Atomic Force Microscopy as a Tool to Assess the Specificity of Targeted Nanoparticles in Biological Models of High Complexity
24
0
0
Texto
Imagem
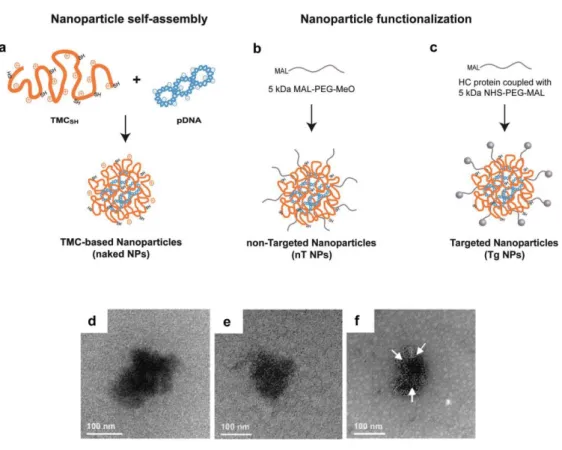

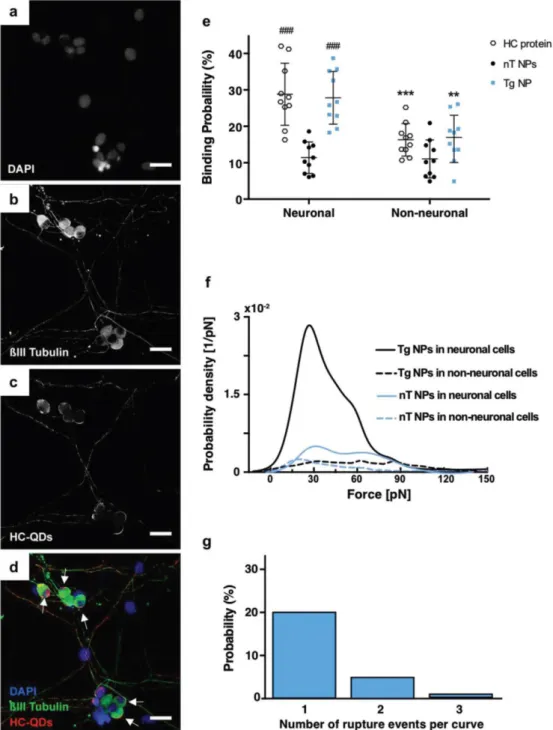
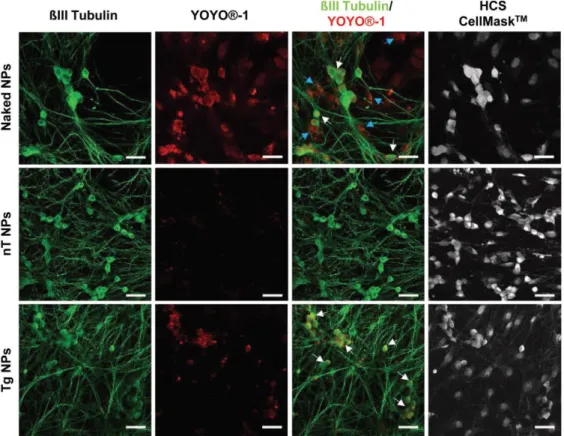
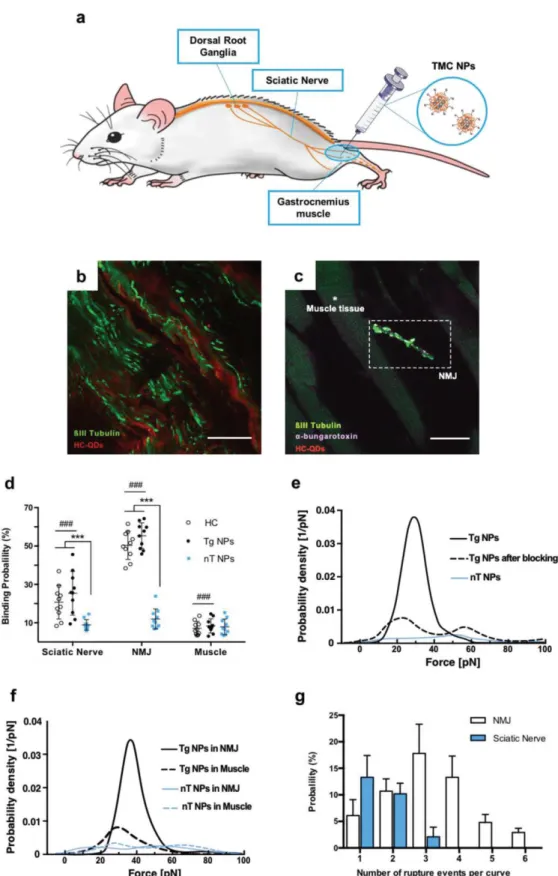
+4
![Table 1. TMC‐based nanoparticles physicochemical characterization Nanoparticle formulation Size [nm] a) Polydispersity index (PdI)a) Zeta‐potential [mV]a)](https://thumb-eu.123doks.com/thumbv2/123dok_br/19247398.974196/24.892.145.685.220.366/table-nanoparticles-physicochemical-characterization-nanoparticle-formulation-polydispersity-potential.webp)
Documentos relacionados