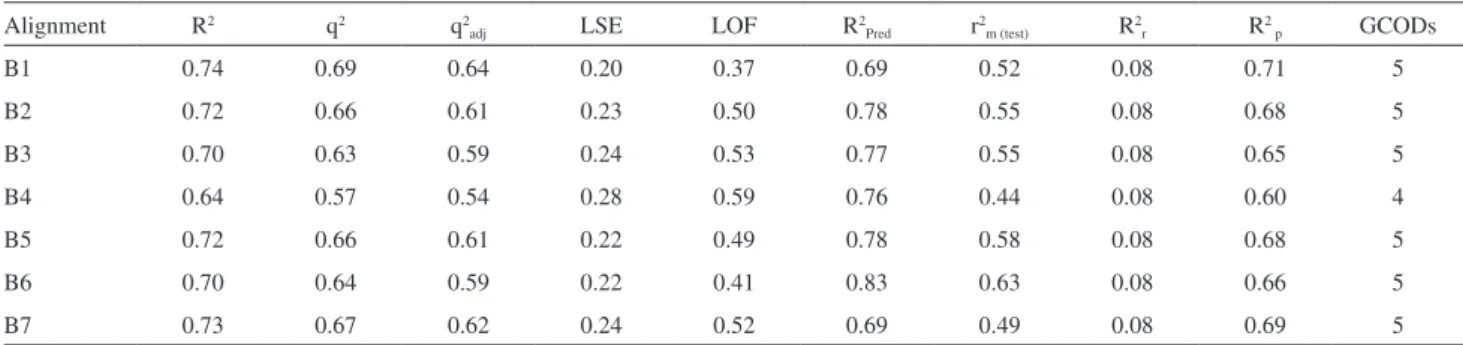
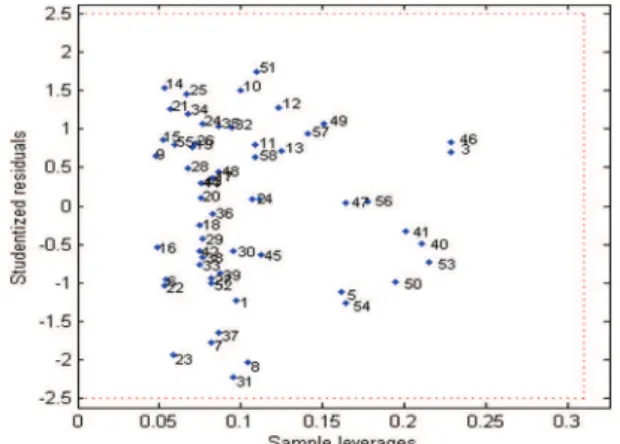
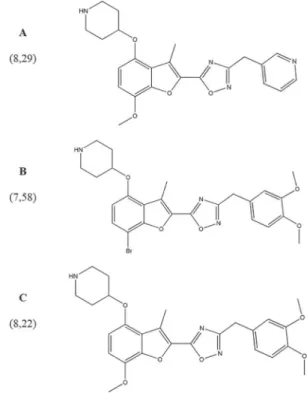
Documentos relacionados
não existe emissão esp Dntânea. De acordo com essa teoria, átomos excita- dos no vácuo não irradiam. Isso nos leva à idéia de que emissão espontânea está ligada à
If for most comercial companies marketing 3.0 is a new concept, cultural, educational and informational structures, especially libraries, have always been
The structure of the remelting zone of the steel C90 steel be- fore conventional tempering consitute cells, dendritic cells, sur- rounded with the cementite, inside of
social assistance. The protection of jobs within some enterprises, cooperatives, forms of economical associations, constitute an efficient social policy, totally different from
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
Besides agar diffusion and MIC values, quantitative evaluation of antimicrobial activity was performed by percent activity values, which reveal the antimicrobial potential of
