Universidade de Brasília
Instituto de Química
Programa de Pós-Graduação em Química
INTERAÇÃO DE VAN DER WAALS NO COMPLEXO
HÉLIO-ANTIHIDROGÊNIO A PARTIR DO ESPECTRO ROVIBRACIONAL
SANDRO FRANCISCO DE BRITO
Orientador : Prof. Dr. HEIBBE CRISTHIAN B.DE OLIVEIRA
Bras´ılia, DF
Universidade de Brasília
Instituto de Química
Programa de Pós-Graduação em Química
INTERAÇÃO DE VAN DER WAALS NO COMPLEXO
HÉLIO-ANTIHIDROGÊNIO A PARTIR DO ESPECTRO ROVIBRACIONAL
SANDRO FRANCISCO DE BRITO
Orientador : Prof. Dr. HEIBBE CRISTHIAN B.DE OLIVEIRA
Dissertac¸ ˜ao apresentada ao programa de p ´os-graduc¸ ˜ao do Instituto de Qu´ımica da Universidade de Bras´ılia como requisito parcial para obtenc¸ ˜ao do t´ıtulo de mestre.
FOLHA DE APROVAÇÃO
Comunicamos a aprovac¸ ˜ao da Defesa de Dissertac¸ ˜ao de Mestrado do (a) aluno (a)Sandro
Fran-cisco de Brito, matr´ıcula no14/0194436, intitulada“Interac¸ ˜ao de van der Waals no complexo H ´elio
- Antihidrog ˆenio a partir do espectro rovibracional”, apresentada no (a) Audit ´orio Verde do Instituto de Qu´ımica (IQ) da Universidade de Bras´ılia (UnB) em 27 de julho de 2016.
Prof. Dr. Heibbe Cristhian Benedito de Oliveira Presidente de Banca (IQ/UnB)
Prof. Dr. Clodoaldo Valverde Membro Titular (UEG)
Prof. Dr. Wiliam Ferreira da Cunha Membro Titular (IF/UnB)
Prof. Dr. Luciano Ribeiro Membro Suplente (UnuCET-UEG)
Agradecimentos
Resumo
Nesta dissertac¸ ˜ao ser ˜ao estudas as propriedades din ˆamicas entre mat ´eria e antim ´ateria, especifi-camente para o sistema h ´elio-antihidrog ˆenio, tais propriedades foram calculadas utilizando a curva de energia potencial obtida por Strasburgeret al(2002, Phys. Rev. Lett. 88(16) 163201). Com esse intuito a equac¸ ˜ao de Schr ¨odinger nuclear para este sistema foi resolvida numericamente e as suas soluc¸ ˜oes combinadas com soluc¸ ˜oes da equac¸ ˜ao da energia rovibracional para sistemas diat ˆomicos. Desta forma foram calculadas pela primeira vez na literatura as constantes espectrosc ´opicas ωe, ωexe, ωeye, αe e
γe. Utilizou - se quatro formas anal´ıticas para representar o potencial efetivo: Rydberg, Rydberg
defor-mada, polin ˆomio em coordenadasBond Ordere Bond Ordemdeformado, todos os potenciais s ˜ao de sexto grau. Foram ajustadas com boa precis ˜ao as curvasab initio, bem como obtivemos valores muito pr ´oximos para as constantes espectrosc ´opicas, independentemente da func¸ ˜ao utilizada. Observou-se que o sistema h ´elio-antihidrog ˆenio possui apenas um ´unico n´ıvel de vibrac¸ ˜ao (ν = 0), com tr ˆes n´ıveis rovibracionais (J = 0,1 e2) para todas as formas anal´ıticas empregadas no trabalho, indicando que existe uma ligac¸ ˜ao extremamente fraca que, portanto, s ´o faz sentido essa ligac¸ ˜ao em temperaturas muito baixas. ´E importante destacar que essas propriedades foram calculadas pela primeira vez na literatura e podem servir de comparac¸ ˜ao com estudos experimentais futuros acerca das propriedades espectrosc ´opicas para o sistema diat ˆomico h ´elio-antihidrog ˆenio.
Abstract
The matter and antimatter properties of helium with antihydrogen were investigated within Born-Oppenheimer approximation using the potential energy curva calculated by Strasburger et al (2002, Phys. Rev. Lett. 88(16) 163201). We have solved the nuclear Schr ¨odinger equation for this system, combined the solutions with a diatomic rovibrational energy equation and determined for the first time, the spectroscopic constants and rovibrational energies as well. Namely, we have evaluatedωe, ωexe,
ωeye,αeandγe. We used four different analytical forms to represent the effective potential: Rydberg,
deformed Rydberg, Bond Order polynomial and deformed Bond Order. We fitted the ab initio curves accuratelly and the spectroscopic constants values are very close independently of the function used. We found that helium-antihidrogen system has only one bound vibration level(ν = 0) with three rotational levels (j= 0,1and2) for all analytical forms, indicating extremely weak binding and existence would be reasonable only at very low temperatures.
Lista de S´ımbolos, Abreviaturas e Acr ˆonimos
γe,αe Constantes de acoplamento rovibracional
ωexe Contribuic¸ ˜ao anarm ˆonica de primeira ordem
ωeye Contribuic¸ ˜ao anarm ˆonica de segunda ordem
cj Coeficientes de expans ˜ao
ωe Contribuic¸ ˜ao harm ˆonica
λj Constante de normalizac¸ ˜ao
λ Peso associado a ordem da perturbac¸ ˜ao
~ Constante de Planck reduzida
h Constante de Planck
B Constante de rotac¸ ˜ao caracter´ıstica do sistema
Be Constante rotacinal de equil´ıbrio
Bν Constante rotacinal com correc¸ ˜ao vibracional
ζ Descreve se os potenciais s ˜ao de curto ou longo alcance no potencial de Morse
Tij Elementos da matriz energia cin ´etica
Vij Elementos da matriz dos potenciais
Sij Elementos da matrizoverlap
ˆ
Te(r) Energia cin ´etica dos el ´etrons
ˆ
Tn(R) Energia cin ´etica dos n ´ucleos
De Energia de dissociac¸ ˜ao
ν Frequ ˆencia de radiac¸ ˜ao
fi(R) Func¸ ˜oes de base conhecidas
φ(r;R) Func¸ ˜ao de onda eletr ˆonica
θ(R) Func¸ ˜ao de onda nuclear
ψ(r,R) Func¸ ˜ao de onda completa
VBO(R) Func¸ ˜ao de coordenadasBond Order
VdBO(R) Func¸ ˜ao de coordenadasBond Orderdeformada
VRyd(R) Func¸ ˜ao de coordenadasRydberg
VdRyd(R) Func¸ ˜ao de coordenadasRydbergdeformada
F(R) Func¸ ˜ao de onda em coordenadas nucleares unidimensionais ˆ
H Hamiltoniano total qu ˆantico
H Hamiltoniano total cl ´assico ˆ
HCM Hamiltoniano do centro de massa
Ym
J (θ, φ) Harm ˆonicos esf ´ericos
ˆ
Hint Hamiltoniano de coordenadas internas
δij Matriz identidade
H Matriz Hamiltoniana
J N ´umero qu ˆantico rotacional
υ N ´umero qu ˆantico vibracional
ηAB Ordem de ligac¸ ˜ao
ˆ
L Operador momento angular
ρ Par ˆametro que denota dist ˆancia entre ´atomos
d Par ˆametro de deformac¸ ˜ao da func¸ ˜ao exponencial
wk Pesos associados ao pontoRkna malha de quadraturas
ˆ
Vnn(R) Potencial de interac¸ ˜ao dos n ´ucleos
ˆ
Vee(r) Potencial de interac¸ ˜ao dos el ´etrons
ˆ
Vne(r,R) Potencial de interac¸ ˜ao dos n ´ucleos e el ´etrons
ˆ
V Termo de perturbac¸ ˜ao
Vm(R) Potencial de Morse
He Atomo de H ´elio´
H Anti-hidrog ˆenio
ABO Aproximac¸ ˜ao de Born-Oppenheimer
CEP Curva de energia potencial
GSA Generelized Simulated Anneling
CERN Organizac¸ ˜ao Europeia para pesquisa Nuclear
F ermilab Fermi Laborat ´orio de Aceleradores Nacional
DV R Representac¸ ˜ao da Vari ´avel Discreta
Lista de Tabelas
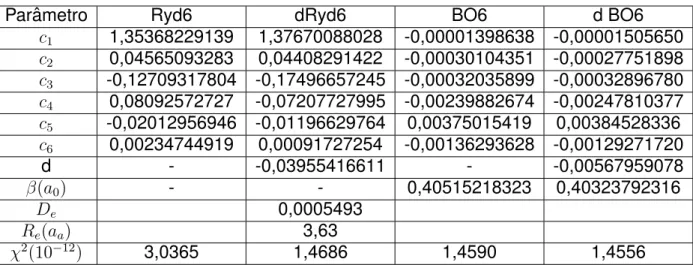
3.1 Tabela de par ˆametros otimizados para todas as CEPs do sistemaHe−H. . . 23 3.2 Constantes espectrosc ´opicas rovibracionais para o sistemaHe−H calculadas usando
Eq.(79) atrav ´es do m ´etodo DVR. Todos os valores est ˜ao apresentados emcm−1. . . . 26
3.3 N´ıveis de energias rovibracionais para o sistemaHe−H (emcm−1) obtidos da soluc¸ ˜ao
Lista de Figuras
1.1 Comparac¸ ˜ao entre a estrutura do ´atomo de hidrog ˆenio usual e seu correspondente for-mado por antipart´ıculas, o anti-hidrog ˆenio . . . 1 1.2 Curva de energia potencial ab initio para o sistema h ´elio anti-hidrog ˆenio. Esta curva foi
calculada por Strasburger e Chojnacki . . . 4
2.1 Sistema de coordenadas moleculares para uma estrutura que representa um sistema qualquer. . . 6 2.2 N ´ucleos que representam um sistema de coordenadas fixos. . . 12
3.1 Comparac¸ ˜ao entre CEP obtida por Strasburgeret ale CEP Ryd6, juntamente com erro ponto-a-ponto do ajuste. . . 23 3.2 Comparac¸ ˜ao entre CEP obtida por Strasburgeret ale CEP dRyd6, juntamente com erro
ponto-a-ponto do ajuste. . . 24 3.3 Comparac¸ ˜ao entre CEP obtida por Strasburgeret al e CEP BO6, juntamente com erro
ponto-a-ponto do ajuste. . . 24 3.4 Comparac¸ ˜ao entre CEP obtida por Strasburgeret ale CEP dBO6, juntamente com erro
Sum´ario
Agradecimentos . . . i
Resumo . . . ii
Abstract . . . iii
S´ımbolos Abreviaturas e Acr ˆonimos . . . iv
Lista de Tabelas . . . vi
Lista de Figuras . . . vii
1 Introduc¸˜ao 1 2 Fundamentac¸˜ao Te ´orica 6 2.1 O Problema Molecular . . . 6
2.2 Formas Anal´ıticas . . . 10
2.2.1 Polin ˆomios em coordenadas Bond Order Generalizada . . . 10
2.2.2 Polin ˆomios em coordenadas f´ısicas de Rydberg . . . 11
2.3 Equac¸ ˜ao de Schr ¨odinger Nuclear . . . 11
2.4 M ´etodo da Representac¸ ˜ao da Vari ´avel Discreta . . . 15
2.5 Propriedades Espectrosc ´opicas Rovibracionais . . . 18
2.5.1 Espectro Rotacional . . . 18
2.5.2 Espectro Vibracional . . . 19
2.5.3 Espectro Rovibracional . . . 20
3 Resultados e Discuss ˜oes 22
4 Conclus ˜oes e Perspectivas 31
Referˆencias Bibliogr´aficas 32
Cap´ıtulo 1
Introduc¸˜ao
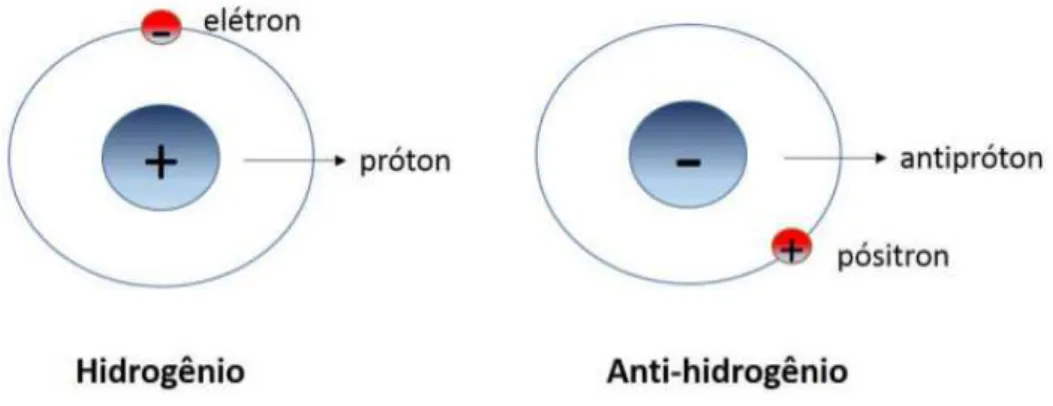
A produc¸ ˜ao artificial de antim ´ateria no CERN [1] e Fermilab [2] no final da d ´ecada de 1990 atraiu de forma sistem ´atica o interesse da comunidade cient´ıfica com o intuito de buscar compreender sua natureza ainda misteriosa. A s´ıntese de ´atomos de antihidrog ˆenio [3] em 2002 foi uma experi ˆencia que permitiu aos cientistas investigarem de forma mais profunda a interac¸ ˜ao entre mat ´eria e antimat ´eria, sendo esse tema de interesse atual especialmente no que diz respeito ao estudo das diferenc¸as entre ambas, e porque mat ´eria ´e t ˜ao mais abundante na natureza do que a antimat ´eria. O sistema formado por antimat ´eria mais simples, analogamente aos ´atomos que estamos habituados a estudar ´e o ´atomo de anti-hidrog ˆenio nos anos recentes muito progresso tem ocorrido nas pesquisas destes sistemas em regimes ultrafinos, como a primeira demonstrac¸ ˜ao de confinamento de anti-hidrog ˆenio em armadilhas magn ´eticas [4] e a s´ıntese destes ´atomos usando a t ´ecnicacusp trap[5]. A estrutura do anti-hidrog ˆenio ´e pictoricamente representada na Figura 1.1, em que o n ´ucleo do sistema possui carga negativa, sendo denominado de antipr ´oton e no lugar do el ´etron h ´a um p ´ositron (det ´em a massa do el ´etron por ´em com carga positiva).
Figura 1.1: Comparac¸ ˜ao entre a estrutura do ´atomo de hidrog ˆenio usual e seu correspondente formado por antipart´ıculas, anti-hidrog ˆenio.
estados mais baixos em que pudessem pemanecer ligados [6]. Este quadro tem se modificado com recentes avanc¸os do ALPHA collaboration reportando o confinamento de ´atomos de anti-hidrog ˆenio por mais de 1000 segundos e indicando que a maioria dos ´atomos confinados alcanc¸avam o estado fundamental [7].
O modelo padr ˜ao da f´ısica de part´ıculas prev ˆe que o anti-hidrog ˆenio deve ter os mesmos n´ıveis de energia que o hidrog ˆenio, sendo que, essa hip ´otese basea-se em postulados do Teorema CPT(”conjunc¸ ˜ao de carga”, ”paridade”e ”invers ˜ao de tempo”) [8]. A quebra da teoria CPT est ´a sendo amplamente es-tudada [9] [10]. Estudos como o de Hori et al [11] bem como os de Kobayashiet al [12] no campo da espectroscopia s ˜ao realizados de forma intensa para verificar quebra de simetria do teorema CPT, isto ´e, realizar medidas espectrosc ´opicas comparando o anti-hidrog ˆenio com o hidrog ˆenio. Portanto, observa - se claramente a import ˆancia das previs ˜oes espectrosc ´opicas em estudos te ´oricos acerca da interac¸ ˜ao entre mat ´eria e antimat ´eria que busquem auxiliar na interpretac¸ ˜ao das medidas experimetais.
Em um trabalho realizado por Strasburger e Chojnacki [13], foi determinado um conjunto de energias eletr ˆonicas para diferentes dist ˆancias internucleares em um conjunto de interac¸ ˜oes entre um ´atomo de antihidrog ˆenio (H) com h ´elio no estado fundamental utilizando a aproximac¸ ˜ao de Born-Oppenheimer. Levando em considerac¸ ˜ao a simetria do sistema ´atomo-anti ´atomo, bem como os fatores que influenciam a interac¸ ˜ao neste sistema, usou-se a teoria da perturbac¸ ˜ao nos estudos realizados pelos autores, pois ´e uma teoria aplicanda nestes sistemas deste `a d ˆecada de 70 [14].Os autores utilizaram um Hamiltoniano com perturbac¸ ˜ao de primeira ordem, na seguinte forma:
ˆ
H = ˆH(0)+λV .ˆ (1.1)
A equac¸ ˜ao (1.1) mostra o termoHˆ(0), que denota o Hamiltoniano n ˜ao perturbado, eVˆ representa
o termo de perturbac¸ ˜ao, usado para corrigir a energia de correlac¸ ˜ao do sistema (Ecorr), que cont ˆem
todas as interac¸ ˜oes interat ˆomicas, tal que:
ˆ
H(0)= ˆHA+ ˆHB. (1.2)
O potencial de perturbac¸ ˜ao ´e completamente explicitado com as seguintes condic¸ ˜oes; A representa o ´atomo, B representa o anti- ´atomo,NA denota o n ´umero de el ´etrons eNB representa o n ´umero de
p ´ositrons. Ent ˜ao o potencial ´e dado como:
ˆ
V = ZAZB
R − NA X i=1 ZB riB + NB X j=1 ZA rjA − NA X i=1 NB X j=1 1 rij , (1.3)
em queZAeZB s ˜ao as cargas do sistema. Na representac¸ ˜ao do potencial foram utilizadas unidades
at ˆomicas.
A energia total do sistema ter ´a a forma:
E=E(0)+λE(1). (1.4)
proprie-dades dos ´atomos isolados. A correc¸ ˜ao de primeira ordem(E(1)) ´e dada como, E(1) = hψ(0)|Vˆ|ψ(0)i,
podendo ser facilmente obtida com as func¸ ˜oes de onda dos ´atomos isolados, ψA eψB. A func¸ ˜ao de
onda n ˜ao perturbada n ˜ao necessita de antissimetrizac¸ao j ´a que as esp ´ecies s ˜ao distinguiv ´eis. Assim a componente de trocaE(1) n ˜ao estar ´a presente neste tipo de sistema, sendo semelhante ao produto de
Hartree.
A func¸ ˜ao de onda empregada por Strasburger e Chojnacki no trabalho ´e composta por part´ıculas le-ves, que denotam uma combinac¸ ˜ao linear de func¸ ˜oes gaussianas explicitamente correlacionadas como uma aproximac¸ ˜ao, baseando-se em resultados muito precisos para mat ´eria ordin ´aria [15] e sistemas positr ˆonicos [16]. Para um sistema formado por um conjunto de Npart´ıculas o formato da func¸ ˜ao de onda ´e:
ψ=
k
X
I=1
CIAeˆ
PN i=1−α
(I)
i (ri−R(iI)) 2−PN
i>jβ (I)
ij (ri−rj)Θ
I. (1.5)
Na func¸ ˜ao de onda dada pela equac¸ ˜ao (1.5),Nrepresenta o n ´umero de part´ıculas leves do sistema, ˆ
A ´e o operador antissimetrizador, que garante as trocas das part´ıculas indistingu´ıveis do sistema,ΘI ´e
a func¸ ˜ao de spin, os expoentesα(iI),β(ijI)s ˜ao os par ˆametros de variac¸ ˜ao n ˜ao-linear, bem como o termo de coordenadaR(I)
i .
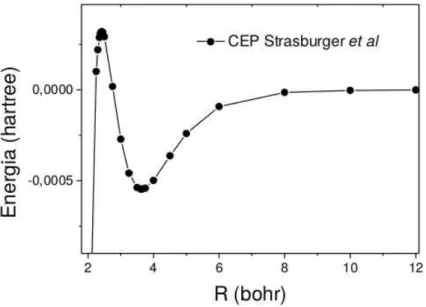
A curva de energia potencial (CEP) no trabalho de Strasburger e Chojnacki foi calculada variacional-mente para o sistema h ´elio-antihidrog ˆenio, cuja dist ˆancia de equil´ıbrio ´ere= 3,63a0. A curva descrita
por Strasburger e Chojnacki possui um m ´aximo em 2,42a0, como representado pela Figura 1.2,
Figura 1.2: Curva de Energia Potencial ab initio para o sistema h ´elio-antihidrog ˆenio. Esta curva foi calculada por Strasburger e Chojnacki.
Devido `a controv ´ersia sobre a exist ˆencia da barreira de potencial na interac¸ ˜ao deste sistema, foi in-vestigada no presente trabalho apenas o poss´ıvel estado fracamente ligado do sistema ”diat ˆomico”He− H, que encontra-se bem localizado no m´ınimo da CEP com propriedades de um sistema de van der Waals. O m´ınimo na curva seria produzido por forc¸as de dispers ˜ao, possibilitando a exist ˆenica do sis-tema molecular em baixas temperaturas. O objetivo principal deste trabalho ´e calcular as propriedades rovibracionais dentro do intervalo na CEP de Strasburger e Chojnacki para dist ˆancias maiores que2,4 bohr (regi ˜ao que caracteriza o poc¸o potencial), ´e necess ´ario resolver a equac¸ ˜ao de Schr ¨odinger do sis-tema, isto ´e, estudar o comportamento dos n ´ucleos e dos el ´etrons (ou n ´ucleo antiprot ˆonico e p ´ositron). A CEPab initioobtida por Strasburgeret alfoi ajustada utilizando quatro formas anal´ıticas,Bond Order, Rydberg e suas respectivas vers ˜oes deformadas: d-Bond Order e d-Rydberg, todas de grau 6. Os par ˆametros foram ajustados utilizando os m ´etodo h´ıbridos como Gradiente Simplex [21], Levenberg-Marquardt[22] [23] eGeneralized Simulated Annealing(GSA)[24]. Ap ´os a execuc¸ ˜ao dos ajustes, foram avaliadas as constantes espectrosc ´opicas rovibracionais relacionadas `a contribuic¸ ˜ao harm ˆonica (ωe), a
contribuic¸ ˜ao anarm ˆonica de primeira ordem(ωeχe) e de segunda ordem (ωeγe) bem como as constantes
de acoplamento rovibracionais (αe, γe) e as energias rovibracionais atrav ´es das soluc¸ ˜oes da equac¸ ˜ao de
Schr ¨odinger nuclear obtidas com o do M ´etodo da Representac¸ ˜ao da Vari ´avel Discreta DVR (do ingl ˆes
Discrete Variable Representation) [25], [26]. A metodologia aqui empregada consiste na combinac¸ ˜ao das energias rovibracionais, obtidas atrav ´es da resoluc¸ ˜ao da equac¸ ˜ao de Schr ¨odinger nuclear com uma equac¸ ˜ao espectrosc ´opica (expans ˜ao de Dunham [27]) para energias rovibracionais em sistemas diat ˆomicos. ´E importante enfatizar que esta metodologia foi empregada com sucesso na descric¸ ˜ao es-pectrosc ´opica de outros sistemas diat ˆomicos como o ´ıon molecularH2+ e a mol ´eculaCl2em diversos
estados eletr ˆonicos.
Cap´ıtulo 2
Fundamentac¸˜ao Te ´orica
2.1 O Problema Molecular
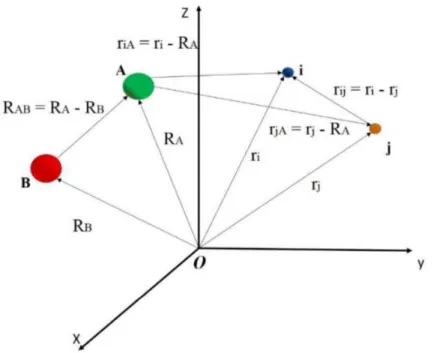
Para descrever a din ˆamica de um sistema poliat ˆomico composto porMn ´ucleos eNel ´etrons, deve-se levar em considerac¸ ˜ao a resoluc¸ ˜ao da equac¸ ˜ao de autovalor de Schr ¨odinger para o estado estacion ´ario deste sistema. Considerando um sistema genericamente representado por dois n ´ucleos e dois el ´etrons em coordenadas moleculares, como o ilustrado pela Figura 2.1.
Figura 2.1: Sistema de coordenadas moleculares para uma estrutura que representa um sistema qual-quer.
Este sistema de coordenadas moleculares tem como objetivo descrever queriA ´e a dist ˆancia entre
oi- ´esimo el ´etron e oA- ´esimo n ´ucleo,RAB ´e a dist ˆancia entre oA- ´esimo e oB- ´esimo n ´ucleos erij s ˜ao
as dist ˆancias entre osi- ´esimo ej- ´esimo el ´etrons.
independente do tempo, denotada por:
ˆ
Hψ(r,R) =Eψ(r,R). (2.1)
onde, ψ(r,R)representa a func¸ ˜ao de onda completa, descrevendo as propriedades din ˆamicas deste sistema molecular. O operador Hamiltaniano (Hˆ) molecular, n ˜ao-relativ´ıstico e independente do tempo
´e denotado como:
ˆ
H =− ~
2
2me N X i=1 ∇2 i− ~2 2 M X A=1 ∇2 A MA + 1 4πǫ0
M X A=1 M X B>A
ZAZB
RAB
+ 1 4πǫ0
N X i=1 N X j>i e2 rij − 1
4πǫ0
M X A=1 N X i=1
ZAZBe2
riA
, (2.2)
Neste operadorMA ´e a raz ˜ao entre a massa do n ´ucleo e a massa do ´eletron,e ´e a carga elementar
do el ´etron,ZAeZB s ˜ao as cargas referentes aos n ´ucleos A e B,~ ´e dado como a constante de Planck
dividida por2π. Escrevendo o Hamiltoniano em unidades at ˆomicas, teremos:
ˆ
H =−
N
X
i=1
∇2i − M
X
A=1
∇2
A
2MA
+ M X A=1 M X B>A
ZAZB
RAB + N X i=1 N X j>i 1 rij − M X A=1 N X i=1
ZAZB
riA
. (2.3)
Denotando a equac¸ ˜ao (2.3) de forma mais simplificada em termos da notac¸ ˜ao de Born-Oppenheimer,
ˆ
H = ˆTe(r) + ˆTn(R) + ˆVnn(R) + ˆVee(r) + ˆVne(r,R), (2.4)
Todos os termos da equac¸ ˜ao (2.4) em unidades at ˆomicas, s ˜ao apresentados de forma explicitida como:
Operador energia cin ´etica dos el ´etrons:Tˆe(r) =−21PNi=1∇2i.
Operador energia cin ´etica dos n ´ucleos:Tˆn(R) =−PMA=1
∇2A
2MA. Interac¸ ˜ao eletrost ´atica dos n ´ucleos:Vˆnn(R) =PMA=1
PM
B>AZRAABZB. Interac¸ ˜ao eletrost ´atica dos el ´etrons:Vˆee(r) =PNi=1
PN
j>ir1ij.
Interac¸ ˜ao eletrost ´atica entre n ´ucleos e el ´etrons:Vˆne(r,R) =−PMA=1
PN
i=1 ZrAiAZB.
Explicitamente a equac¸ ˜ao que deve ser resolvida para demonstrar a din ˆamica do sistema em estudo ´e dada por:
" − N X i=1 ∇2 i − M X A=1 ∇2 A
2MA
+ M X A=1 M X B>A
ZAZB
RAB + N X i=1 N X j>i 1 rij − M X A=1 N X i=1
ZAZB
riA
#
ψ(r,R) =Eψ(r,R). (2.5)
Em mec ˆanica qu ˆantica, em um dos seus teoremas temos a afirmativa que, caso um hamiltoniano seja separ ´avel sua autofunc¸ ˜ao total pode ser escrita como um produto das autofunc¸ ˜oes individuais para cada termo do hamiltoniano, sendo o autovalor total a soma dos autovalores individuais. Esse teorema por ´em n ˜ao pode ser aplicado em tal situac¸ ˜ao, na resoluc¸ ˜ao da equac¸ ˜ao (2.5), pois o termo de interac¸ ˜ao n ´ucleo el ´etronVˆne(r,R)impede o desacoplamente dos movimentos nucleares e eletr ˆonicos, e assim,
Considerando que a equac¸ ˜ao (2.5) s ´o pode ser resolvida para sistemas monoeletr ˆonicos, para sis-temas maiores, como o diat ˆomico, devem ser feitas aproximac¸ ˜oes para conhecer sua soluc¸ ˜ao. Portanto uma maneira muito utilizada para resoluc¸ ˜ao deste problema encontra-se na aproximac¸ ˜ao de Born-Oppenheimer (ABO). Essa aproximac¸ ˜ao leva em considerac¸ ˜ao a ideia f´ısica de que a massa do pr ´oton ´e maior que a massa do el ´etron e, portanto, um n ´ucleo movimenta-se mais lentamente que os el ´etrons.
Sendo assim, os el ´etrons ajustam seu movimento rapidamente ao movimento dos n ´ucleos. Logo, usando a expans ˜ao adiab ´atica, pode-se considerar que variac¸ ˜oes na func¸ ˜ao de onda eletr ˆonica justam-se de maneira quajustam-se instant ˆanea a mudanc¸as em coordenadas nucleares. Pela expans ˜ao adiab ´atica a func¸ ˜ao de onda total pode ser escrita como:
ψ(r,R) =φ(r;R)θ(R). (2.6)
Nessa equac¸ ˜aoφ(r;R)denota a func¸ ˜ao de onda eletr ˆonica que depende explicitamente de coorde-nadas eletr ˆonicas e parametricamente de coordecoorde-nadas nucleares e θ(R)descreve a func¸ ˜ao de onda nuclear, que s ´o depende de coordenadas nucleares.
A equac¸ ˜ao (2.5) pode ser reescrista de acordo com a aproximac¸ ˜ao adiab ´atica com a seguinte forma:
"
−
N
X
i=1
∇2i − M
X
A=1
∇2
A
2MA
+ M X A=1 M X B>A
ZAZB
RAB + N X i=1 N X j>i 1 rij − M X A=1 N X i=1
ZAZB
riA
#
φ(r;R)θ(R) =Eφ(r;R)θ(R). (2.7)
Distribuindo os termos, obtemos:
−
N
X
i=1
∇2i[φ(r;R)θ(R)]− M
X
A=1
∇2
A[φ(r;R)θ(R)]
2MA
+ M X A=1 M X B>A
ZAZB
RAB
[φ(r;R)θ(R)]+
+ N X i=1 N X j>i
φ(r;R)θ(R)
rij − M X A=1 N X i=1
ZAZB
riA
φ(r;R)θ(R) =Eφ(r;R)θ(R), (2.8)
´
E necess ´ario explicitar o laplaciano em coordenadas nucleares na equac¸ ˜ao (2.8), para obter a se-guinte relac¸ ˜ao:
∇2A[φ(r;R)θ(R)] = (∇2Aφ(r;R)θ(R) + 2∇Aφ(r;R)∇Aθ(R) +φ(r;R)(∇2Aθ(R)), (2.9)
Pode-se considerar fisicamente que a func¸ ˜ao de onda eletr ˆonica sofre variac¸ ˜oes adiab ´aticas em relac¸ ˜ao a coordenada nuclear (R). Portanto: ∇2Aφ(r;R) ≈ 0 e ∇Aφ(r;R)∇Aθ(R) ≈ 0. Logo, a
aproximac¸ ˜ao de Born-Oppenheimer (ABO) ´e considerada da seguinte forma:
∇2A[φ(r;R)θ(R)]≈φ(r;R)(∇2Aθ(R)), (2.10)
de coordenadas eletr ˆonicas e param ˆetricas de coordenadas nucleares e assim dividindo toda equac¸ ˜ao porφ(r;R)θ(R), obtem-se:
− 1
2φ(r;R)
N
X
i=1
∇2iφ(r;R)− 1
θ(R)
M
X
A=1
∇2
Aθ(R)
2MA
+ M X A=1 M X B>A
ZAZB
RAB + N X i=1 N X j>i 1 rij − M X A=1 N X i=1 ZA riA
=E, (2.11)
Rearranjando a equac¸ ˜ao (2.11), obtemos:
− 1
θ(R)
M
X
A=1
∇2
Aθ(R)
2MA
+ M X A=1 M X B>A
ZAZB
RAB
−E= 1
2φ(r;R)
N
X
i=1
∇2
iφ(r;R)+ M X A=1 N X i=1 ZA riA − N X i=1 N X j>i 1 rij . (2.12)
A igualdade torna-se verdadeira quando os dois lados da equac¸ ˜ao s ˜ao uma constante que depen-dam de R. Definiremos essa constante como−ε(R).
Desta forma, s ˜ao obtidas duas equac¸ ˜oes desacopladas, que s ˜ao:
− 1
θ(R)
M
X
A=1
∇2
Aθ(R)
2MA
+ M X A=1 M X B>A
ZAZB
RAB
−E=−ε(R), (2.13)
e
1 2φ(r;R)
N
X
i=1
∇2iφ(r;R) + M X A=1 N X i=1 ZA riA − N X i=1 N X j>i 1 rij
=−ε(R). (2.14)
Multiplicando a equac¸ ˜ao (2.13) porθ(R)e a equac¸ ˜ao (2.14) por−φ(r;R), tem-se que:
" −1 2 M X A=1 ∇2
Aθ(R)
MA + M X A=1 M X B>A
ZAZB
RAB
−E
#
θ(R) =−ε(R)θ(R), (2.15)
e " −1 2 N X i=1
∇2iφ(r;R)− M X A=1 N X i=1 ZA riA + N X i=1 N X j>i 1 rij #
φ(r;R) =ε(R)φ(r;R). (2.16)
Rearranjando a equac¸ ˜ao (2.15), encontramos:
" −1 2 M X A=1
∇2Aθ(R)
MA + M X A=1 M X B>A
ZAZB
RAB
+ε(R)
#
θ(R) =Eθ(R), (2.17)
onde chama-se a soma abaixo de ”curva de energia potencial”, curva que descreve como os n ´ucleos se movem sob a ac¸ ˜ao de um campo produzido por el ´etrons.
V(R) =
M X A=1 M X B>A
ZAZB
RAB
+ε(R). (2.18)
" −1 2 M X A=1 ∇2
Aθ(R)
MA
+V(R)
#
Observando a equac¸ ˜ao (2.16), desacopla-se o movimento eletr ˆonico do movimento nuclear utili-zando a ABO, sendo esta equac¸ ˜ao definida como equac¸ ˜ao de Schr ¨odinger eletr ˆonica estacion ´aria o seu conjunto de soluc¸ ˜oes fornece as energias eletr ˆonicas para cada configurac¸ ˜ao nuclear. O evento ex-traordin ´ario que devemos notar ´e o fato do conjunto de autovalores dados pela equac¸ ˜ao de Schrodinger eletr ˆonica, ou seja, suas energiasε(R)serem usados para calcular a equac¸ ˜ao de autovalores nuclear (2.17), esta equac¸ ˜ao descreve os movimentos de rotac¸ ˜ao, vibrac¸ ˜ao e translac¸ ˜ao.
2.2 Formas Anal´ıticas
Na construc¸ ˜ao do trabalho foram utilizadas quatro func¸ ˜oes anal´ıticas para descrever a curva ab initiopara o sistemaHe−H, de tal forma que essas curvas descrevam um ajuste com o menor erro poss´ıvel na obtenc¸ ˜ao das propriedades de interesse. As curvas s ˜ao: Bond Order de grau 6, Rydberg generalizada de grau 6 e suas correspondentes formas deformadas de mesmo grau.
2.2.1 Polin ˆomios em coordenadas Bond Order Generalizada
O conceito de ordem de ligac¸ ˜ao (BO) fora introduzido em estudos te ´oricos pelo qu´ımico Linus Pauling, ajustando esse conceito como um par ˆametro para classificar a forc¸a de ligac¸ ˜ao de sistemas diat ˆomicos, sendo ent ˜ao a ordem de ligac¸ ˜ao dessas estruturas representadas, como:
ηAB =e
−βAB(R−Re)ρ, (2.20)
em que:
ρ= (RAB−Re). (2.21)
A coordenadaRABdenota a dist ˆancia entre o ´atomo A e B, comResendo a dist ˆancia de equil´ıbrio e
βABum par ˆametro que relaciona a forc¸a de interac¸ ˜ao entre os ´atomos da estrutura diat ˆomica. Tomando
essa definic¸ ˜ao quando a dist ˆancia de equil´ıbrio ´e igual a separac¸ ˜ao entre os ´atomos, ou seja,RAB =
Re, a ordem da ligac¸ ˜ao torna-se igual a unidade, assim ηAB = 1. Se separarmos os ´atomos que
participam da ligac¸ ˜ao com uma dist ˆancia que tenda ao infinito, a ordem da ligac¸ ˜ao tender ´a a zero, caso os ´atomos se aproximem, a ordem da ligac¸ ˜ao ir ´a aumentar at ´e o limite estabelecido pela relac¸ ˜ao;
e−βAB(R−Re)
ρ. Portanto as interac¸ ˜oes at ˆomicas que s ˜ao descritas pelo modeloBond Order encontram-se em um intervalo que varia de zero at ´e o valor limite dee−βAB(R−Re)ρ
A func¸ ˜ao de coordenadas Bond Order ´e definida como uma combinac¸ ˜ao linear de exponenciais, denotadas como:
VBO(R) = N
X
j=1
cjηj (2.22)
separac¸ ˜ao interat ˆomica que leva a uma condic¸ ˜ao de equil´ıbrio.
Adicionadamente, foi utilizada no ajuste da CEP a func¸ ˜ao anal´ıtica deformada das coordenadas BO deformadas (dBO) de grau 6, dada como:
VdBO(R) =
6
X
i=1
ci(expd(−βρ))i, (2.23)
em que
expd(−βρ) = [1−dβρ]
1
d. (2.24)
A equac¸ ˜ao (2.24) representa a func¸ ˜ao d-exponencial, uma generalizac¸ ˜ao da func¸ ˜ao exponencial convencional de acordo com a definic¸ ˜ao de Euler baseada na termo-estat´ıstica de Tsallis. No caso particular em que d aproxima-se de zero a func¸ ˜ao exponencial de Euler ´e recuperada. O polin ˆomio
Bond Order ´e de grau 6, eρ= (R−Re),ci,β e d s ˜ao par ˆametros ajust ´aveis.
2.2.2 Polin ˆomios em coordenadas f´ısicas de Rydberg
Neste trabalho utilizou-se para descrever uma CEP a func¸ ˜ao de Rydberg, que ´e denotada explicita-mente na forma:
VRyd(R) =−D e " 1 + 6 X i=1
ci(R−Re)i
#
e−ci(R−Re)
. (2.25)
O potencial (2.25) denotaDecomo a energia de dissociac¸ ˜ao do um sistema diat ˆomico, ci s ˜ao os
coeficientes ajust ´aveis, R ´e denota a separac¸ ˜ao intermolecular eRe ´e a separac¸ ˜ao de equil´ıbrio
(com-primentos de ligac¸ ˜ao). A forma deformada deste potencial, a func¸ ˜ao d-Rydberg, ´e mostrada como:
VdRyd(R) =−D e " 1 + 6 X i=1
ci(R−Re)i
#
[1−dc1(R−Re)]
1
d. (2.26)
Todos os par ˆametros foram ajustados utilizando os m ´etodos GSA [24], Gradiente Simples [28] e Levenberg Marquardt [22] [23].
2.3 Equac¸˜ao de Schr ¨odinger Nuclear
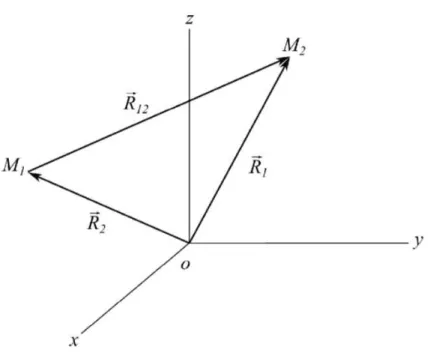
Considerando o m ´etodo utilizado para se resolver a parte nuclear da equac¸ ˜ao de Schr ¨odinger em um sistema gen ´erico como o representado pela Figura 2.2, em queM1eM2representam as massas
nucleares em suas respectivas posic¸ ˜oesR1 R2, dado um refer ˆencia fixa. J ´a R12denota a dist ˆancia entre os n ´ucleos 1 e 2.
Figura 2.2: N ´ucleos que representam um sistema de coordenadas fixos.
realizado se dedica a descever um sistema de dois corpos e o Hamiltoniano cl ´assico desse sistema pode ser escrito como:
H = 1
2M1
P1P1+ 1 2M2
P2P2+V(R). (2.27)
O operador acima descreve as energias cin ´eticas dos n ´ucleos 1 e 2, respectivamente, e V(R) denota a interac¸ ˜ao dos dois n ´ucleos. Buscando tornar o sistema com resoluc¸ ˜ao mais simples, ´e poss´ıvel escrever as energias cin ´eticas em termos da posic¸ ˜ao do centro de massa e a posic¸ ˜ao do n ´ucleo 2 em relac¸ ˜ao ao n ´ucleo 1, tal como:
RCM= M1R1+M2R2
M1+M2
, (2.28)
e
R12=R2−R1. (2.29)
Considerando a relac¸ ˜ao do momento linear, substituindo as relac¸ ˜oes (2.28) e (2.29) em (2.27) o operador Hamiltoniano cl ´assico em func¸ ˜ao das coordenadas do centro de massa, e tomando as cor-respond ˆencias entre as vari ´aveis que descrevem a din ˆamica de um sistema na mec ˆanica cl ´assica e os operadores hermitianos na mec ˆanica qu ˆantica a nova relac¸ ˜ao ser ´a representada por:
ˆ
H =− 1 2M∇
2
CM−
1 2µ∇
2
12+V(R12). (2.30)
Ap ´os realizadas um conjunto de operac¸ ˜oes, nota-se que surge naturalmente a massa reduzida do sistema formado por dois corposµ= M1M2
M1+M2
func¸ ˜ao do centro de massa e da dist ˆancia relativa entre os n ´ucleos, de forma que pode-se definir.
ˆ
HCM ≡ −
1 2M∇
2
CM,
ˆ
Hint≡ −
1 2µ∇
2
12+V(R).
O Hamiltoniano do centro de massa (HˆCM) descreve o movimento de translac¸ ˜ao, enquanto que o
Hamiltoniano de coordenadas internas (Hˆint) indica os movimentos de vibrac¸ ˜ao e rotac¸ ˜ao no n ´ucleo.
A equac¸ ˜ao de Schr ¨odinger que leva em considerac¸ ˜ao os Hamiltonianos do centro de massa e dos movimentos de coordenadas internas ´e dada por:
[ ˆHCM+ ˆHint]θ(RCM,R) =Eθ(RCM,R). (2.31)
Assim, pela equac¸ ˜ao (2.31) pode-se notar claramente que essa equac¸ ˜ao ´e separ ´avel, e por esse argumento existe uma soluc¸ ˜ao do tipo,θ(RCM,R) =η(RCM)ϕ(R). Com a utilizac¸ ˜ao da separac¸ ˜ao de vari ´aveis obt ˆem-se duas equac¸ ˜oes desacopladas:
ˆ
HCMη(RCM) =−
1 2M∇
2
CMη(RCM) =Etransη(RCM), (2.32)
e
ˆ
Hintϕ(R) =
− 1
2µ∇
2
12+V(R)
ϕ(R) =Eintϕ(R). (2.33) A equac¸ ˜ao (2.33) ´e a equac¸ ˜ao de Schr ¨odinger para uma part´ıcula livre cujas soluc¸ ˜oes dependem das condic¸ ˜oes de contorno e descreve o movimento de translac¸ ˜ao do sistema diat ˆomico regido pela posic¸ ˜ao do centro de massa. Supondo que este sistema n ˜ao esteja sujeito a forc¸as externas, pode-se considerar queEtrans= 0. Assim, a energia total ser ´a obtida exclusivamente pela equac¸ ˜ao (2.33) que descreve os
movimentos de vibrac¸ ˜ao e rotac¸ ˜ao. O potencialV(R)em (2.33) ´e dito de forc¸a central, isto ´e, depende apenas das dist ˆancias entre as part´ıculas interagentes de modo que ´e invariante `a orientac¸ ˜ao espacial a qual o di ´atomo encontra-se, tendo portanto simetria esf ´erica. A simetria do potencial indica que deve-se modificar o sistema de coordenadas do laplaciano∇2
12para coordenadas esf ´ericas, reescrevendo a
equac¸ ˜ao (2.33), obtemos a seguinte equac¸ ˜ao:
− 1
2µ
"
∂2
∂R2 +
2 R
∂ ∂R−
ˆ
L2
R2 −2µV(R)
#
ϕ(R) =Eintϕ(R), (2.34)
em queLˆ2 ´e o operador momento angular ao quadrado. Pela equac¸ ˜ao (2.34) pode-se observar que a vibrac¸ ˜ao e a rotac¸ ˜ao est ˜ao desacopladas, de forma que podemos separar a func¸ ˜ao de onda em que a parte radial que est ´a em func¸ ˜ao de (R) e outra parte, angular, que est ´a em func¸ ˜ao de (θ eφ). Logo, podemos escrever a func¸ ˜aoϕ(R)como o produto de duas func¸ ˜oes na forma:
ϕ(R) =ψ(R)Ym
reconhecendo que,
ˆ
L2Ym
J (θ, φ) =J(J+ 1)YJm(θ, φ). (2.36)
A func¸ ˜aoYm
J (θ, φ)na equac¸ ˜ao (2.36) denota os harm ˆonicos esf ´ericos, e a partir das equac¸ ˜oes (2.35)
e (2.36), pode-se expressar a equac¸ ˜ao de Schr ¨odinger nuclear unidimensional, como segue:
− 1
2µ
d2ψ(R)
dR2 +
2 R
dR
dR−
J(J+ 1)ψ(R)
R2 −2µV(R)ψ(R)
=Eintψ(R). (2.37)
Buscando denotar uma simplificac¸ ˜ao para equac¸ ˜ao de Schr ¨odinger nuclear unidimensional, faremos a seguinte substituic¸ ˜ao: F(R) =Rψ(R), e finalmente chega-se a relac¸ ˜ao:
− 1
2µ
d2F(R)
dR2 +
J(J+ 1)
2µR2 +V(R)
F(R) =EintF(R), (2.38) ou
− 1
2µ
d2F(R)
dR2 +Vef(R)F(R) =EintF(R). (2.39)
O potencial efetivoVef(R) = J2(µJR+1)2 +V(R)formado pela curva de energia potencial unidimensi-onal e da correc¸ ˜ao de distorc¸ ˜ao centr´ıfuga determinada pelo estado rotaciunidimensi-onal. A equac¸ ˜ao (2.39) ´e a equac¸ ˜ao de Schr ¨odinger nuclear radial na qual o sistemaHe−H se reduziu e que deve ser resolvida para determinarmos completamente a func¸ ˜ao de onda nuclear. A equac¸ ˜ao (2.39) n ˜ao possui soluc¸ ˜ao anal´ıtica, logo, ´e necess ´ario utilizar m ´etodos de aproximac¸ ˜ao para denotar uma soluc¸ ˜ao, e para propor soluc¸ ˜oes usaremos o m ´etodo de Rayleigh-Ritz.
O m ´etodo variacional de Rayleigh-Ritz ´e aplicado para determinar o valor esperado de observ ´aveis f´ısicas no contexto da mec ˆanica qu ˆantica. Uma forma de caracterizar o m ´etodo de maneira breve, ´e multiplicar a equac¸ ˜ao (2.38) porF∗
(R)e integra-se em todo espac¸o acess´ıvel ao sistema aplicando as condic¸ ˜oes de contorno. Em seguida, aproximamos a func¸ ˜ao de onda exataF(R)por func¸ ˜oes conheci-das, isto ´e, fazemos uma combinac¸ ˜ao linear denfunc¸ ˜oes de base conhecidas,fi(R):
F(R) =
n
X
j=1
cjfi(R), (2.40)
sendo oscj o conjunto de coeficientes da expans ˜ao a serem determinados. Com este procedimento
troca - se o problema de se determinar quais func¸ ˜oes F(R)satisfazem a equac¸ ˜ao diferencial de se-gunda ordem com coeficientes vari ´aveis, equac¸ ˜ao (2.39), por um problema de encontrar um conjunto de coeficientescj, que denota a resoluc¸ ˜ao de um problema alg ´ebrico. A partir desse m ´etodo pode-se
resolver o problema tranformando a (2.39) por uma equac¸ ˜ao matricial, tal que:
Hc=ESc. (2.41)
autovalores ece matriz coluna dos coeficientes. Os elementos destas matrizes sao dados por:
Tij =
1 2µ
Z ∞
0
Z ∞
0
df∗
i(R)
dR
dfi(R)
dR dR, (2.42)
Vij =
Z ∞
0
f∗
i(R)Vef(R)fi(R)dR, (2.43)
e
Sij=
Z ∞
0
f∗
i(R)(R)fi(R)dR. (2.44)
Neste trabalho, cada elemento deHser ´a numericamente avaliado atrav ´es do M ´edoto da Representac¸ ˜ao da Vari ´avel Discreta (DVR). Utilizando-se func¸ ˜oes de bases ortonormais de modo queS=I, ser ´a a ma-triz identidade, isto ´eSij=δij.
2.4 M´etodo da Representac¸˜ao da Vari´avel Discreta
O m ´etodo DVR, que teve como um de seus pioneiros Light et al [29], se apresenta como uma ferramenta extremamente poderosa para resolver problemas qu ˆanticos tanto para estados estacion ´arios como para estados dependentes do tempo. Neste trabalho, o m ´etodo DVR foi aplicado na obtenc¸ ˜ao dos autovalores e autofunc¸ ˜oes da equac¸ ˜ao de Schr ¨odinger radial satisfazendo o princ´ıpio variacional. O m ´etodo DVR ´e baseado na expans ˜ao da func¸ ˜ao de onda em um conjunto de bases ortonormaisφi(Ri);
i= 1,2, ..., Ne a utilizac¸ ˜ao de regras de quadraturas gaussianas para computar as integrais envolvidas [30]. Estas func¸ ˜oes de base tem a seguinte propriedade:
φj(Rk) =δjk(j, k= 1,2, ..., n), (2.45)
As func¸ ˜oes de base cont´ınuas s ˜ao indexadas em valores discretos das vari ´aveis em um grade de pontos no espac¸o com coordenadasRk (quadratura gaussiana). Sendo queRk representa os pontos
da quadratura gaussiana em que as func¸ ˜oes de base ser ˜ao avaliadas. Caso seja expandida a soluc¸ ˜ao
F(Ri)como uma combinac¸ ˜ao de func¸ ˜oes de baseφi(R), logo:
F(R)≈
N
X
j=1
cjφj(R), (2.46)
ondecjs ˜ao os coeficientes da expans ˜ao a serem determinados, e as func¸ ˜oes representam a discretizac¸ ˜ao
da vari ´avelR.
Portanto as func¸ ˜oes discretizadas s ˜ao obtidas a partir de um conjunto de func¸ ˜oes cont´ınuas primiti-vas, que s ˜ao conhecidas, associando a estas uma quadratura gaussiana.
Pode-se escreverφj(R)como a projec¸ ˜ao deφjsobreRna notac¸ ˜ao de Dirac:
inserindo a relac¸ ˜ao de completeza, dada por:
n
X
i=1
|iihi|= 1, (2.48)
na equac¸ao (2.47), obt ˆem-se:
φj(R) = n
X
i=1
hR|fiihfi|φji, (2.49)
As integrais dos elementos da matrizhfi|φjipodem ser calculadas utilizando quadraturas
gaussia-nas,
hfi|φji ≈ n
X
k=1
wkf
∗
i(Rk)φj(Rk), (2.50)
em que wk s ˜ao os pesos associados ao pontoRk na malha de quadraturas. Substituindo (2.50) em
(2.51), obt ˆem - se:
φj(R) = n X i=1 n X k=1
fi(R)wkf
∗
i(Rk)φj(Rk). (2.51)
Como as func¸ ˜oes de baseφj(Rk)s ˜ao ortogonais, ent ˜ao:
φj(R) =wj n
X
i=1
fi(Rk)f
∗
i(Rk), (2.52)
Assim, escolhendo um pontoRjqualquer dentro da malha das quadraturas, teremos que:
φj(Rj) =wj n
X
i=1
fi(Rk)f
∗
i(Rk), (2.53)
Considerando que as func¸ ˜oes de base escolhidas est ˜ao normalizadas podemos afirmar que:
n
X
i=1
fi(Rj)f
∗
i(Rj) = 1, (2.54)
de modo que o peso associado `a cada ponto da quadratura ´e:
wj=
1
Pn
i=1fi(Rj)fi∗(Rj)
. (2.55)
No entanto, as func¸ ˜oes de base n ˜ao est ˜ao normalizadas e, portanto, ser ´a mecess ´ario normaliz ´a -las, expressando a seguinte substituic¸ ˜ao, por:
¯
φj(R) =λjφj(R), (2.56)
em que λj ´e a constante de normalizac¸ ˜ao de modo que hφ¯j|φ¯ji = 1, ou melhor, λj2hφ¯j|φ¯ji = 1.
λ2j n
X
i=1
wkφ
∗
j(Rk)φj(Rk) = 1. (2.57)
De acordo com a equac¸ ˜ao (2.45) as func¸ ˜oes primitivas s ˜ao ortonormais, assim poderemos conside-rar que (2.57) ser ´a:
λ2jwj= 1, (2.58)
ou
λj=
1
w12
j
. (2.59)
Substituindo a constante de normalizac¸ ˜ao, equac¸ ˜ao (2.59) conjuntamente com a equac¸ ˜ao (2.52) na equac¸ ˜ao (2.56), obteremos a func¸ ˜ao de base discreta e normalizadaφ¯j(R):
¯
φj(Rj) =wj
1 2
n
X
i=1
fi(Rk)f
∗
i(Rk), (2.60)
Desta maneira o conjunto que descreve a soluc¸ ˜ao da equac¸ ˜ao de Schr ¨odinger na equac¸ ˜ao (2.46) utilizando as func¸ ˜oes de bases discretizadas (2.60) ´e denotado como:
F(R)≈
n
X
j=1
cjφ¯j(Rj), (2.61)
Substituindo (2.61) em (2.43), e utilizando os conceitos de quadratura gaussiana, teremos a seguinte equac¸ ˜ao:
Vij= n
X
k=1
φ∗
i(Rk)Vef(Rk)φ
∗
j(Rk)wk, (2.62)
ondeφ∗
j(Rk) =δjk. Portanto a matriz energia potencialVtorna-se diagonal sendo este resultado uma
das principais caracter´ısticas do m ´etodo DVR. Sendo ent ˜ao Rk os pontos da quadratura gaussianan que s ˜ao os valores esperados da matriz composta pelos elementos:
Rij =hfi|Rˆ|φji. (2.63)
A matriz energia cin ´etica T pode ser calulada atrav ´es de quadraturas gaussianas com um con-junto de pontos igualmente espac¸ados, tomando esses pontos pertencentes a um certo intervalo [a,b], teremos que:
Ri=a+
b−a
N i, (2.64)
Tij= N−1
X
n=1
hRi|Tˆ|fnihfn|Rii. (2.65)
Considerando a soma anal´ıtica sobre todos os ´ındices n, os elementos ser ˜ao:
Tij =
1 2µ
1 (b−a)2
π2
2
1
sin2hπ(j−k) 2N
i−
1
sin2hπ(j+k) 2N
i
, (2.66)
parai6=j, e:
Tij=
1 2µ
1 (b−a)2
π2
2 2N2
3 − 1
sin2πi
N
!
, (2.67)
parai=j. Portanto o DVR diagonaliza o potencial da matriz hamiltoniana, e permite que os elemen-tos da matriz de energia cin ´etica sejam obtidos analiticamente por express ˜oes simples dadas pelas equac¸ ˜oes (2.66) e (2.67).
2.5 Propriedades Espectrosc ´opicas Rovibracionais
Vimos anteriormente que a equac¸ ˜ao de Schr ¨odinger ´e separada em uma parte eletr ˆonica e ou-tra nuclear por meio da aproximac¸ ˜ao de Born-Oppenheimer e que est ´a ´ultima pode ser largamente simplificada para sistemas diat ˆomicos. Nesta simplificac¸ ˜ao conseguimos desacoplar o movimento translacional regido pelo centro de massa, dos movimentos internos. Os movimentos internos, por sua vez, s ˜ao descritos pela vibrac¸ ˜ao e rotac¸ ˜ao do sistema de dois corpos. Usualmente utilizam-se duas aproximac¸ ˜oes para tratar estes movimentos, s ˜ao elas: aproximac¸ ˜ao do oscilador harm ˆonico para vibrac¸ ˜oes, e aproximac¸ ˜ao do rotor r´ıgido para rotac¸ ˜oes. Experimentalmente estas aproximac¸ ˜oes justificam-se ao observar que o espectro rotacional encontra-se em geral na regi ˜ao de micro-ondas e o espectro vibracional na regi ˜ao do infravermelho [32].
2.5.1 Espectro Rotacional
A rotac¸ ˜ao de sistemas diat ˆomicos pode ser aproximada por um sistema ideal denominado ro-tor r´ıgido, a qual assume que a revoluc¸ ˜ao de uma mol ´ecula de massa reduzida µ ocorre com uma separac¸ ˜ao internuclear fixa igual `a dist ˆancia de equil´ıbrioRe. Quando se resolve a equac¸ ˜ao de Schr
¨odin-ger para um rotor r´ıgido as energias permitidas nesse tipo de sistema s ˜ao denotadas da seguinte forma:
EJ =~2
J(J+ 1)
2I , (2.68)
em que, J = 0,1,2, ..., p e I = µRe2 ´e o momento de in ´ercia do sistema denotado. A diferenc¸a de
energia entre dois n´ıveis rotacionais ´e dada por:
ou
∆E= h
2
4π2I(J+ 1). (2.70)
Levando em conta os termos de frequ ˆencia, atrav ´es da condic¸ ˜ao de frequ ˆencia de Bohr∆E =hν, teremos:
ν= 2B(J+ 1), (2.71)
em queB =h/8π2I ´e a constante rotacional caracter´ıstica do sistema em estudo. De acordo com a
equac¸ ˜ao (2.71) nota-se que o modelo de um rotor r´ıgido para sistemas diat ˆomicos prev ˆe que o espec-tro rotacional consiste em uma s ´erie de linhas igualmente espac¸adas com separac¸ ˜oes2B cm−1 [33].
Contudo, em sistemas caracterizados por serem diat ˆomicos n ˜ao s ˜ao verdadeiramente um rotor r´ıgido devido vibrac¸ ˜oes simult ˆaneas, ainda que de pequena amplitude. Consequentimente o espac¸amento das linhas espectrais de rotac¸ ˜ao n ˜ao ser ˜ao exatamente constantes.
2.5.2 Espectro Vibracional
O modelo vibracional de um sistema diat ˆomico poder ´a ser aproximado pelo modelo do oscilador harm ˆonico qu ˆantico. As soluc¸ ˜oes permitidas das equac¸ ˜ao de Schr ¨odinger para o oscilador harm ˆonico s ˜ao restringidas a valores discretos dados por:
Eυ=hc
υ+1 2
ωe, (2.72)
em queυ= 0,1,2,3, ..., peωe ´e a frequ ˆencia vibracional fundamental. A diferenc¸a de energia entre dois
n´ıveis adjacentes ´e dada como:
∆E=Eυ+1−Eυ, (2.73)
ou
∆E=hcωe. (2.74)
Como
os sucessivos estados de energia do oscilador harm ˆonico s ˜ao separados pela mesma energia, este modelo prediz que o espectro vibracional consiste em apenas uma linha espectral cuja frequ ˆencia ´e dada porωe. Esta aproximac¸ ˜ao s ´o ´e v ´alida nos estados vibracionais mais baixos, pois para dist ˆancias
internucleares fora da separac¸ ˜ao de equil´ıbrio, o espac¸amento entre os n´ıveis se aproxima cada vez mais devido `a anarmonicidade da curva de energia potencial.
Vm(R) =De(1−e
−ζ(R−Re))2, (2.75)
em queDedenota a energia de dissociac¸ ˜ao eζdetermina se o potencial ´e de curto ou de longo alcance.
Fazendo uma expans ˜ao em s ´erie de Taylor, o termo exponencial da equac¸ ˜ao (2.75) em torno deR=Re
e utilizando uma aproximac¸ ˜ao em primeira ordem, obtemos:
Vm(R) =Deζ2(R−Re)2. (2.76)
Utilizando esse potencial na equac¸ ˜ao de Schr ¨odinger para o oscilador harm ˆonico, obteremos os seguintes autovalores emcm−1:
Eυ=
υ+1 2
ωe−
υ+1 2
2
ωeχe+
υ+1 2
3
ωeγe+... (2.77)
em queωeχeeωeγes ˜ao denotadas de constantes anarm ˆonicas de vibrac¸ ˜ao do sistema diat ˆomico. Com
essa correc¸ ˜ao o espac¸amento entre os n´ıveis diminui quando os estados vibracionais est ˜ao mais ele-vados. Para estados vibracionais pr ´oximos da dist ˆancia de equil´ıbrio, o espac¸amento ´e muito pr ´oximo aos descritos pela equac¸ ˜ao (2.74), j ´a que as correc¸ ˜oes anarm ˆonicas s ˜ao pequenas se comparadas `a
ωe.
2.5.3 Espectro Rovibracional
As aproximac¸ ˜oes descritas pelo oscilador harm ˆonico qu ˆantico e rotor r´ıgido, permitem tratar o pro-blema dos movimentos internos separadamente. No entanto, os movimentos vibracionais e rotacionais n ˜ao ocorrem de maneira independente, dando origem a uma estrutura fina nas bandas vibracionais.
Se os movimentos est ˜ao acoplados uma maneira mais simples para estudar o espectro rovibracional seria utilizar o modelo do oscilador harm ˆonico conjuntamente com o modelo do rotor r´ıgido, conside-rando que o hamiltoniano do sistema diat ˆonico ´e composto pelas duas contribuic¸ ˜oes, ou seja:
ˆ
Hrov= ˆHvib+ ˆHrot. (2.78)
Pela equac¸ ˜ao (2.78), tem-se que os autovalores descritos pelo hamiltoniano rovibracional s ˜ao a soma dos autovalores individuais de cada hamiltoniano separado, o que pode-se denotar como:
Erov(υ, J) =
υ+1 2
ωe+J(J+ 1)Be, (2.79)
em queBe=~2/2hcIe ´e a constante rotacional de equil´ıbrio. O acoplamento rovibracional surge quando
o sistema encontra-se em rotac¸ ˜ao, por ´em a dist ˆancia internuclearR−Resofre flutuac¸ ˜oes que dependem
da func¸ ˜ao de onda vibracional e do n ´umero qu ˆanticoυ. PortantoBepossui uma depend ˆencia deυque
pode ser expressa como uma perturbac¸ ˜ao, que quando expandida em torno de υ+12
, obtemos:
Bυ=Be−
υ+1 2
αe+
υ+1 2
2
em queBυ ´e a correc¸ ˜ao vibracional da constante rotacional,αeeγes ˜ao denominadas constantes de
acoplamento rovibracionais [34].
A energia rovibracional de um determinado n´ıvel(υ, J), deve levar em considerac¸ ˜ao todas as contribuic¸ ˜oes vibracionais (harm ˆonicas e anarm ˆonicas) e rotacionais(r´ıgidas e n ˜ao r´ıgida). A nova relac¸ ˜ao que deve ser escrita para expressar todas as contribuic¸ ˜oes, ´e dada como:
Eυ,J =
υ+1 2
ωe−
υ+1 2
2
ωeχe+
υ+1 2
3
ωeγe+...
+
"
Be−
υ+1 2
αe+
υ+1 2
2
γe+...
#
J(J+ 1) +... (2.81)
Nesta dissertac¸ ˜ao, as constantes espectrosc ´opicas foram calculadas combinando as energias rovi-bracionaisEυ,J obtidas apartir da soluc¸ ˜ao da equac¸ ˜ao de Schr ¨odinger nuclear com a equac¸ ˜ao (2.81).
Desta combinac¸ ˜ao, ´e obtido um sistema de equac¸ ˜oes fechadas para constantes espectrosc ´opicas,tal como:
ωe=
1
24[14(E1,0−E0,0)−93(E2,0−E0,0) + 23(E3,0−E1,0)]
ωeχe=
1
4[13(E1,0−E0,0)−11(E2,0−E0,0) + 3(E3,0−E1,0)]
ωeγe=
1
6[3(E1,0−E0,0)−3(E2,0−E0,0) + 3(E3,0−E1,0)] (2.82)
αe=
1
8[−12(E1,1−E0,1) + 4(E2,1−E0,1) + 4ωe−23αe]
γe=
1
Cap´ıtulo 3
Resultados e Discuss ˜oes
Nesta sec¸ ˜ao ser ˜ao apresentados os resultados das propriedades din ˆamicas do sistemaHe−H, cu-jas energias lept ˆonicas (el ´etron e p ´ositron) foram obtidas por Strasburger e Chojnacki [13]. Os autores calcularam um conjunto de energias lept ˆonicas para diferentes dist ˆancias internucleares para interac¸ ˜ao
He−H utilizando a aproximac¸ ˜ao de Born-Oppenheimer. Esse trabalho os autores utilizaram como func¸ ˜ao de onda para part´ıculas leves uma combinac¸ ˜ao linear de func¸ ˜oes gaussianas explicitamente correlacionadas como uma aproximac¸ ˜ao, baseando-se em resultados muito precisos para mat ´eria or-din ´aria [15] e sistemas positr ˆonicos [16] . No trabalho de Strasburger e Chojnacki foi calculada variaci-onalmente a CEP, para o sistemaHe−H cuja dist ˆancia de equil´ıbrio ´ere= 3,63a0. Na Figura 1.2 foi
apresentada a CEP obtida pelos autores mostrando um barreira na regi ˜ao repulsiva da CEP. Segundo os autores, este m ´aximo poderia ser explicado pela leve predomin ˆancia da repuls ˜ao eletrost ´atica entre o ´atomo e o anti- ´atomo perante `as forc¸as atrativas. Em [13] Strasburgeret alchamou a atenc¸ ˜ao para a possibilidade de existir um estado fracamente ligado na regi ˜ao pr ´oxima a 3.64a0 e que se trata de
um estado excitado para este sistema e que a relaxac¸ ˜ao seria suprimida pela barreira na CEP. Conse-quentemente, esta barreira diminuiria a taxa de aniquilac¸ ˜ao el ´etron-p ´ositron de modo que este sistema pode apresentar um tempo de vida relativamente longo para ser detectado. De fato, esta possibilidade pode ser bem razo ´avel quando se observa que nos experimentos de gerac¸ ˜ao e aprisionamento de an-tihidrog ˆenio (H), estes est ˜ao imersos em um ambiente de h ´elio l´ıquido ultraresfriado e que um fator limitante ao tempo de vida dos anti- ´atomos est ´a nas colis ˜oes com gases formados por ´atomos usuais, como o h ´elio.
O interesse deste trabalho ´e investigar as propriedades din ˆamicas do sistemaHe−H, na regi ˜ao da CEP que viabiliza a exist ˆencia de um poss´ıvel estado ligado. Para isto, constru´ımos o potencial efetivo da equac¸ ˜ao de Schr ¨odinger nuclear (2.39) ajustando a CEP ab initio de Strasburger et alutilizando quatro diferentes formas anal´ıticas:Bond Order(BO), d-Bond Order(dBO),Rydberg(Ryd) e d-Rydberg
(dRyd), todas de grau 6. Utilizou-se os m ´etodos h´ıbridos Gradiente Simplex, Levenberg-Marquardt e Generalized Simulated Anneling(GSA) para otimizar os par ˆametros das func¸ ˜oes anal´ıticas que est ˜ao apresentados na Tabela 3.1.
sistema He−H com as func¸ ˜oes Ryd6, dRyd6, BO6 e dBO6. A energia de dissociac¸ ˜ao ´e dada em hartree e a dist ˆancia de equil´ıbrio est ´a em raio de bohr, sendo fixadas durante o ajuste.
Par ˆametro
Ryd6
dRyd6
BO6
d BO6
c
11,35368229139
1,37670088028
-0,00001398638
-0,00001505650
c
20,04565093283
0,04408291422
-0,00030104351
-0,00027751898
c
3-0,12709317804
-0,17496657245
-0,00032035899
-0,00032896780
c
40,08092572727
-0,07207727995
-0,00239882674
-0,00247810377
c
5-0,02012956946
-0,01196629764
0,00375015419
0,00384528336
c
60,00234744919
0,00091727254
-0,00136293628
-0,00129271720
d
-
-0,03955416611
-
-0,00567959078
β(a
0)
-
-
0,40515218323
0,40323792316
D
e0,0005493
R
e(a
a)
3,63
χ
2(10
−12)
3,0365
1,4686
1,4590
1,4556
Tabela 3.1: Tabela de par ˆametros otimizados para todas as CEPs do sistemaHe−H.
As figuras de 3.1-3.4 mostram as curvas ajustadas com os par ˆametros otimizadas na Tabela 3.1. Nessas figuras s ˜ao demonstrados os erros ponto-a-ponto.
Figura 3.2: Comparac¸ ˜ao entre CEP obtida por Strasburger et ale CEP dRyd6, juntamente com erro ponto-a-ponto do ajuste.
Figura 3.4: Comparac¸ ˜ao entre CEP obtida por Strasburger et ale CEP dBO6, juntamente com erro ponto-a-ponto do ajuste.
As CEPs ajustadas pelos par ˆamentros otimizados na Tabela 3.1 s ˜ao comparadas aos pontosab initiode Strasburgeret al(SCEP) nas Figuras 3.1-3.4, para os potenciais Ryd6, dRyd6, BO6 e dBO6 respectivamente. Analizando todas as figuras bem como a Tabela 3.1 nota-se que o erro χ2 para
qualquer uma das formas anal´ıticas ´e da ordem de 10−7
Hartree, indicando uma boa concord ˆancia entre a CEP ajustada e a calculada por Strasburgeret al. Pelas Figuras apresentadas, os desvios que indicam os m ´aximos e m´ınimos encontrados para as energiasab initioe as energias ajustadas foram de8,37×10−7(8,63×10−4) Hartree para dBO6. Enquanto os desvios para BO6, dRyd6 e Ryd6 foram:
8,09×10−7(2,00×10−8),7,21×10−7(2,69×10−8) e8,25×10−7(2,05×10−8) Hartree, respectivamente.
Todos os ajustes aqui apresentados forneceram uma excelente descric¸ ˜ao para as curvas de energia potencial que buscam denotar a interac¸ ˜ao do sistema He−H, pois os desvios est ˜ao abaixo do erro qu´ımico aceit ´avel (abaixo de 1 kcal/mol ou 0,0015 Hartree) em todas as regi ˜oes da CEP calculada tanto nas zonas harm ˆonicas quanto nas zonas anarm ˆonicas (assint ´otica, atrativa e repulsiva). Por uma an ´alise mais sofisticada observa-se que os ajustes que envolvem as func¸ ˜oes deformadas (dRyd6 e dBO6) foram ligeiramente superiores. Podemos justificar esse fato comparando a soma doscj para as
formas das func¸ ˜oes dBO6 e BO6, j ´a que a soma desses coeficientes deve ser igual `a dissociac¸ ˜aoDe=
5,493×10−4
Hartree, e os resultados foram deDe= 5,47081×10
−4
para dBO6 eDe= 6,45998×10
par ˆametro ddas func¸ ˜oes deformadas dRyd6 e dBO6 s ˜ao razoavelmente pequenos: -0.03955416611 e -0.00567959078, respectivamente. Contudo as func¸ ˜oes deformadas s ˜ao extremamente sens´ıveis ao par ˆametrod.
As constantes espectrosc ´opicas rovibracionais obtidas atrav ´es das soluc¸ ˜oes da Equac¸ ˜ao de Schr ¨odin-ger nuclear, via Eq.(2.79) ser ˜ao apresentadas na Tabela 3.2 para todas as formas analit´ıcas estudadas. Para calcular estas constantes utilizou-se a massa do anti-hidrog ˆenio como sendo igual `a massa do hidrog ˆenio comum, com o intuito de se determinar a massa reduzidaµdo sistemaHe−H. Os valores dessas massas foram de 4,00386 a.m.u para o ´atomo de h ´elio e 1,008132 a.m.u para o anti-hidrog ˆenio, retirados da refer ˆencia [34].
Tabela 3.2: Constantes espectrosc ´opicas rovibracionais para o sistema He−H calculadas usando Eq.(79) atrav ´es do m ´etodo DVR. Todos os valores est ˜ao apresentados emcm−1
.
Potencial
ω
eω
eχ
eω
eγ
eα
eγ
eB
eRyd6
105,77
36,16
6,12
10,68
2,87
5,674
dRyd6
105,51
36,05
6,11
10,65
2.86
5,674
BO6
107,70
37,00
6,24
10,62
2,85
5,674
dBO6
106,63
36,54
6,18
10,67
2,87
5,674
Na Tabela 3.2 podemos observar que os valores obtidos para as constantesωe,ωeχe,ωeγe,αeeγe,
est ˜ao relativamente pr ´oximas para todas as func¸ ˜oes anal´ıticas empregadas neste estudo.
Devido ao fato da energia de dissociac¸ ˜ao e a dist ˆancia de equil´ıbrio serem fixadas nos potenciais Rydberg e Rydberg deformada, seus ajustes expressaram valores ligeiramente melhores que os resul-tados obtidos usando BO6, pois este potencial apresenta o valor da energia de dissociac¸ ˜ao como um par ˆametro que n ˜ao pode ser fixado, assim, apresenta valores n ´umericos relativamente maiores para frequ ˆencia fundamental ωe e para primeira correc¸ ˜ao anarm ˆonica ωeχe. Devido a fraca interac¸ ˜ao do
sistema He−H, sua CEP tem um aspecto ”raso”, no sentido de que as correc¸ ˜oes anarm ˆonicas cor-respondem a uma frac¸ ˜ao consider ´avel sobre a aproximac¸ ˜ao harm ˆonica. Notemos, por exemplo, que os valores de ωeχeeωeγecalculados usando a func¸ ˜ao Ryd6 correspondem a valores ditos altos quando
comparados a sistemas que s ˜ao considerados ligados, nos quais essas constantes s ˜ao muito menores que estes valores aqui apresentados. O valor deBefoi obtido usando a equac¸ ˜ao (2.71) e substituindo
a massa reduzida e a dist ˆancia de equil´ıbrio do parHe−H.
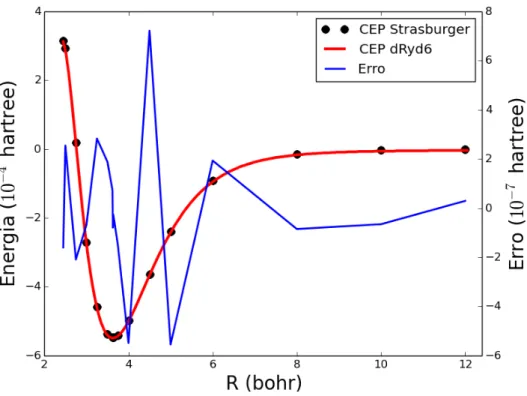
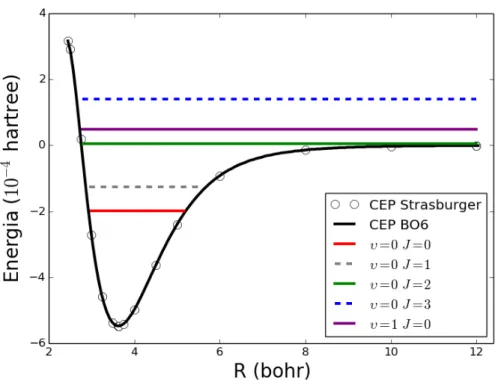
Na Tabela 3.3 est ˜ao listadas o conjunto de energias rovibracionais obtidas atrav ´es da soluc¸ ˜ao da equac¸ ˜ao de Schr ¨odinger nuclear com o n ´umero qu ˆantico vibracional (υ) variando de 0 a 4, e o n ´umero qu ˆantico rotacionalJ variando entre 0 e 3. Pode-se verificar pela Tabela 3.3 que, independentemente da forma anal´ıtica empregada, apenas um n´ıvel vibracional, o estado fundamental (υ= 0), e dois n´ıveis rotacionaisJ = 0,1encontram-se abaixo do limite de dissociac¸ ˜aoDe= 120,03cm−1. Esse fato indica
que o sistemaHe−H ´e formado por uma interac¸ ˜ao de van der Waals.
Tabela 3.3: N´ıveis de energias rovibracionais para o sistemaHe−H (emcm−1) obtidos da soluc¸ ˜ao da
equac¸ ˜ao de Schr ¨odinger nuclear pelo m ´etodo DVR para os potenciais Ryd6, d-Ryd6, BO6 e dBO6.
υ
J
Ryd6
dRyd6
BO6
dBO6
0
77,83
77,93
77,09
77,50
1
131,18
131,20
131,08
131,12
2
0
167,34
167,36
167,23
167,27
3
223,06
223,10
222,99
222,96
4
296,09
296,12
296,06
295,98
0
93,74
93,85
92,98
93,44
1
137,23
137,27
137,14
137,19
2
1
175,01
175,03
174,87
174,92
3
231,51
231,55
231,42
231,41
4
305,04
305,07
305,00
304,93
0
122,29
122,34
121,55
122,81
1
149,28
149,35
149,16
149,25
2
2
190,61
190,61
190,41
190,48
3
248,66
248,70
248,54
248,55
4
323,17
323,21
323,11
323,04
0
151,79
151,82
151,48
151,71
1
172,28
172,36
171,83
172,18
2
3
214,97
214,98
214,64
214,81
3
274,94
274,98
274,74
274,81
4
350,83
350,87
350,72
350,70
fundamental, j ´a foram reportados na literatura estados excitados ligados. De acordo com o teorema CPT, todas as propriedades da antimat ´eria devem ser iguais `a mat ´eria usual correspondente. Pode-mos, pois, apresentar a ideia de que se para mat ´eria usual o sistema He−H encontra-se ligado em seus estados excitados, ent ˜ao o sistemaHe−Htamb ´em pode estar ligado em algum estado excitado. No trabalho de Jonsellet al[17], os autores realizaram um estudo sobre o processo colisional entre
He−Hpartindo da CEP de Strasburger e Chojnacki. Levaram em conta os estados fracamente ligados com tempo de vida relativamente longo, cuja amplitude encontra-se quase que totalmente dentro do poc¸o, pr ´oximo de2,42a0. Verificaram ent ˜ao que para n ´umero qu ˆanticos rotacionaisJ ≤3, o sistema
He−H suporta estados ligados. Os resultados apresentados na Tabela 3.3 denotam n´ıveis rovibracio-nais ligados nos casos em queυ= 0eJ = 0e assim concordam com dados apresentados por Jonsell
et al. A t´ıtulo de comparac¸ ˜ao, as energias obtidas neste trabalho atrav ´es do m ´etodo DVR e os n´ıveis obtidos por Jonsellet al foram: 3,5221×10−4 hartree e 3,043×10−4 hartree paraJ = 0
respectiva-mente. Uma diferenc¸a significativa aparece quando ´e consideradoJ = 3, em nosso trabalho obtemos 6,92×10−4
que esta fora do limite da ligac¸ ˜ao e 5,39×10−4
Hartree que est ´a abaixo da energia de dissociac¸ ˜ao.
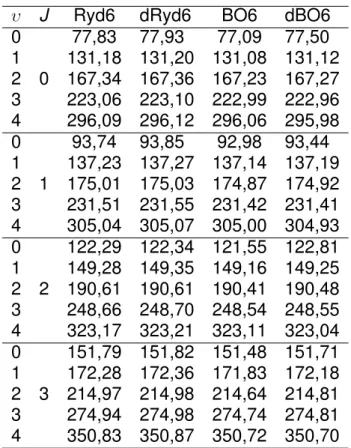
Figura 3.5: N´ıveis de Energia Rovibracionais para o sistemaHe−H com a forma anal´ıtica Ryd6.
Figura 3.7: N´ıveis de Energias Rovibracionais para o sistemaHe−H com a forma anal´ıtica BO6.
Figura 3.8: N´ıveis de Energias Rovibracionais para o sistemaHe−H com a forma anal´ıtica dBO6.
mol ´eculas diat ˆomicas heteronucleares. O m´ınimo da CEPab initioassemelha-se a estruturas que inte-ragem por forc¸as dispersivas como mol ´eculas de van der Waals. Caso seja considerado, por exemplo, a interac¸ ˜ao do sistemaHe−Hecomo um sistema diat ˆomico fraco, sua forma dim ´erica somente ´e formada
`a temperatura de10−3
K [35]. O sistemaHe2apresenta energia de dissociac¸ ˜aoDe= 3,4728×10
−5
Ha
comRe= 5,61a0[34], demonstrando que o sistemaHe−Hest ´a por uma ordem de grandeza mais
for-temente ligado (De= 5,5469×10
−4Hartree eR
e= 3,63a0). Poc¸os de potencial com um leve m´ınimo,
como os sistemasHe−H eHe−He, s ˜ao denominados de mol ´eculas de van der Waals devido sua baixa energia de interac¸ ˜ao. Isto leva a crer que exista um estado ligado para este sistema descrito pelas energias rovibracionais obtidas neste trabalho. Adicionalmente, ´e importante enfatizar que a interac¸ ˜ao
Cap´ıtulo 4
Conclus ˜oes e Perspectivas
Neste trabalho foram determinas pela primeira vez na literatura as energias e constantes espec-trosc ´opicas rovibracionais para interac¸ ˜ao He−H usando a CEP ab initio obtida por Strasburger e Chojnacki [13]. Quatro formas anal´ıticas foram utilizadas para ajustar os pontos ab initio do poten-cial interat ˆomico e os par ˆametros foram otimizados utilizando os m ´etodos h´ıbridosGradiente Simplex,
Levenberg - MarquardteGeneralized Simulated Annealing (GSA). O procedimento de ajuste foi consi-derado satisfat ´orio embasando-se no erro ponto-a-ponto n ˜ao superou o erro qu´ımico aceit ´avel (abaixo de 1 kcal/mol∼0,0015 hartree) em todas as regi ˜oes da CEP obtida.
As constantes espectrosc ´opicas aqui obtidas apresentaram valores muito pr ´oximos para todas as formas anal´ıticas empregadas com diferenc¸a m ´axima que n ˜ao passou de7%paraωe,13%paraωeχe,
8% para ωeγe, 2% paraαe e 1% paraγe, quando utilizou-se a func¸ ˜ao Bond Order de grau 6. Estas
pequenas diferenc¸as mostram que os ajustem apresentam qualidade similar. A exist ˆencia de estados ligados ´e poss´ıvel para os n´ıveis rovibracionais,υ= 0eJ = 0,1. Esta interac¸ ˜ao se assemelha aquelas que ocorrem com mol ´eculas de van der Waals com baixa energia de dissociac¸ ˜ao e dist ˆancia internu-clear relativamente grande. A precis ˜ao das propriedades din ˆamicas apresentadas neste trabalho est ˜ao intimamente ligadas `a qualidade da CEPab initiode Strasburgeret al, a qual ´e considerada de altam-mente precisa. Desta forma acredita-se que os resultados obtidos possam servir de comparac¸ ˜ao com estudos futuros acerca das propriedades espectrosc ´opicas para o sistema diat ˆomicoHe−H.
Como perspectivas futuras pretendemos confrontar estes resultados utilizando outra metodologia para a determinac¸ ˜ao de constantes espectrosc ´opicas, tais como o m ´etodo de Dunham [27]. Este m ´etodo ´e baseado em f ´ormulas derivadas da teoria da perturbac¸ ˜ao, no qual o potencial (func¸ ˜ao anal´ıtica)