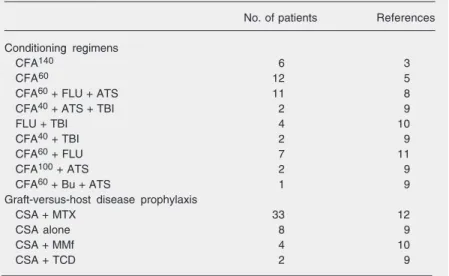
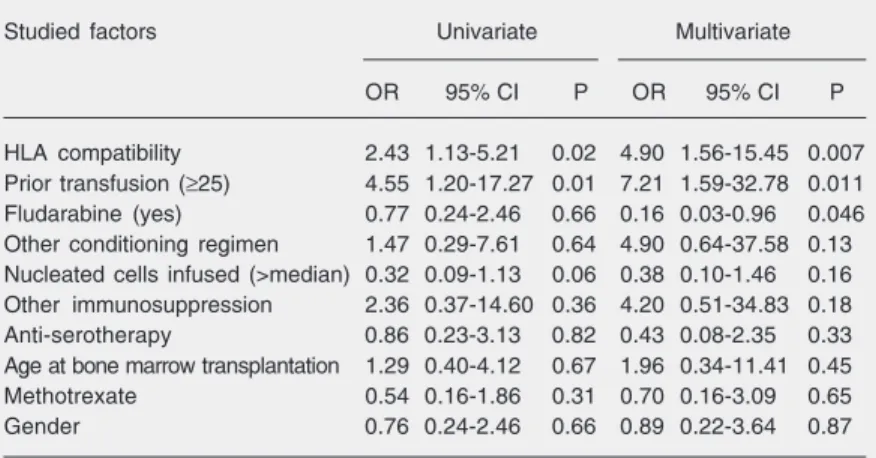
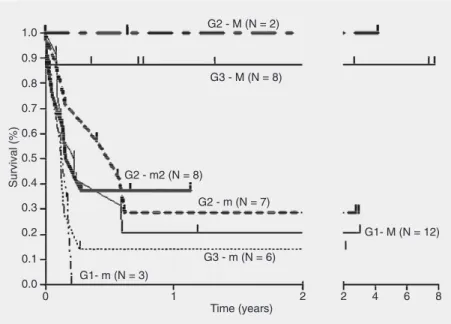
Allogeneic hematopoietic stem cell transplantation from an alternative stem cell source in Fanconi anemia patients: analysis of 47 patients from a single institution
Texto
Imagem

Documentos relacionados
feasibility of bone marrow mononuclear cell autologous (stem cell) transplantation in patients with acute stroke. -Functional outcome
Le Dieu de Leibniz connaît, avant de créer, la série complète de tous les événements du monde, et Il préfère celle qui s’impose, dans la combinatoire des compossibilités,
A intoxicação por cloreto de sódio é considerada uma causa de polioencefalomalacia, pois, animais acometidos por esse distúrbio apresentam necrose laminar
Autologous stem cell transplantation in elderly patients (> or = 60 years) with diffuse large B-cell lymphoma: an analysis based on data in the European Blood and
ease, and a second related donor allogeneic hematopoietic stem cell transplantation..
Hematopoietic stem cell transplantation in children with acute leukemia: similar outcomes in recipients of umbilical cord blood versus marrow or peripheral blood stem cells from
Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma.. Biol Blood
Use of tyrosine kinase inhibitors to prevent relapse after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute
