Bioprocess design of canine adenovirus vectors
for gene therapy applications
Paulo David Machado dos Santos Fernandes
Dissertation presented to obtain a Ph.D. degree in Engineering and Technology Sciences, Biotechnology
Instituto de Tecnologia Química e Biológica António Xavier
Universidade Nova de Lisboa
Supervisor: Paula Marques Alves
Co-supervisor: Ana Sofia Coroadinha
ii Bioprocess design of canine adenovirus vectors for gene therapy applications
By Paulo Fernandes
First edition:
Cover: MDCK cells in microcarriers infected with CAV-2 vectors.
ITQB-UNL/iBET Animal Cell Technology Unit
Instituto de Tecnologia Química e Biológica/
Instituto de Biologia Experimental e Tecnológica
Av. da República EAN, 2780-157 Oeiras, Portugal
http://tca.itqb.unl.pt
http://www.itqb.unl.pt
http://www.ibet.pt
Copyright ©2015 by Paulo Fernandes
All rights reserved
iii Supervisors:
Dr. Paula Marques Alves, CEO of iBET, Instituto de Biologia Experimental e Tecnológica (iBET), Oeiras, Portugal; Invited associate professor at Faculdade de Ciências e Tecnologia, NOVA university of Lisbon; Principal investigator and Director of Animal Cell Tecnology Unit at Instituto de Tecnologia Química e Biológica (ITQB/UNL), Oeiras, Portugal
Dr. Ana Sofia Coroadinha, Auxiliary investigator and Head of Cell line development and molecular biotechnology laboratory, Animal Cell Technology Unit, iBET and ITQB/UNL, Oeiras, Portugal
Jury:
Dr. Otto-Wilhelm Merten, Head of the Applied Vectorology and Innovation group of Généthon, Généthon, Évry, France
Dr. Rubén Hernández-Alcoceba, Investigator of CIMA – Center for Applied Medical Research, University of Navarra, Pamplona, Spain
Dr. Michael Parkhouse, Head of the Infections & Immunity group, IGC, Oeiras, Portugal
iv Foreword
This thesis is the result of four years of research at the Animal Cell Technology Unit of ITQB-UNL/iBET, Oeiras, Portugal, under the supervision of Paula Marques Alves and Ana Sofia Coroadinha.
v Acknowledgements
I would like to express my gratitude to all the people that contributed to this thesis and to the hosting institutions, iBET and ITQB, for the excellent working conditions.
To my supervisor, Dr Paula M. Alves, for the opportunity to let me grow as a scientist in animal cell technology unit (ACTU) and develop this work, for the example of dedication, for the confidence, guidance, support, pragmatism and for granting me all the means to work.
To my co-supervisor, Dr Ana Sofia Coroadinha, for the guidance, confidence and support, for making me feel a peer and for pushing my enthusiasm to science.
To Dr Eric J. Kremer, from IGMM, Montpellier, France, for providing the materials necessary to start the area of CAV-2 vectors at ACTU, for the time, enthusiasm, inspiration and discussions during this journey.
To the financial support provided by Fundação para a Ciência e Tecnologia (SFRH/BD/70810/2010) and by European Commission, through the FP7 project BrainCAV.
To the key persons in any bioprocess developed at ACTU – Marcos Sousa (upstream process) and Dr. Cristina Peixoto (downstream process) – for the guidance and support.
To former and current colleagues from ACTU for the technical help and great times I have spent with you in these years. In particular, to Vanessa Bandeira, Carina Brilha, Rute Castro, Sofia Rebelo, Filipa Rodrigues, Marta Silva, Daniel Simão and Hugo Soares.
Aos meus amigos de longa data, Francisca, André, Sofia e Mafalda, por todos os momentos e aventuras que ajudam a recuperar energias e resolver problemas mesmo sem falar deles. Em especial à Francisca pela companhia nesta caminhada de PhD students, por me obrigar a apanhar ar, pelas conversas e gargalhadas importantes em não perder o foco e continuar em frente. Ao grupo das sessões pós-laboratório no jardim do Cego – Francisca, André e Raquel.
vii Abstract
The potential of human adenovirus vectors as vehicles for gene transfer with clinical applications in vaccination, cancer treatment and in many monogenic and acquired diseases has been demonstrated in several studies and clinical trials. However, the clinical use of these vectors can be limited by pre-existing humoral and cellular anti-capsid immunity. One way to circumvent this bottleneck while keeping the advantages of using adenovirus vectors is using non-human viruses such as Canine Adenovirus type 2 (CAV-2). Moreover, CAV-2 vectors present attractive features to develop potential treatment of neurodegenerative and ocular disorders. While the interest in CAV-2 vectors increases, scalable and robust production processes are required to meet the need for preclinical and possibly clinical uses.
This PhD thesis aimed to develop and optimize the production process of CAV-2 vectors, namely E1-deleted (ΔE1) and helper-dependent vectors (HDVs), by i) addressing technical aspects to enable large-scale production compliant with the guidelines for manufacturing clinical material and ii) understanding cell physiological state and virus replication behind high production titers and product quality.
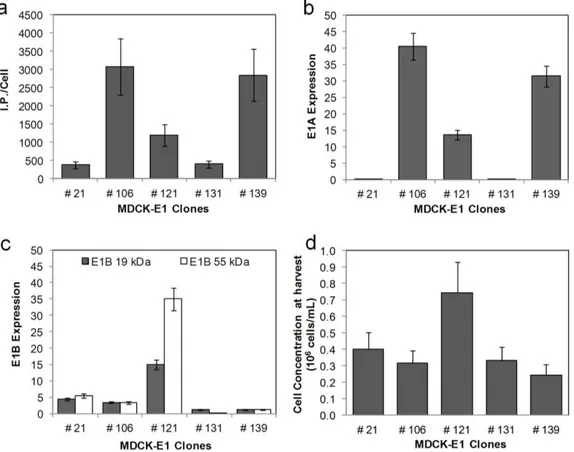
Being MDCK cells already approved by the regulatory agencies (FDA and EMA), the use of MDCK-based cell lines can facilitate the acceptance of CAV-2 clinical grade production. Accordingly, MDCK-E1 and MDCK-E1-Cre cell lines were established in Chapter II, with titers comparable to those described for human adenovirus producing cells. In addition, a clear correlation between E1 (mostly E1A) expression levels and CAV-2 production was observed during clone characterization, corroborating the importance of evaluating E1 levels when screening and developing adenovirus producer cells.
viii vectors availability at larger scales, leveraging the manufacturing process of CAV-2 vectors to the level of human adenovirus vectors.
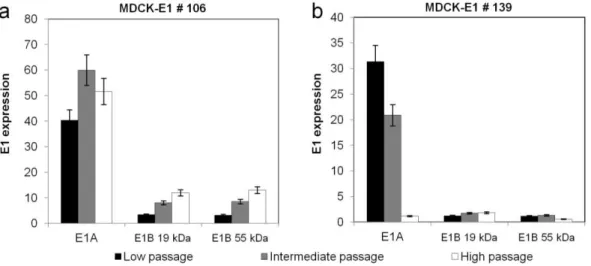
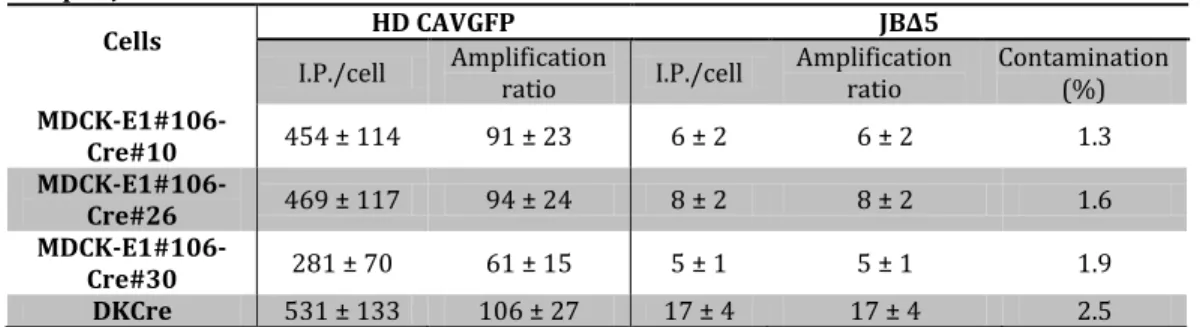
The HDV production system, that comprises the use of Cre recombinase-expressing cells and infection schemes with two vectors (HDV and helper vector (HV)), is more complex than ΔE1 vectors system. Therefore, special attention was paid to the propagation of HDVs. HDV production was optimized by evaluating MDCK-E1-Cre cell line stability and infection conditions in Chapter IV. MDCK-E1-Cre cells specific productivity decreased with the increase of cell culture passage. Best infection conditions (defined MOI ratio between HDV and HV) were sufficient to minimize HV contamination and surpass the need of Cre-expressing cells. The amplifications performed under a defined MOI of HDV and HV can be thus moved to MDCK-E1 cells, which presents a robust performance even under high passage number (Chapter II and Chapter IV). Despite these advances, HDV production yields were still lower than ΔE1 vectors. This was further explored in Chapter V by monitoring HDV replication steps to understand what factors were still limiting production yields. Replication progression was intensified during HDV production, as genome replication occurred faster during HDV production, leading to higher and earlier expression of structural proteins. Such high viral protein content was consistent with increased cell death observed during production and contributed to the low volumetric titers. The low infectivity of the HDVs generated suggested a defective maturation process and/or increased levels of defective particles. Altogether, this study identifies genome replication, viral protein synthesis and maturation as features to be modulated in the pursuit of an optimal HDV production.
ix Resumo
Os vectores adenovirais humanos têm demonstrado elevado potencial como veículos para a transferência de genes. As suas aplicações clínicas são vastas, passando pelo desenvolvimento de vacinas, tratamento de cancro e doenças monogénicas. No entanto, o uso clínico destes vectores pode ser condicionado pela imunidade humoral e celular pré-existente em pacientes, resultante da exposição a este tipo de vírus ao longo da vida. Este problema pode ser ultrapassado com o uso de adenovírus não-humanos, tais como os adenovírus caninos tipo 2 (CAV-2), que mantêm todas as vantagens associadas ao uso de vectores adenovirais. Mais ainda, os vectores CAV-2 possuem características promissoras para serem usados no desenvolvimento de tratamentos contra doenças neurodenegerativas e oculares. Como tal, o interesse pelos vectores CAV-2 tem aumentado, o que torna imperativo o desenvolvimento de processos de produção robustos e escaláveis que possam ir de encontro à quantidade e qualidade necessárias de material biológico para aplicação em ensaios pré-clínicos e clínicos.
Esta tese de doutoramento teve como objectivo desenvolver e optimizar a produção de vectores CAV-2, nomeadamente vectores com delecção na região E1 (ΔE1) e vectores
helper-dependent (HDV), de modo a i) respeitar os aspectos técnicos que facilitam a produção em larga-escala e/ou de material com grau clínico e ii) compreender a fisiologia das células produtoras e a replicação viral para desenvolver um bioprocesso com produtividade máxima.
As células MDCK, já usadas para produção de outros vírus e aprovadas pelas autoridades reguladoras de produtos para uso humano (FDA e EMA), foram usadas no Capítulo II para estabelecer linhas celulares produtoras de vectores CAV-2 (MDCK-E1 e MDCK-E1-Cre). Os títulos de produção de CAV-2 com estas novas linhas celulares foram semelhantes aos descritos para células produtoras de adenovírus humanos. Durante este trabalho foi também observada uma correlação entre os níveis de E1 (principalmente E1A) e produção de CAV-2, o que revela a importância da avaliação dos níveis de E1 no rastreio e selecção de linhas celulares produtoras de vectores adenovirais.
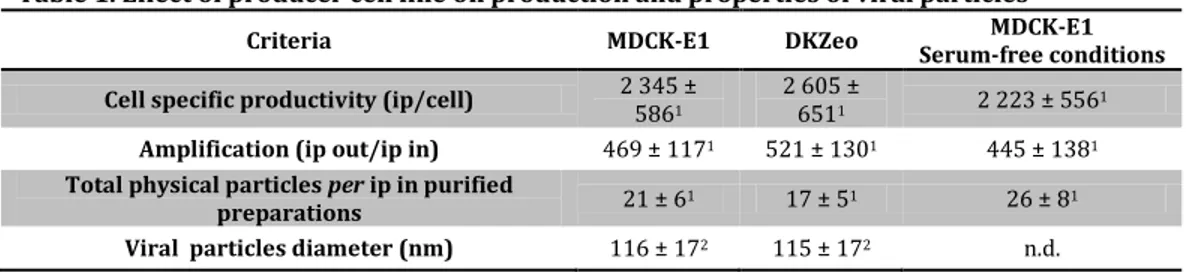
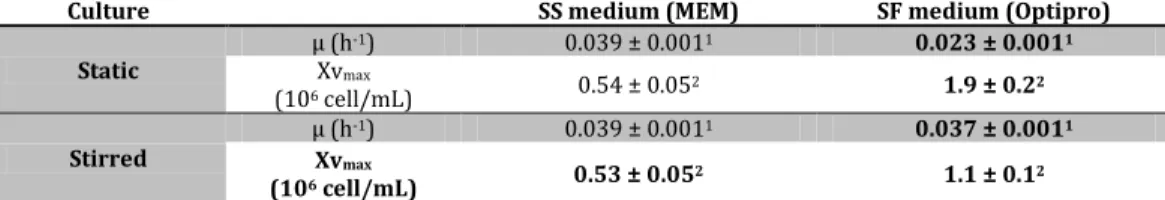
x agitado (parte A) e ii) adaptação de células aderentes a crescimento em suspensão (parte B). A produtividade de vectores CAV-2 no bioprocesso baseado em microcarriers foi semelhante à obtida em culturas estáticas, no entanto foi necessário suplementar o meio de cultura com soro para permitir a adesão das células aos microcarriers. O bioprocesso com células adaptadas a suspensão ocorreu em condições livres de soro, embora tenha resultado numa ligeira diminuição dos títulos volumétricos. Ambos os bioprocessos permitem a produção de vectores CAV-2 a escalas maiores, tornando o processo de manufactura equivalente ao usado para adenovírus humanos.
xii Thesis publications
Published:
1. Fernandes P, Almeida AI, Kremer EJ, Alves PM, Coroadinha AS. 2015. Canine helper-dependent vectors production: implications of Cre activity and co-infection on adenovirus propagation. Sci Rep. accepted(in press)
2. Fernandes P, Simão D, Guerreiro M, Kremer EJ, Coroadinha AS, Alves PM. 2015. Impact of Adenovirus life cycle on the generation of canine helper-dependent vectors. Gene Ther. Jan;22(11):40-9. http://dx.doi.org/10.1038/gt.2014.92
3. Fernandes P, Peixoto C, Santiago VM, Kremer EJ, Coroadinha AS, Alves PM, 2013. Bioprocess development for canine adenovirus type 2 vectors. Gene Ther. Apr;20(4):353-60. http://dx.doi.org/10.1038/gt.2012.52
4. Fernandes P, Santiago VM, Rodrigues AF, Tomás H, Kremer EJ, Alves PM, Coroadinha AS. 2013. Impact of E1 and Cre on Adenovirus Vector Amplification: Developing MDCK CAV-2-E1 and E1-Cre Transcomplementing Cell Lines. PLoS One. 2013;8(4):e60342. http://dx.doi.org/10.1371/journal.pone.0060342
Submitted:
5. Fernandes P, Silva AC, Coroadinha AS, Alves PM. Propagation of Adenovirus vectors. In D.T. Curriel (ed.) Adenoviral Vectors for Gene Therapy, 2nd edition, Academic Press (submitted)
In preparation:
6. Fernandes P, Sousa MFQ, Kremer EJ, Coroadinha AS, Alves PM. Suspension MDCK-E1 cell line for the manufacturing of CAV-2 vectors (in preparation)
Other publications in the scope of virus production:
7. Silva AC, Roldão A, Teixeira A, Fernandes P, Sousa MFQ, Alves PM 2015. Cell immobilization for the production of viral vaccines. In: Al-Rubeai, Mohamed (Ed.), Animal Cell Culture, Cell Engineering, Vol. 9, Springer. http://dx.doi.org/10.1007/978-3-319-10320-4_17
xiii List of contents
Chapter I – Introduction ………. 1
Chapter II – Developing MDCK CAV-2-E1 and E1-Cre transcomplementing cell lines …. 31
Chapter III – Upstream bioprocess development for E1-deleted CAV-2 vectors …………. 55
Part A – Production with adherent cells using microcarrier technology………... 57
Part B – Production with cells adapted to suspension ………... 73
Chapter IV – Implications of Cre activity and co-infection on canine helper-dependent vector production ……….. 91
Chapter V –Impact of adenovirus life cycle on the generation of canine helper-dependent
vectors ………... 113
Chapter VI – Discussion and perspectives ………. 141
Chapter I
Introduction
This chapter is adapted from the book chapter:
Chapter I
2
Contents
1. Gene therapy ... 3
2. Adenovirus vectors for gene therapy ... 3
2.1. Structure and genome ... 4
2.2. Infection and replication cycle ... 4
3. Manufacturing of adenovirus vectors ... 5
3.1. Adenovirus vectors and producer cells ... 5
3.1.1. 1st generation vectors ... 6
3.1.2. Conditionally-replicating vectors ... 7
3.1.3 2nd generation vectors ... 7
3.1.4. 3rd generation vectors ... 8
3.1.5. Novel adenovirus vectors ... 9
3.2. Upstream process for adenovirus vectors ... 10
3.2.1. Cell culture and adenovirus production process ... 11
3.2.2. Adenovirus seed stocks ... 12
3.2.3. The infection process and harvest strategy ... 13
3.2.4. Productivity of Adenovirus vector manufacturing ... 14
3.2.5. Adenovirus production at high cell density ... 14
3.2.6. Physical parameters and process monitoring... 15
3.3. Production process of helper-dependent vectors ... 16
3.4. Downstream process for adenovirus vectors ... 17
4. Concerns in the manufacturing of adenovirus vectors for product release ... 18
4.1. Extraneous agents ... 18
4.2. Replication competent adenovirus ... 19
4.3. Vector genetic stability ... 19
4.4. Quantity and potency ... 20
5. Scope of the thesis ... 20
6. Author contribution ... 21
Introduction
3 1. Gene therapy
Gene therapy is defined as the transfer of genetic material into an individual’s cells or tissues, treating or preventing a disease. After three decades of research, gene therapy is
currently considered solid (1), showing success in the clinic with an emphasis on rare,
inherited disorders including hemophilia (2), Leber Congenital Amaurosis (3-5) and
X-linked severe combined immunodeficiency (6). Moreover, a critical milestone was
achieved with the market authorization from European Medicines Agency of Glybera
(alipogene tiparvovec), an adeno-associated virus based product to treat deficiency in
lipoprotein lipase in patients with a severe form of hyperglyceridemia (7). Included in
gene therapy is also the sub-field of oncolytic virotherapy. In fact, most of the clinical
trials in gene therapy have been aimed at the treatment of cancer. Oncolytic virotherapy
utilises viruses capable of specifically targeting and replicating in tumor cells and causing
cell lysis, thereby killing the infected tumor cells (8). In some cases, these viruses are
modified to deliver suicide genes or immunostimulatory cytokines to enhance the
immune response against cancer.
Although nonviral approaches are becoming increasingly common to perform gene
delivery (9), viral vectors remain by far the most popular approach, having been used in
more than 70% of the trials performed to date (http://www.abedia.com/wiley/). Among
these, the top most used are based on: i) adenovirus, ii) retrovirus, iii) vaccinia virus, iv)
adeno-associated virus, v) poxvirus, vi) lentivirus and vii) herpes simplex virus. Worth
mentioning the emerging interest on vectors from adeno-associated viruses and
lentivirus, as depicted by the research dedicated to these vectors and growth rate of use
(9).
2. Adenovirus vectors for gene therapy
Viral vectors are currently the most efficient tools for in vivo gene transfer. Due to their in
vivo efficiency, adenovirus vectors (AdV) are used more often than any other vector in
clinical trials, in which the human adenovirus vector (HAdV) serotypes 5 and 2 are the
most characterized ones among all other serotypes of the same family (10, 11).
Adenovirus wide cell tropism in quiescent and non-quiescent cells, its inability to
integrate the host genome and its high production titre (12) make AdV very good
Chapter I
4
2.1. Structure and genome
Adenoviruses are non-enveloped, icosahedral viruses of 60 to 90 nm with a linear double
stranded DNA genome of ~36 kbp. The genome can be divided in two sets of
transcriptional units: early genes that are expressed before the onset of viral DNA
replication, and late genes which are preferentially expressed after viral DNA replication
(Figure 1). This classification also defines the early and late phase of the infectious cycle.
E1A is the first gene to be expressed following infection and is involved in the
transcription of the other viral early genes. The mRNAs of major late transcription unit
are grouped into five families (L1-L5) which are dependent on the activation of major late
promoter during viral DNA replication.
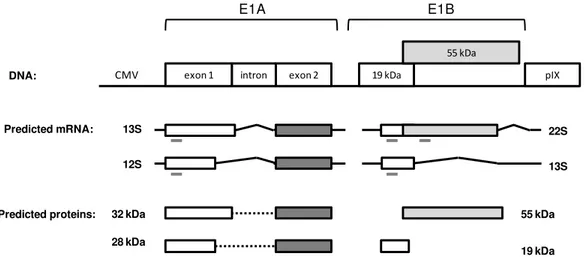
Figure 1. Schematic representation of wild-type adenovirus genome and the different generations of non-replicating vectors. E1 to E4: early region transcript units; L1 to L5: late region transcprits unit; ITR: inverted terminal repeats; MLP: major late promoter; Ψ: packaging signal; GOI: gene-of-interest.
2.2. Infection and replication cycle
The sequential uptake process relies on an initial contact with cellular receptors
responsible for attachment (13) and internalization of the virus particle (14).
Internalization occurs by receptor-mediated endocytosis (15). Once in the cytosol, the
virion is transported via microtubuli towards the nucleus. Meanwhile, the particle is
dismantled by an ordered elimination of structural proteins so that when it reaches the
nuclear membrane, only the core particle is left. Adenovirus uncoating culminates with
the release of the viral DNA into the nucleus via nuclear pore complexes (16). The early
genes are responsible for expressing mainly non-structural, regulatory proteins (17). ITR
E1A/E1B MLP
E3 L1 to L5
E2B E2A E4ITR
Ψ ITR ∆E1 ∆E3 Ψ GOI ITR ITR ∆E1 ∆E3 Ψ GOI ITR
∆E2B ∆E2A ∆E4
ITR Ψ
GOI and stuffer DNA ITR Wild-type
1stgeneration
2ndgeneration
Introduction
5 These proteins alter the expression of host proteins that are necessary for DNA synthesis,
activate other early genes (such as the virus-encoded DNA polymerase) and avoid
premature death of the infected cell by the host-immune defenses. During viral genome
replication, late phase transcription is activated (17). This infection phase is mainly
focused on producing capsid proteins and packaging the replicated viral genomes;
structural proteins are assembled into virions, viral DNA is packaged and viruses are
released from the cell as a result of virus induced cell lysis (Figure 2).
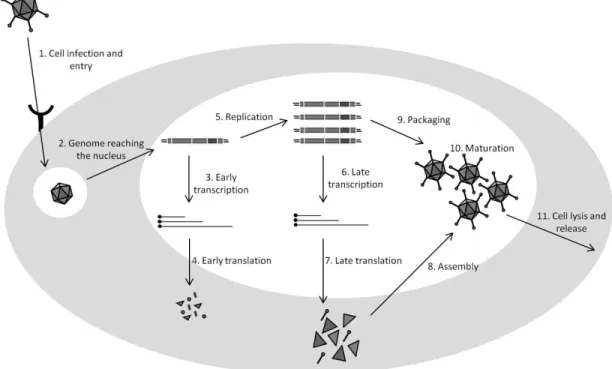
Figure 2. Adenovirus replication cycle. Briefly, after cell binding (1), the mature viral particle is internalized and transported towards the nucleus (2). During this transport, the particle is dismantled by an ordered elimination of some structural proteins and culminates with the delivery of the viral genome into the nucleus via nuclear pore complexes. Once in the nucleus, early genes are expressed (3, 4) and viral DNA replication starts (5). Late phase of infection is then activated and structural proteins expressed (6, 7). These proteins are likely assembled into empty virions (8), followed by the packaging of viral genome (9). Finally, viral particles are subjected to a maturation process becoming infectious (10) and cell lysis is accomplished (11).
3. Manufacturing of adenovirus vectors
3.1. Adenovirus vectors and producer cells
The development of adenovirus vectors is based on modifications of wild type genome
Chapter I
6
interest (GOI). While these viral genes are essential for virus replication, to propagate
adenovirus vectors it is necessary the establishment of producer cell lines, i.e. cells
expressing the viral elements deleted from the vector genome. The adenovirus vectors
that are non-replicating in a clinical setting and strictly used for gene transfer can be
categorized as first, second or third generation vectors, depending on the extension of
viral genes removed (Figure 1). The conditionally-replicating vectors are another
category of adenovirus vectors and are designed to specifically target, propagate and
deliver gene(s) in cancer cells.
3.1.1. 1st generation vectors
The majority of adenovirus vectors used for gene therapy or many other therapeutic
purposes are non-replicating, in which the E1 region is deleted from the genome often in
combination with E3, providing space for the insertion of expression cassettes. Such
adenovirus vectors are denominated as first generation or E1-deleted (∆E1) vectors. E1 region can be divided in E1A and E1B subunits and codes for products involved in the
activation of other early and late genes expression and in making host cells more
amenable to initiate virus propagation (i.e. inhibition of antiviral response and apoptosis)
(18-24). Therefore, to produce ∆E1 vectors a producer cell line containing adenovirus E1 sequences is necessary to complement these functions. On the other hand E3 region,
which is involved in antagonizing host defense mechanisms, is not essential for viral
amplification in vitro (25). Together, the deletion of E1 and E3 permits a transgene insert
capacity up to 8.2 kb. HEK 293 represents the traditional cell line used to
trans-complement the lack of E1 and produce adenovirus vectors. Because of the significant
homology of first-generation adenovirus vectors with the HEK 293 DNA, the main
disadvantage in using HEK293 is its potential to generate replication-competent
adenoviruses (RCA), raising safety concerns in a therapeutic product (26-30). To avoid
this, several cell lines were established under the rational of reducing the homology of
viral DNA sequences incorporated in cell line with viral vectors genome (reviewed in
(31)). Following this effort, PER.C6 represents the first cell line in which the generation of
RCA is abolished and typical adenovirus yields are ensured (32).
HEK293 cell line was generated with sheared HAdV-5 DNA, however the current strategy
to generate producer cell lines for E1 complementation is through the incorporation of
Introduction
7 to minimize any recombination between viral genome and cell DNA were further
developed and involve the integration of E1A and E1B into producer cell genome at
separate locations (33, 34). E1 complementing cell lines are the backbones for the
remaining producer cells used for other adenovirus vectors.
3.1.2. Conditionally-replicating vectors
Conditionally-replicating adenovirus vectors, being also denominated oncolytic
adenoviruses, have been employed for cancer treatment to specifically target and
replicate in cancer cells (8, 35, 36). The lytic nature of adenovirus replication directly kills
tumor-infected cells, releasing the associated antigens. On the other hand, progeny
viruses can be spread throughout a tumor, further infecting and destroying other cancer
cells.
The construction of oncolytic adenoviruses is based on the deletion or modification of
viral gene functions that are critical to viral replication in normal cells, but dispensable in
tumor cells. This includes insertion of mutations in E1A region (37, 38), or deletion of
E1B (39, 40) from wild-type genome, to target cancer cells with defects in the
retinoblastoma (Rb) and p53 pathways, respectively, as these pathways are defective in
most human tumors. Another strategy in targeting oncolytic adenoviruses replication in
cancer cells involves the control of E1 transcription by using tumor or tissue specific
promoters such as prostate-specific enhancer/promoter for prostate cancer (41), or
E2F-I for cancer cells with a defective Rb pathway (42). E2F-In addition to these modifications,
these vectors can further incorporate genes encoding immune stimulatory factors to
boost the anti-tumor immunity (reviewed in (8)), giving rise to the so-called
armed-vectors and to one of the most promising gene delivery systems for cancer therapy.
Production of oncolytic adenoviruses is similar to first generation vectors, in which E1
functions are provided by the producer cell line. Therefore, cell lines already established
for first generation vectors are used to the manufacture of these vectors and include HeLa
(43), A549 (44), HEK293 and PER.C6 (45).
3.1.3 2nd generation vectors
Although the removal of E1 region renders the virus replication-defective, the delivery of
high doses of first generation vectors and/or the presence of E1-like factors in many cells
Chapter I
8
strong immune response, reducing the efficacy of these vectors. To circumvent that,
further deletions in E2 and/or E4 regions were explored, leading to the establishment of
2nd generation vectors. The E2 region (E2A and E2B subunits) codes for three proteins
essential for viral replication: DNA-binding protein (DBP) transcribed from E2A subunit,
terminal protein and viral DNA polymerase from E2B subunit (49). E4 products modulate
transcription, the cell-cycle, cell signaling and DNA repair and are essential for productive
virus infection (50), but only one of the ORF3 or ORF6 is required for successful virus
production in cell culture (51, 52).
Given the toxicity associated with E2 and E4 viral products, most of the cell lines for 2nd
generation vectors rely on the use of an inducible system (reviewed in (31)). Similarly to
1st generation vectors, the majority of E1/E2 and E1/E4 complementary producer cells
lines use HEK 293 as parental cell line.
Despite the sophisticated systems available for 2nd generation vectors manufacturing,
including vector construct and incorporation of inducible systems in the producer cell
lines, the use of these vectors remains without advances. In general, reduced production
yields are obtained for vectors with multiple deletions. In fact, when compared to first
generation vectors, the yields of these vectors can be reduced, and no major
improvement in toxicity is observed in vivo (31, 53-58). Moreover, the transgene expression of these vectors is also described as unstable, which probably made 3rd
generation vectors a more suitable choice to that end (reviewed in (59)).
3.1.4. 3rd generation vectors
3rd generation vectors, also known as gutted or gutless, high capacity or
helper-dependent vectors (HDVs), are the most advanced adenovirus vectors. They are devoid of
all viral coding genes, only harbouring on their genome the essential cis-acting elements:
inverted terminal repeats (ITRs) and packaging signal. This allows the insertion of
therapeutic gene or genes up to ~37 kb. Additional stuffer DNA is included to render
vector genome size similar to wild-type and maintain viral particle stability (60). Apart of
their increased safety, the use of these vectors result in long-term transgene expression
(reviewed in (59)). To produce these vectors, while E1 functions are provided by the
producer cell line, the remaining functions are provided by an helper vector (HV). In fact,
the need of a HV increases the complexity of production system and it is the main
Introduction
9 production of both HDV and HV, raising the need to reomove HV contamination from the
final product. To date, the most elegant way to prevent HV propagation is the approach
described by Parks et al. in which the HV packaging signal is flanked by loxP sites, and
under the expression of Cre-recombinase the HV genome is cleaved (hampering its
encapsidation) and HV particles production is minimized (61). Thus, besides E1,
producer cell lines for HDV have to further express Cre recombinase. Following same
approach, FLPe recombinase system is also used to minimize HV contamination (62, 63).
Similarly to E1 transcomplementing cell lines, the most common producer cell lines using
recombinase system are derived from HEK 293 cells, although PER.C6 cells are also used
to that purpose (reviewed in (31)). Despite these efforts in HDV production system, two
major bottlenecks are still found when considering the use of HDV in patients: i) HV
contamination is still not fully eliminated and ii) HDV production yield still faces the need
of multiple amplification steps and inconsistency in infectious particles yields (discussed
in section 3.3). Despite these bottlenecks, the increased cassette incorporation, improved
expression and lower immunity is still pushing their further development (59).
3.1.5. Novel adenovirus vectors
While most research on vector development is based on the utilization of human
adenovirus serotypes 5 and 2, over 80% of the adult population has been naturally
exposed to these viruses (64). Therefore, pre-existing humoral and cellular immunity
may preclude efficient gene transfer when these adenovirus vectors are used (65-70).
Apart of limitations in therapeutic efficacy (71), immune responses against the vector
may result in a number of undesirable side effects, including liver toxicity (72) and
systemic inflammatory response syndrome due to repeated vector administration (73).
Eliminating liver tropism and the epitopes involved in viral proteins recognition by
neutralizing antibodies has been proposed to reduce the immune response. Alternatively,
vectors based on low seroprevalence AdVs can be also be used to circumvent the
pre-existing immunity. In addition, the diversity of AdV protein isoforms and their variety of
ligand-receptor interactions found in the different serotypes can also be the basis to
target different cell types (70). Adenovirus vectors derived from alternative human and
non-human serotypes, to which the human population has a lower or no prevalence of
Chapter I
10
Human subgroup B adenoviruses, and in particular serotypes 11 and 35, are being used
to develop adenovirus vectors (reviewed in (31)). Typically, E1 (both E1A and
E1B)-deleted vectors from these serotypes cannot replicate in regular E1 producer cell lines
already established for serotype 5 vectors, although E1A-deleted vectors can be
propagated in PER.C6 cells (74). These vectors are therefore produced by modifying the
typical 1st generation cell lines already available (75-78) or replacing vectors genes by
HAdV5 sequences to permit viral propagation on unmodified cell lines, such as PER.C6
(79).
Several non-human adenoviruses derived from bovine, simian, porcine, ovine, murine
and canine sources have been used as backbone to develop E1-deleted vectors for gene
therapy or vaccine purposes (reviewed in (70)). For the majority of non-human vectors,
specialized E1 producer cells have been developed to propagate these vectors, such as
bovine kidney or fetal retinal cells expressing Bovine or human adenovirus E1 sequences
(80), or canine kidney cells expressing the canine adenovirus type 2 (CAV-2) E1 (81, 82).
Chimpanzee derived vectors of subgroup E can be produced on HEK293 cells already
developed (83, 84). However, similarly to HAdV11 and HAdV35, chimpanzee derived
vectors of subgroup B cannot be propagated on these cells, and a chimeric strategy is
used to allow propagation in HEK293 cells (85, 86). CAV-2 derived vectors are probably
the best described and advanced non-human vectors (87). The ability to preferentially
transduce neurons combined with a remarkable capacity of axonal transport, make
CAV-2 vectors candidates for the treatment of neurodegenerative diseases (88). Furthermore,
3rd generation CAV-2 vectors were already developed and tested in animal models,
confirming their potential for gene transfer based therapies (89, 90).
3.2. Upstream process for adenovirus vectors
An adenovirus production process starts by growing the cell line of choice to the desired
cell density for infection followed by the inoculation of an adenovirus stock to initiate the
infection and virus production cycle (Figure 3) (91). The need for significant amounts of
clinical grade adenovirus vectors, which in some cases may reach 1013 total
particles/patient (1011 infectious particles/patient), requires efficient and robust
processes for production and purification at large-scale compliant with good
manufacturing practices (GMP)(12). From a scalable point-of-view, the production of
Introduction
11 undertaken to maximize bioprocess yields and reduce production costs. In this section,
process considerations for the adenovirus production will be discussed mostly focusing
on first generation vectors, as the main bioprocess advances have been achieved with
these vectors.
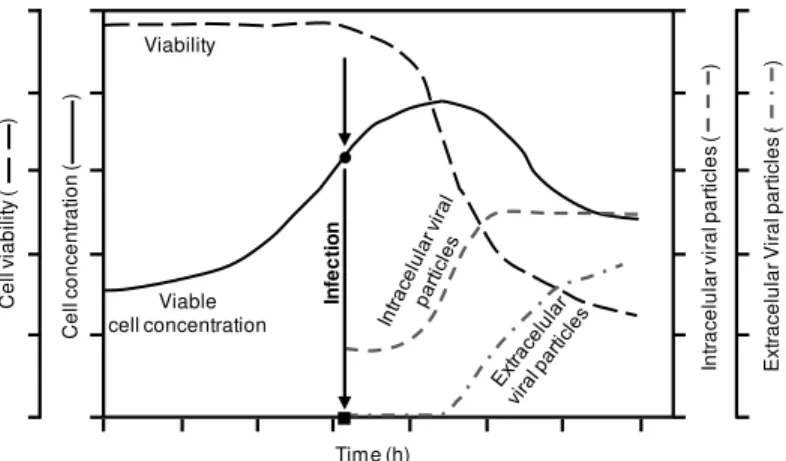
Figure 3. Typical profile of an adenovirus production process, including cell growth, viability and infection kinetics. After inoculation, cells grow exponentially and are usually infected at the end of exponential growth phase (represented by an arrow). Viral particles are assembled intracellularly. Due to the lytic nature of adenovirus propagation, cell lysis is induced after the production of viral particles. As a result, cell concentration and viability decreases and viral particles released to the supernatant. ●Cell concentration at infection (CCI); ■ time of infection (TOI). Adapted from (91).
3.2.1. Cell culture and adenovirus production process
Adenovirus production process can be performed in static or stirred cultures. From a
scalable point-of-view, stirred cultures are preferred. The use of microcarriers represents
the simplest approach to transfer adherent cells to stirred culture systems. The initial
steps of microcarriers preparation and cell seed from adherent cultures represent the
main drawback when using this system (92), since cell manipulation is cumbersome
when working at larger scales. Most of the production processes for adenoviruses
reported in literature use cells growing in suspension (reviewed in (93)). Although
adaptation of cells to grow in suspension can be time-consuming at the expense of
lowering cell specific productivity, suspension cell lines are preferred as they greatly
facilitate large-scale productions. Furthermore, the adaptation of cells to suspension
involves transference to serum-free culture medium, which represents an additional
advantage for biopharmaceuticals production.
In general, the medium is first chosen and optimized for its ability to support cell growth.
Chapter I
12
medium components is increased, and need to be taken into account when defining the
final medium formulation. Also, cells cultured with media containing animal serum
should be avoided. The undefined composition and high batch-to-batch variability of
serum, together with its potential source of contaminations, raises safety concerns and
hinders the standardization of cell culture processes for the production of
biopharmaceuticals (94). Almost all media manufacturers commercialize serum-free
formulations designed and optimized for specific cell lines and/or final product.
The majority of scalable adenovirus manufacturing processes are performed in
stirred-tank bioreactors both for microcarriers and suspension cultures (93). Technological
advances in disposable equipments, such as single-use Wave and stirred tank bioreactors,
have been shown by several companies (e.g. GE Healthcare, Sartorius, PBS). The use of
such systems poses several advantages for GMP production by avoiding the need of
cleaning and sterilization validation and alleviating facility requirements. Therefore, the
increasing use of disposable equipments for adenovirus manufacturing is anticipated.
3.2.2. Adenovirus seed stocks
AdV viral particles must be rescued from the initial vector genome construct. This is
usually performed by linearization of vector genome from the plasmid, and transfection
of producer cells. Viruses are then harvested once cytopathic effect is evident or, as
alternative, 2-3 days after transfection. This procedure is typically performed in static
culture conditions. When working with producer cells easily transfectable and first
generation vectors, the rescue of viral particles after this transfection step is relatively
high, and one or two more production rounds are performed to amplify the amount of
viral vectors and establish a purified viral seed stock. In such cases, scalable production
processes are usually established once a purified viral seed stock is obtained. When
developing viral vectors that typically present low productivities, such as HDV, process
development is defined from the transfection step to the final viral seed stock production
to maximize working-scale and volumetric productivity in all steps and minimize the
number of amplifications required to produce sufficient viral material (91) (discussed in
section 3.3.).
For GMP production, the purified viral seed stock must be certified and tested to confirm
the absence of adventitious agents (see section 4.1). On the other hand, the use of well
Introduction
13 reproducibility between different production batches. Therefore, determination of viral
particles, physical to infectious particles ratio and selection of best storage conditions are
carefully undertaken to ensure that particles properties from viral seed stock are
maintained (95-99).
3.2.3. The infection process and harvest strategy
Production process starts once producer cells are growing, being infected afterwards and
the newly produced adenoviruses harvested and the end of process (Figures 2 and 3).
Defining best process-related parameters, such as multiplicity of infection (MOI), cell
concentration at infection (CCI), time of infection (TOI) and time of harvest (TOH) is
determinant to ensure an optimal production process. Because adenovirus infections are
relatively fast and lytic, processes are established as single-round infections with MOI > 1
to ensure that all cells are infected. Although MOI > 1 needs to be established, infecting
the cells with more virus than those optimally required may have a negative effect on
cells viability, compromising virus productivity. TOI or CCI is usually selected when cells
are at exponential growth phase. For adherent, static cultures, cells are infected when
60-80% confluence is achieved, while for stirred cultures the typical cell densities range used
for adenovirus production is 0.5-1 x 106 cells/mL in batch mode with medium exchange
at infection. To produce adenovirus above this cell density and maintain cell
specific-productivity, fed-batch or perfusion modes must be added to the bioprocess (discussed in
section 3.2.5.).
After infection, the cell growth is arrested within 24 hpi. Viral DNA replication and virus
assembly occurs between 10-24 hpi and 20-48 hpi, respectively. As virus production
progresses, cell viability decreases after 24 hpi. Typically, at 48 hpi, when the volumetric
virus productivity reaches its plateau, the cell viability is around 40-80%. At this point, a
percentage of the virus (between 10-50%) has already been released from lysed cells, but
the rest of the virus remains intracellular. The cultivation process can proceed further
with no significant increase in virus production, but with a significant increase in virus
found in the culture medium. Harvest timing and method can be tailored to the entire
bioreaction bulk, or only to the intra- or extracellular fractions. The remaining cells are
Chapter I
14
3.2.4. Productivity of Adenovirus vector manufacturing
One of the most attractive features for adenovirus vectors, namely ∆E1-deleted vectors, is the high production yields obtained when compared to other viral vectors. Typically, titers of ∆E1-deleted vectors range from 103 to 104 IP per cell, whereas genome-containing particles are one log higher (reviewed in (93)). Considering the usual CCI used
for adenovirus production, this corresponds to a volumetric productivity of 109 – 1010 IP/mL or 1010 – 1011 PP/mL. Worth mentioning that such yields are dependent on adenovirus vector construct and culture conditions. Some production processes for HDV
are described as holding relatively low specific (102 IP/cell) and volumetric (108 IP/mL)
titers (104, 105) or inconsistent PP:IP ratios (106) (see section 3.3). In addition, cell
specific productivity under batch mode is limited to CCI below 1x106 cells/mL; producing
adenoviruses above this cell concentration results in lower specific yields (see section
3.2.5). Finally, cell physiological state can also impact the infectivity of the newly
produced adenoviruses. Indeed, when producing adenoviruses with MOI higher than the
optimal, the resulting PP:IP ratio tends to increase.
3.2.5. Adenovirus production at high cell density
In a batch operation mode, the cell density at infection is a very important parameter as it
impacts virus production. In fact, the narrow range for an optimal infection cell density
between 0.5-1.0x106 cells/mL is well documented (reviewed elsewhere (91)): a drop in
specific productivity at higher cell densities occurs even though medium formulations
allow cell growth up to 5 x 106 cells/mL. This is often referred to as the “cell density effect”. While this phenomenon is not totally understood, the decrease in productivity is thought to be due to limitations in a nutrient critical for virus propagation, accumulation
of inhibitory byproducts, or a combination of both (107). Following this hypothesis, the
typical approach to maintain cell-specific productivity at increased cell densities consists
of exchanging medium at infection time. This operation mode, widely used for viral vector
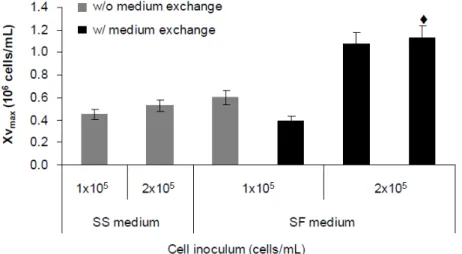
production, enables maximum cell-specific productivity up to 2x106 cells/mL. The main
limitation of such approach is the incorporation of a medium exchange/cell separation
step. While at scales of few liters, this is relatively easy to perform by centrifugation,
when dealing with hundreds or thousands of liters such manipulation is highly
Introduction
15 Several fed-batch approaches were devoted to adenovirus production based on the
control or addition of glutamine, glucose and aminoacids (108-110). Despite the
simplicity to implement, most of the fed-batch strategies towards maintaining
cell-specific productivity have failed.
Perfusion processes are capable of achieving high cell densities, through a continuous
renewal of fresh medium, providing new nutrients and diluting/removing
inhibiting-byproducts. Therefore, perfursion must be such that nutrient supply and byproducts
removal rates are sufficient to ensure robust cell performance at increasing cell
concentration. The perfusion rates described for adenovirus production can range from
0.5 to 3 culture volumes per day (V/day), being 2 V/day the rate mostly used (43,
111-113). When compared to low cell density batch productions, specific productivity of 293
cells infected at 3 – 6x106 cells/mL was maintained, which constituted a 5-fold increase in the final production yield (111, 113). The highest cell density tested to successfully
produce adenoviruses under perfusion mode is described in (43) using Hela cells at 107
cells/mL.
Other efforts were made to better understand the cell density effect. The specific
metabolic demands of producer cells during growth and virus production have been
analyzed through a metabolic flux analysis approach, showing that a favorable metabolic
state for adenovirus production should have an increase in glycolytic and TCA fluxes
(113, 114), and in ATP production rates upon infection. This state is also extended to
perfusion modes (113). Furthermore, it was demonstrated that a decrease in the
proportion of cells in S phase was related to a decrease in specific productivity at high cell
densities (115). Following this observation, the synchronization of HEK293 cells
chemically or by lowering temperature led to an enrichment of cells in S phase and up to
a 7.3 fold increase in AdV cell specific titer (116). Despite these insights, perfusion modes
still remain the best strategy to successfully produce AdV at high cell densities.
3.2.6. Physical parameters and process monitoring
Physical parameters such as temperature, pH and dissolved oxygen are usually well
established when manufacturing AdV at larger scales. The impact of such parameters on
AdV production has been previously reviewed (91, 93), therefore only optimal values
Chapter I
16
Production of adenoviruses is typically set to cell growth temperature of 37°C, although
previous works showed that by decreasing the temperature to 35°C an improvement in
virus production can be obtained. The common pH values for adenoviruses have been
established as 7.2 – 7.3. On the other hand, production of canine adenoviruses in stirred tank bioreactor at pH 7.4 showed production yields similar to human adenoviruses.
Despite the lack of reports addressing the effect of dissolved oxygen (DO) on adenovirus
production, most of the controlled processes set DO to values higher than 30% (92, 93).
Small-scale productions are typically monitored with offline sampling. While such offline
methods can be used to monitor some parameters, such as cell concentration and pH,
online methods are very useful for stirred-tank bioreactor productions at large-scale,
specially to select TOI and TOH. The cell/infection status can be monitored through a
variety of noninvasive online measurements. In particular, cell density can be estimated
by the oxygen uptake rate and the capacitance levels (113, 117, 118), as they can also be
used to monitor infection kinetics and confirm the maximum productivity point and
harvest timing.
3.3. Production process of helper-dependent vectors
Despite the potential of HDV, extensive use of such vectors for gene transfer experiments
has been restrained by two main bottlenecks: production yields and HV contamination.
Similarly to first generation vectors, HDV particles need to be rescued from linearized
plasmid after transfecting producer cells. To provide all the viral elements required for
HDV replication, cells must be also infected with HV (see section 3.1.4.). Typically, low
yields from this initial step are obtained and/or multiple rounds of HDV production are
needed until the desired vectors titer is achieved. Reducing the number of amplifications
is a primary condition to establish a robust production process. This also minimizes the
possibility of recombination between HV/HDV and producer cell line, avoiding the
occurrence of RCA, packaging-competent or recombinant HV, which have a propagation
advantage when compared to HDV (119, 120). Therefore, the most significant advances
for scalable production of HDV are designed early in the transfection step to maximize
volumetric productivity and set amplification rounds to its minimum (105).
The importance of recombinase system, such as Cre/loxP, with high levels of Cre to avoid
HV propagation is unquestionable (121). In fact, considering that the remaining HV
Introduction
17 excision (121), major advances were made to increase the levels of recombinase during
HDV production (106). Moreover, the definition of optimal MOI ratio showed a critical
impact either in maximizing HDV propagation, but also in reducing HV contamination.
The use of high HV MOI, besides being unnecessary and even negative for HDV
production (104), implicates the development of alternative designs to attain higher
levels of Cre than those supported by the cell line to reduce HV contamination (106).
Further, this also leads to the accumulation of higher levels of excised helper DNA
molecules (after Cre recombinase excision), increasing the chance of recombination
between viral vectors. In fact, some authors showed that these helper DNA molecules,
being prone to rearrangements, contributed to the generation of recombination between
viral vectors, in which helper vectors with rearranged genomes had a growth advantage
(120). In addition, some studies show a relatively high and/or great inconsistency in
maintaining PP:IP ratios among different preparations of same HDV (106, 122). Special
attention must be paid to this, as the PP:IP ratio of clinical grade adenoviruses is limited
by Food and Drug Administration (see section 4.4.).
3.4. Downstream process for adenovirus vectors
Since downstream processing is not in the scope of this work, only a brief overview is
presented. Traditional purifications of adenovirus vectors are performed by cesium
chloride (CsCl) density gradient ultracentrifugation. While useful for preparations at
laboratory scales, scalable purification of adenovirus vectors relies on membrane and
chromatographic processes (91, 123, 124). The main aim of downstream processing is to
eliminate contaminants, either process related (e.g., bovine serum albumin, Benzonase,
extractables, and leachables) or product related (e.g., host cell proteins, DNA,
proteoglycans, and glycosaminoglycans); other product-related impurities include free
proteins, aggregates, and empty capsids (125). The ultimate goal is to obtain a product
with high purity, potency, and quality, which can meet the stringent guidelines of the
regulatory authorities, such as the FDA and EMA. After harvesting and prior purification
two main steps are employed to production bulk: cell lysis, to release intracellular
adenoviruses and increase the yield, and genomic DNA breakdown, to facilitate DNA
removal. Purification can then be divided in three major steps: clarification,
concentration/purification and polishing. A suitable clarification step should remove cell
Chapter I
18
laboratory scales, or continuous flow centrifugation and microfiltration at industrially
relevant scales (91, 123, 126, 127). The concentration/purification step aims to reduce
the stream volume and facilitate the upfront equipment and materials. In this step, low
molecular weight proteins, fragmented DNA and other impurities are further removed
and ultrafiltration or chromatography columns are usually employed (91, 123, 128). The
step of polishing is usually performed using chromatographic processes and applied to
remove remaining impurities closely related to the product of interest (91, 128). The final
step for the production of GMP-grade adenovirus product is a filtration using a 0.2 μm sterile membrane. In the final product, host cell DNA and protein levels should be bellow
the specifications set by the European Pharmacopoeia (Ph. Eur 5.2.3) and the World
Health Organization (WHO expert Committee on Biological Standardization, WHO
Technical Report Series 878, 47th report, 1998) (124).
4. Concerns in the manufacturing of adenovirus vectors for product release
Commonly to vaccines for human use, release of adenovirus vectored products includes
tests for potency, general safety, sterility, purity, identity, and constituent materials. In
addition, screening for bacterial endotoxins and pyrogens are added when the product is
intended for use by injection (U.S. Pharmacopeial Convention and USP-NF bulletin,
http://www.usp.org). Every biopharmaceutical has its own characteristics that are
considered when developing and qualifying these tests. Therefore this section is focused
in the tests connected to the upstream manufacturing process of adenovirus vectors:
extraneous agents, RCAs, vector genetic stability and quantity/potency.
4.1. Extraneous agents
Viral inactivation and clearance steps are generally performed in the production of
inactivated vaccines which contribute in removing potential extraneous agents from final
product. However, these procedures cannot be applied in adenovirus vectors for gene
therapy, as their bioactivity is critical to gene transfer. Safety strategies need to be thus
implemented to lower the risk of introduction and carryover of contaminants, such as
extraneous agents, adventitious viruses, and transmissible spongiforme encephalopathy
causing agents. According to good manufacturing practices, a track record of all the
materials used in construction and manufacturing of adenovirus vectors must be kept.
Introduction
19 absence of adventitious viruses according to the ICH Q5A and Q5D guidelines. The use of
animal-derived components should be avoided as much as possible, as it represents a
potential source of contamination (94). In situations where the use of animal components
is unavoidable, excellent traceability and testing are performed to discard any risk of
enter and transfer of extraneous agents to the manufacturing process. The screening for
adventitious has relied on the use of in vitro infectivity assays, in vivo studies and specific polymerase chain reaction (PCR) tests. More recently, massive parallel sequencing has
been also applied to this end (129).
4.2. Replication competent adenovirus
Contamination of clinical batches with RCA is an important safety concern. Possible
consequences range from increased local inflammatory responses and tissue damage due
to uncontrolled systemic replication in immune-compromised individuals. Although the
issue of RCA formation by homologous recombination was solved by the introduction of
new cell lines like PER.C6 (see section 3.1), regulatory agencies still require testing to
confirm their absence in clinical batches (124). The maximum contamination level is set
to 1 RCA per 3x1010 VP and is based on the current FDA guidelines for HAdV5 vectors
(FDA Gene Therapy Letter, 2000). The detection of replicating virus is based on the
screening for cytopatic effect (CPE) after inoculating non-complementing cell lines. CPE
positive results are then confirmed by a more specific assay such as PCR or
immunofluorescence (130).
4.3. Vector genetic stability
To confirm product stability throughout the manufacturing process, genetic stability of
the viral vector is tested. To demonstrate this, viral vector is propagated for a number of
passages beyond the level used in production, generally 5 passages (more information at
U.S. Pharmacopeial Convention and USP-NF bulletin, http://www.usp.org). Mutation
frequency in replication-deficient adenovirus is considered rare, however such analysis is
important to evaluate that transgene region and corresponding expression is maintained,
and critical when considering the use of oncolytic adenoviruses, which, unlike gene
transfer-strict vectors, are intended to further propagate in a clinical setting (45).
Chapter I
20
have a replication advantage over the target vector. For this purpose, PCR detection
combined with sequence analysis of PCR fragments can be used for screening any
variations in transgene sequence that might occur (124).
4.4. Quantity and potency
Quantification of viral particles is important for monitoring yields during process
development, but also in final product to control of the amount of viral protein injected
within an acceptable safety window. Typically, absorvance measurements are used to
quantify physical particles based in the correlations between adenovirus preparations
and absorvance at 260 nm described by Maizel et al. (1968). While these measurements
require purified and concentrated preparations, alternative methods have been
developed to allow adenovirus quantification during process development and,
simultaneously, improve the limit of detection (see bellow) (91).
Adenovirus vectors exert their clinical effect by transducing target cells. Therefore,
quantification of infectious particles is required during the overall production process, for
product release, and stability assessments of viral preparations. In fact, a parameter
considered to represent the quality of preparations is the ratio between the amount of
physical and infectious particles. For gene therapy programs using adenoviruses, PP:IP
ratio must be <30:1 (FDA Gene Therapy Letter, 2000).
Standard plaque assays or tissue culture infectious dose (TCID50) assays using
complementing cell lines can be employed to quantify infectious titers. To improve
accuracy and precision of these assays, alternative methods can be applied which are
based in quantitative PCR techniques or in the detection of the transgene expression.
5. Scope of the thesis
The central objective of this PhD thesis was to develop a scalable production process for ∆E1 and HD CAV-2 vectors using MDCK derived cell lines as producer cells, assuring optimal cell performance and maximum productivity. Critical cell culture and virus
infection parameters were studied and combined with a better understanding of cell
physiological state and viral cycle steps behind a successful production process.
To accomplish this, the work was organized according to the following:
Introduction
21 Since MDCK cells are accepted in Food and Drug Administration (FDA) and European
Medicine Agency (EMA) and used for the production of many viral vaccines, the use of an
MDCK-based cell lines would facilitate the regulatory approval for the clinical grade
production of CAV-2 vectors. Accordingly, MDCK-E1 and MDCK-E1-Cre cell lines were
established (Chapter II).
(II) Develop a scalable production process for CAV-2 vectors
The need for significant amounts of clinical grade adenovirus vectors, which in some
cases may reach 1013 total particles/patient or 1011 infectious particles/patient, requires
efficient and robust processes for production and purification at large-scale compliant
with good manufacturing practices (GMP) (12). Therefore, current lab-scale protocols
must be adapted and transferred to a scalable stirred culture system (Chapter III).
(III) Establish best infection conditions for HD CAV-2 production
Process optimization for ΔE1 vectors is relatively straightforward. However, given the production system complexity, special attention must be paid to HDV propagation. Using
the Cre/loxP system, producer cells must express Cre-recombinase, which can be toxic to
cells (131), and production is performed under co-infection. Therefore, cell line stability
and co-infection conditions must be well defined to ensure production process
reproducibility (Chapter IV).
(IV) Identify the limitations in HD CAV-2 vectors production from an adenovirus replication cycle perspective.
Despite the potentialities of HDV for gene therapy, one important bottleneck is the yields,
which compromises process scale-up, viral particles quality and its availability to pre and
clinical trials. Still, the low productive viral cycle of HDV has been somehow neglected
(Chapter V).
6. Author contribution
Chapter I
22
7. References
1. Naldini, L. 2009. Medicine. A comeback for gene therapy. Science 326:805-806.
2. Nathwani, A. C., E. G. Tuddenham, S. Rangarajan, C. Rosales, J. McIntosh, D. C. Linch, P. Chowdary, A. Riddell, A. J. Pie, C. Harrington, J. O'Beirne, K. Smith, J. Pasi, B. Glader, P. Rustagi, C. Y. Ng, M. A. Kay, J. Zhou, Y. Spence, C. L. Morton, J. Allay, J. Coleman, S. Sleep, J. M. Cunningham, D. Srivastava, E. Basner-Tschakarjan, F. Mingozzi, K. A. High, J. T. Gray, U. M. Reiss, A. W. Nienhuis, and A. M. Davidoff. 2011. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365:2357-2365.
3. Bainbridge, J. W., A. J. Smith, S. S. Barker, S. Robbie, R. Henderson, K. Balaggan, A. Viswanathan, G. E. Holder, A. Stockman, N. Tyler, S. Petersen-Jones, S. S. Bhattacharya, A. J. Thrasher, F. W. Fitzke, B. J. Carter, G. S. Rubin, A. T. Moore, and R. R. Ali. 2008. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med 358:2231-2239.
4. Maguire, A. M., F. Simonelli, E. A. Pierce, E. N. Pugh, Jr., F. Mingozzi, J. Bennicelli, S. Banfi, K. A. Marshall, F. Testa, E. M. Surace, S. Rossi, A. Lyubarsky, V. R. Arruda, B. Konkle, E. Stone, J. Sun, J. Jacobs, L. Dell'Osso, R. Hertle, J. X. Ma, T. M. Redmond, X. Zhu, B. Hauck, O. Zelenaia, K. S. Shindler, M. G. Maguire, J. F. Wright, N. J. Volpe, J. W. McDonnell, A. Auricchio, K. A. High, and J. Bennett. 2008. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 358:2240-2248.
5. Testa, F., A. M. Maguire, S. Rossi, E. A. Pierce, P. Melillo, K. Marshall, S. Banfi, E. M. Surace, J. Sun, C. Acerra, J. F. Wright, J. Wellman, K. A. High, A. Auricchio, J. Bennett, and F. Simonelli. 2013. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology 120:1283-1291.
6. Hacein-Bey-Abina, S., J. Hauer, A. Lim, C. Picard, G. P. Wang, C. C. Berry, C. Martinache, F. Rieux-Laucat, S. Latour, B. H. Belohradsky, L. Leiva, R. Sorensen, M. Debre, J. L. Casanova, S. Blanche, A. Durandy, F. D. Bushman, A. Fischer, and M. Cavazzana-Calvo. 2010. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 363:355-364.
7. Bryant, L. M., D. M. Christopher, A. R. Giles, C. Hinderer, J. L. Rodriguez, J. B. Smith, E. A. Traxler, J. Tycko, A. P. Wojno, and J. M. Wilson. 2013. Lessons learned from the clinical development and market authorization of Glybera. Human gene therapy. Clinical development 24:55-64.
8. Choi, I. K., and C. O. Yun. 2013. Recent developments in oncolytic adenovirus-based immunotherapeutic agents for use against metastatic cancers. Cancer gene therapy 20: 70-76.
9. Ledley, F. D., L. M. McNamee, V. Uzdil, and I. W. Morgan. 2014. Why commercialization of gene therapy stalled; examining the life cycles of gene therapy technologies. Gene Ther 21:188-194.
10. McConnell, M. J., and M. J. Imperiale. 2004. Biology of adenovirus and its use as a vector for gene therapy. Hum Gene Ther 15:1022-1033.
11. Tatsis, N., and H. C. Ertl. 2004. Adenoviruses as vaccine vectors. Mol Ther 10:616-629. 12. Dormond, E., M. Perrier, and A. Kamen. 2009. From the first to the third generation
adenoviral vector: what parameters are governing the production yield? Biotechnol Adv 27:133-144.
13. Soudais, C., S. Boutin, S. S. Hong, M. Chillon, O. Danos, J. M. Bergelson, P. Boulanger, and E. J. Kremer. 2000. Canine adenovirus type 2 attachment and internalization: coxsackievirus-adenovirus receptor, alternative receptors, and an RGD-independent pathway. J Virol 74:10639-10649.