Lipoprotein-Induced Adhesion Molecule Expression via
Smad3 Protein
Beidong Chen, Wendong Wang, Tao Shen, Ruomei Qi*
Key Laboratory of Geriatrics, Beijing Institute of Geriatrics & Beijing Hospital, Ministry of Health, Beijing, China
Abstract
Atherosclerosis is a chronic inflammation disease that is initiated by endothelial cell injury. Oxidized low-density lipoprotein (ox-LDL) is directly associated with chronic vascular inflammation. To understand whether thioredoxin1 (Trx1) participates in an antiinflammatory defense mechanism in atherosclerosis, we investigated the effect of Trx1 on the expression of two adhesion molecules, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), in human umbilical vein endothelial cells (HUVECs). Thioredoxin1 and dominant-negative mutant thioredoxin1 (TD) were transiently overexpressed using adenovirus vector gene transfer. Our data showed that Trx1 overexpression suppressed ox-LDL-induced adhesion molecule expression in HUVECs. The overexpression of Trx1 promoted ox-LDL-ox-LDL-induced Smad3 phosphorylation and nuclear translocation. A co-immunoprecipitation assay indicated that Smad3 continued to interact with Trx1 with or without ox-LDL stimulation. These results suggest that Trx1 inherently suppresses VCAM-1 and ICAM-1 expression in vascular endothelia and may prevent the initiation of atherosclerosis by attenuating adhesion molecule expression. The enhancement of Smad3 phosphorylation and nuclear expression appears to be primarily responsible for the Trx1-induced downregulation of adhesion molecules.
Citation:Chen B, Wang W, Shen T, Qi R (2013) Thioredoxin1 Downregulates Oxidized Low-Density Lipoprotein-Induced Adhesion Molecule Expression via Smad3 Protein. PLoS ONE 8(9): e76226. doi:10.1371/journal.pone.0076226
Editor:Alberico Catapano, University of Milan, Italy
ReceivedNovember 13, 2012;AcceptedAugust 22, 2013;PublishedSeptember 23, 2013
Copyright:ß2013 Chen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding:This work was supported by grants from the National Natural Science Foundation of China (no. 30900627, 81270379, 81200221). The funding agency had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests:The authors have ceclared that no competing interests exist. * E-mail: [email protected]
Introduction
Oxidized low-density lipoprotein (ox-LDL) is well known to play a crucial role in the initiation and progression of atherosclerosis, which can be considered an inflammatory disease [1]. Modified LDL can induce endothelial cell activation and the expression of adhesion molecules that facilitate the firm adhesion and activation of leukocytes and platelets, thereby enhancing inflammatory processes that underlie atherosclerosis [2]. Smad transcription factors are specific downstream mediators of the transforming growth factor b (TGF-b) signaling pathway. TGF-b is a multifunctional cytokine that regulates cell proliferation, differen-tiation, apoptosis, and extracellular matrix accumulation [3]. Smad3 belongs to receptor-regulated Smads (R-Smads) and can be activated by TGF-b and activin receptors. TGF-b has an antiatherogenic effect, in which it prevents the ox-LDL-induced expression of adhesion molecules [4] and contributes to plaque stabilization [5]. Additionally, the disruption of TGF-bsignaling in T cells accelerates atherosclerosis in apolipoprotein E knockout mice [6]. In vascular cells, cholesterol suppressed TGF-bsignaling by increasing lipid rafts and the caveolae accumulation of TGF-b
receptors [7]. Although the TGF-b/Smad pathway has been shown to have protective, antiinflammatory effects on cells that are important to atherosclerotic lesion formation [8], remaining unclear is how Smad3 contributes to ox-LDL stimulation in human umbilical vein endothelial cells (HUVECs).
used HUVECs to establish cells that overexpressed Trx or dominant-negative Trx and investigated the effects of Trx on Smad3 and adhesion protein expression in HUVECs.
Materials and Methods
Ethics statement
According to the Declaration of Helsinki, umbilical cords were donated by cesarean section patients, from whom we received written informed consent. The study was approved by the ethics committee of the Beijing Institute of Geriatrics, Ministry of Health (approval no. 2006-05).
Reagents
SIS3(S0447), 5,59-dithiobis (2-nitrobenzoic acid; DTNB), thior-edoxin reductase (TrxR) from rat liver, phenylmethanesulfonyl fluoride (PMSF), Protease Inhibitor Cocktail, and type I collage-nase were purchased from Sigma-Aldrich (St. Louis, MO, USA). 29,79-Dichlorodihydrofluorescein diacetate (DCFH-DA) was ob-tained from the Beyotime Institute of Biotechnology (Shanghai, China). Anti-rabbit biotinylated antibody was obtained from Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd (Beijing, China). The Trx1 expression vector pcDNA3-Trx1 and redox-inactive dominant-negative mutant Trx (TD; Cy-s32RSer32/Cys35RSer35[C32S/C35S]) expression vector pcDNA3-TD were kindly provided by Dr. J. Yodoi (Institute for Virus Research, Kyoto University, Kyoto, Japan). NE-PER nuclear and cytoplasmic extraction reagents and the BCA assay kit were purchased from Pierce (Rockford, IL, USA). All of the other reagents were of analytical grade.
Preparation and culture of HUVECs
Freshly isolated umbilical cords were obtained from healthy donors. Primary HUVECs were isolated from the umbilical vein using type I collagenase and then cultured in Dulbecco Modified Eagle Medium supplemented with 20% fetal bovine serum, endothelial cell growth factor, 100 U/ml penicillin, 100 U/ml streptomycin, and 1% glutamine in a humidified incubator at 37uC and 5% CO2. HUVECs at passages 2–4 were used in the present study.
Construction of Trx and dominant-negative mutant thioredoxin (TD) adenovirus
The ViraPower Adenoviral Gateway Expression system from Invitrogen (Carlsbad, CA, USA) was used to construct green fluorescent protein (GFP), Trx, and TD adenovirus expression vectors. The entry vector, pENTR/D-TOPO, was kindly provided by Dr. Jianping Cai (Beijing Institute of Geriatrics). The DNA restriction enzymes KpnI and XbaI were purchased from TaKaRa Bio Company (Otsu, Shiga, Japan). T4 DNA Ligase and PacI were purchased from Promega (Madison, WI, USA) and New England BioLabs (Ipswich, MA), respectively. We constructed adenovirus expression vectors according to the manufacturer’s protocols. Briefly, target fragments were digested from a pcDNA3.0 vector and inserted into the Entry vectors. The gateway technique was used to recombine and generate adeno-virus expression vectors. After the identification of the adenoadeno-virus expression vectors, these plasmids were purified and digested using PacI. The 293A cell line (provided by the ViraPower Adenoviral Gateway Expression Kit, Invitrogen) was used to package the adenovirus. After 8–10 days of transfection, the viruses were harvested. HUVECs were infected with an adenovirus that contained GFP (Ad-GFP) as a control group, Trx (Ad-Trx), and
TD (Ad-TD) for 60 h to overexpress GFP, Trx, and TD in HUVECs, respectively.
Knockdown of Trx1 by siRNA
The sequence of the small-interfering RNA (siRNA) used against Trx1 was 59-AUGACUGUCAGGAUGUUGCdTdT-39 [19]. The scramble oligonucleotide 59 -UUCUCCGAACGUGU-CACGUTT-39 was used as a negative control (NC). The cells were seeded in six-well plates and cultured overnight and transfected with 50 nM siRNA or NC oligonucleotide, respec-tively, using Effectene Transfection Reagent (Qiagen), according to the manufacturer’s instructions. All of the experiments with Trx1-knockdown cells were performed 60 h after transfection.
Assay of Trx activity in HUVECs
The insulin disulfide reduction assay was essentially performed as described elsewhere [20], with a slight modification. Transiently transfected cells were lysed in lysis buffer (20 mM HEPES [pH 7.9], 100 mM KCl, 300 mM NaCl, 10 mM ethylenedi-aminetetraacetic acid [EDTA], and 0.1% Nonidet P-40 plus protease inhibitors). Cell extracts (20mg) were preincubated at 37uC for 20 min with 2ml of dithiothreitol (DTT) activation buffer that consisted of 50 mM HEPES (pH 7.6), 1 mM EDTA, 1 mg/ ml bovine serum albumin (BSA), and 2 mM DTT in a total volume of 70ml to reduce Trx. The final concentration of DTT was 57mM. Afterward, 40ml of reaction mixture (256 mM HEPES [pH 7.6], 10 mM EDTA, 2.05 mg/ml nicotinamide adenine dinucleotide phosphate [NADPH], and 6.4 mg/ml insulin) was added. The reaction began with the addition of 10ml of rat TrxR (100 A412 U/ml), and incubation continued for 20 min at 37uC. The reaction was stopped by the addition of 0.5 ml of 6 M guanidine-HCl and 1 mM DTNB (3-carboxy-4-nitrophenyl disulfide), and absorbance was measured at 412 nm.
Isolation and oxidation of LDL
Human LDL was isolated from the plasma of healthy donors by sequential ultracentrifugation using a previously described method [21]. The concentration of LDL protein was determined using a UV-160A ultraviolet-visible spectrum spectrophotometer (Shi-madzu, Kyoto, Japan). For oxidation, LDL (3–5 mg/ml) was incubated with 5mM CuSO4as the oxidant for 12–16 h at room temperature and quenched by the addition of 2 mM EDTA. The ox-LDL preparation was sterilized through sterile 0.22-mm Millex syringe-driven filters. The LDL oxidation level (0.1mM mal-ondialdehyde (MDA)/mg protein) was determined by the thiobarbituric acid reactive substance (TBARS) assay.
Western blot analysis
Monocyte-endothelial cell adhesion assay
U937 monocytes (16107cells/ml) were incubated with 10mM 29,79-bis-(2-carboxyethyl)-5-(and-6)-carboxy-fluorescein acetoxy-methyl ester (BCECF-AM; Beyotime Institute of Biotechnology, Shanghai, China) for 30 min at 37uC in RPMI-1640 medium. HUVECs (56105cells/well) were seeded in six-well plates and stimulated with 100mg/ml ox-LDL for 6 h. The HUVECs were then washed with PBS three times to remove ox-LDL. The fluorescent-labeled U937 monocytes were added to the stimulated HUVECs and incubated for a further 2 h. After washing out the unbound U937 three times, monocyte adhesion was measured by detecting the fluorescent intensity using a Fluoroskan (Thermo Scientific). Wells that contained HUVECs alone were used as blanks.
Immunoprecipitation and immunoblotting
Lysates (500mg protein) were immunoprecipitated with 2mg of Trx antibody overnight at 4uC. After incubation with protein A and protein G-sepharose (GE Healthcare, Munich, Germany) for 2 h at 4uC, the resulting beads were washed and boiled in PAGE sample buffer, and the proteins were resolved by SDS-PAGE. Immunoblotting was performed with antibodies directed against Smad3 and pSmad3. To avoid the influence of the heavy chain in the homology antibody, a specific secondary antibody to rabbit IgG light chain (HRP) was used, and the proteins were then detected using an ECL kit. Mouse monoclonal (SB62a) secondary antibody to rabbit IgG light chain (HRP) was purchased from Abcam (Boston, MA, USA).
Detection of reactive oxygen species generation in cells DCFH-DA was used to detect intracellular reactive oxygen species (ROS) generation (49). Briefly, GFP, Trx, and Ad-TD cells were cultured overnight and then loaded with 10mM DCFH-DA for 30 min. The ROS indicator in the medium was then washed off. After three additional washes, the cells were digested with trypsin. The cells were then harvested and determined at an excitation wavelength of 480 nm and emission wavelength of 520 nm on an F-4500 Fluorescence Spectropho-tometer (Hitachi).
Data analysis
The data are expressed as mean6SEM. Statistical comparisons were made using one-way analysis of variance (ANOVA) followed by the Bonferroni test for multiple-group comparisons. Values of p,0.05 were considered statistically significant.
Results
Thioredoxin downregulated VCAM-1 and ICAM-1 expression in HUVECs
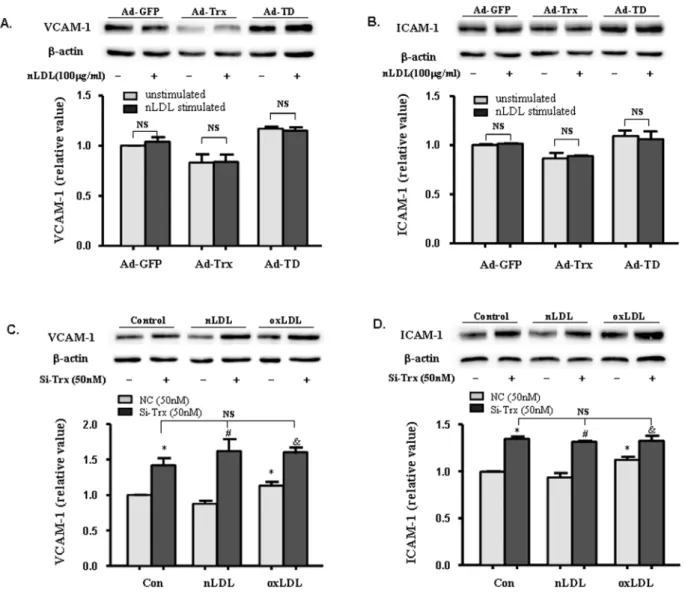
To determine whether Trx plays a role in the regulation of VCAM-1 and ICAM-1 expression in HUVECs, the expression of these two adhesion molecules was analyzed in cells that overexpressed Trx and dominant-negative Trx. The immunoblot-ting analysis showed that the protein levels of Trx in the Ad-Trx and Ad-TD groups were increased compared with the Ad-GFP control group. As expected, the insulin reduction-based assays showed that Trx activity increased in the Ad-Trx group but decreased in the Ad-TD group compared with the Ad-GFP group (Fig. 1A). Functional Trx eliminated intracellular ROS by providing electrons to the peroxiredoxin-catalyzed reduction of ROS; therefore, the amount of intracellular ROS could reflect Trx activity. As shown in Fig. 1B, ROS generation was inhibited in the
Ad-Trx group. In contrast, enhanced ROS production in HUVECs was found in the TD group. These results indicate that Ad-Trx overexpressed functional Trx, whereas Ad-TD only enhanced redox-dysfunctional Trx. To determine whether Trx affects the expression of ICAM-1 and VCAM-1 in HUVECs, adenovirus-infected HUVECs were treated with or without ox-LDL for 6 h, and protein levels were detected by Western blot. As shown in Fig. 1C and D, Trx overexpression inhibited ICAM-1 and VCAM-1 expression under both basal and ox-LDL-stimulat-ed conditions, whereas TD overexpression did not have this protective effect and even significantly enhanced VCAM-1 expression. To verify the functional effects of Trx in HUVECs, a monocyte-endothelial cell adhesion assay was performed. As shown in Fig. 1E and F, consistent with the Western blot results, Trx overexpression inhibited cell adhesion to ox-LDL-stimulated HUVECs, whereas TD enhanced adhesion. Additionally, the effect of Trx on VCAM-1 and ICAM-1 expression was investigated in cells that had their endogenous Trx1 knocked down by siRNA (siTrx). As shown in Fig. 1G, VCAM-1 and ICAM-1 expression was significantly enhanced in siTrx cells under basal conditions.
Native LDL failed to increase adhesion molecule expression in HUVECs
Native LDL (nLDL) is a critical control of ox-LDL. We detected VCAM-1 and ICAM-1 expression in nLDL-stimulated Ad-GFP, Ad-Trx, and Ad-TD cells. As shown in Fig. 2A, nLDL did not significantly enhance adhesion molecule expression in the three HUVECs groups. VCAM-1 and ICAM-1 expression was also detected in nLDL- and ox-LDL-stimulated Trx knock-down cells. Unexpectedly, Trx knock-down greatly increased the expression of adhesion molecules to such an extent that nLDL and ox-LDL failed to further enhance their expression.
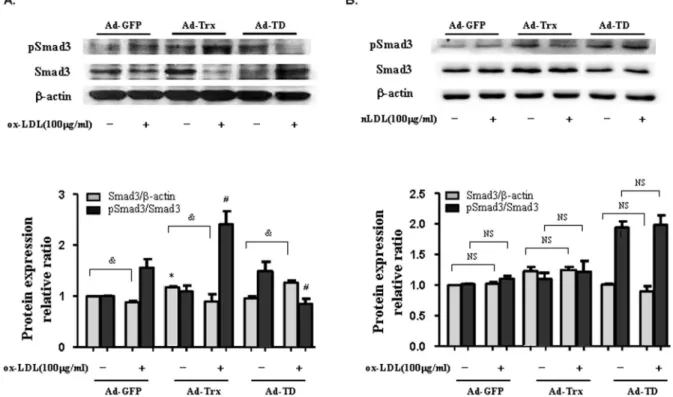
Thioredoxin induced Smad3 phosphorylation in ox-LDL-treated HUVECs
To ascertain whether the antiinflammatory effect of Trx interacts with the Smad3 pathway, the expression levels of pSmad3 and Smad3 were detected in the three groups of cells under basal and ox-LDL-stimulated conditions. As shown in Fig. 3A, ox-LDL stimulation decreased Smad3 expression in the GFP and Trx groups but had an opposite effect in the Ad-TD group. Trx overexpression further enhanced Smad3 phos-phorylation and TD overexpression decreased Smad3 phosphor-ylation compared with the Ad-GFP control group after ox-LDL stimulation in HUVECs. Additionally, nLDL was used as a control for ox-LDL. As shown in Fig. 3B, no significant difference was observed compared with the unstimulated groups after nLDL stimulation in HUVECs. These results indicate that Trx plays an important regulatory role in Smad3 expression and phosphoryla-tion.
SIS3, a specific inhibitor of Smad3 phosphorylation, reversed the Trx-induced inhibition of ICAM-1 and VCAM-1 expression
ICAM-1 and VCAM-1 expression in the Ad-GFP and Ad-Trx groups after ox-LDL stimulation. These data indicate that the Smad3 pathway may be involved in the Trx-induced inhibition of adhesion molecules in HUVECs.
Trx had a constitutive interaction with Smad3 and pSmad3 in HUVECs
Co-immunoprecipitation was performed to investigate the interaction between Trx and Smad3 in HUVECs with and Figure 1. Thioredoxin downregulates VCAM-1 and ICAM-1 expression in HUVECs.(A)Top, Immunoblot of Trx1 in HUVECs infected by GFP adenovirus (Ad-GFP), Trx adenovirus (Ad-Trx), and TD adenovirus (Ad-TD).Bottom, Trx1 activity in Ad-GFP, Ad-Trx, and Ad-TD cells, determined by insulin reduction-based assay. (B) Reactive oxygen species production in HUVECs. After 2 h stimulation of ox-LDL (100mg/ml), ROS production was
analyzed by measuring the mean fluorescence intensity using flow cytometry. (C,D) Immunoblot of VCAM-1 and ICAM-1 in GFP, Trx, and Ad-TD cells under basal conditions and after 4 h ox-LDL (100mg/ml) stimulation. Relative VCAM-1 and ICAM-1 expression was determined by
densitometric analysis. In all of the histograms, each value represents the mean6SEM (n= 3 independent measurements). *p,0.05, compared with unstimulated Ad-GFP cells;#p
,0.05, compared with ox-LDL-stimulated Ad-GFP cells. (E) U937 monocyte adhesion assay in ox-LDL-stimulated Ad-GFP, Ad-Trx, and Ad-TD cells. U937 cells that adhered to HUVECs were observed under a fluorescent microscope at 2006magnification. (F) The
intensity of fluorescence-labeled adherent U937 monocytes was measured with a fluorometer (excitation wavelength, 485 nm; emission wavelength, 530 nm). The intensity was normalized to that of control cells in each group. The data are expressed as mean6SEMs (n= 5). *p,0.05, compared with ox-LDL-treated Ad-GFP group;#
p,0.05, compared with unstimulated Ad-GFP group. (G)Top, Immunoblot of Trx1, VCAM-1, and ICAM-1 in wildtype HUVECs (Wt) and HUVECs transfected by negative control siRNA (NC) or Trx siRNA (si-Trx).Bottom, Relative Trx, VCAM-1, and ICAM-1 expression was determined by densitometric analysis. In all of the histograms, each value represents the mean6SEM (n= 3 independent measurements). *p,0.05, compared with Wt cell.
doi:10.1371/journal.pone.0076226.g001
Figure 2. Effect of native LDL (nLDL) on the expression of VCAM-1 and ICAM-1 in Trx-overexpressing and -knock-down HUVECs.(A, B) Immunoblot of VCAM-1 and ICAM-1 in Ad-GFP, Ad-Trx, and Ad-TD cells under basal conditions and after 4 h nLDL (100mg/ml) stimulation. (C, D)
Immunoblot of VCAM-1 and ICAM-1 in NC and si-Trx cells under basal conditions and after 4 h nLDL (100mg/ml) or ox-LDL (100mg/ml) stimulation.
Relative VCAM-1 and ICAM-1 expression was determined by densitometric analysis. In all of the histograms, each value represents the mean6SEM (n= 3 independent measurements). *p,0.05, compared with unstimulated NC cells;#p
,0.05, compared with nLDL-stimulated NC cells;&p,0.05, compared with ox-LDL-stimulated NC cells.
Figure 3. Effect of Trx on the expression of pSmad3 and Smad3 in HUVECs stimulated with ox-LDL and nLDL.(A) Immunoblot of pSmad3 and Smad3 expression in ox-LDL-stimulated Ad-GFP, Ad-Trx, and Ad-TD HUVECs. (B) Immunoblot of pSmad3 and Smad3 expression in nLDL-treated Ad-GFP, Ad-Trx, and Ad-TD HUVECs. Relative pSmad3 and Smad3 content was determined by densitometric analysis. The data from three separate experiments are expressed as mean6SEM. *p,0.05, compared with unstimulated Ad-GFP cells (Smad3/b-actin);#p
,0.05, compared with ox-LDL-stimulated Ad-GFP cells (pSmad3/Smad3);&p
,0.05, unstimulated cells compared with ox-LDL stimulated cells (Smad3/b-actin). doi:10.1371/journal.pone.0076226.g003
Figure 4. Role of SIS3 in VCAM-1 and ICAM-1 expression in HUVECs stimulated with ox-LDL.(A) Overexpression of Trx suppressed ox-LDL-induced VCAM-1 expression. The pretreatment of Ad-Trx cells with SIS3 for 1 h completely reversed the inhibitory effect of Trx on VCAM-1 expression. (B) The overexpression of Trx inhibited ox-LDL-induced ICAM-1 expression. The pretreatment of Ad-Trx cells with SIS3 reversed the inhibitory effect of Trx on ICAM-1 expression. The relative expression of VCAM-1 and ICAM-1 was determined by densitometric analysis. The data from three separate experiments are expressed as mean6SEM. *p,0.05, compared with unstimulated Ad-GFP cells;#p
,0.05, compared with ox-LDL-stimulated Ad-GFP cells;$p,0.05, compared with ox-LDL-stimulated Ad-Trx cells.
without ox-LDL stimulation. Polyclonal rabbit anti-Trx antibody was used to pull down the Trx interaction protein, and anti-Smad3 and pSmad3 antibodies were used to immunoblot the Trx-pull-down proteins. Normal rabbit IgG served as a negative control. As shown in Fig. 5A and B, Trx interacted with Smad3 and pSmad3 under both basal and ox-LDL-stimulated conditions. The interaction between Trx and pSmad3 was enhanced by ox-LDL stimulation. To determine whether the interactions were regulated by the Trx redox site and how the Trx redox site affected the interactions between Trx and Smad3 protein with ox-LDL stimulation, the Ad-Trx and Ad-TD groups were used to perform co-immunoprecipitation with and without ox-LDL stimulation. The data suggested that the interaction between Trx and Smad3/ pSmad3 was unaffected by the Trx redox site under basal and ox-LDL stimulation conditions, ox-ox-LDL stimulation increased the interaction between Trx1 and pSmad3/Smad3 in both the Ad-Trx and Ad-TD groups (Fig. 5C and D).
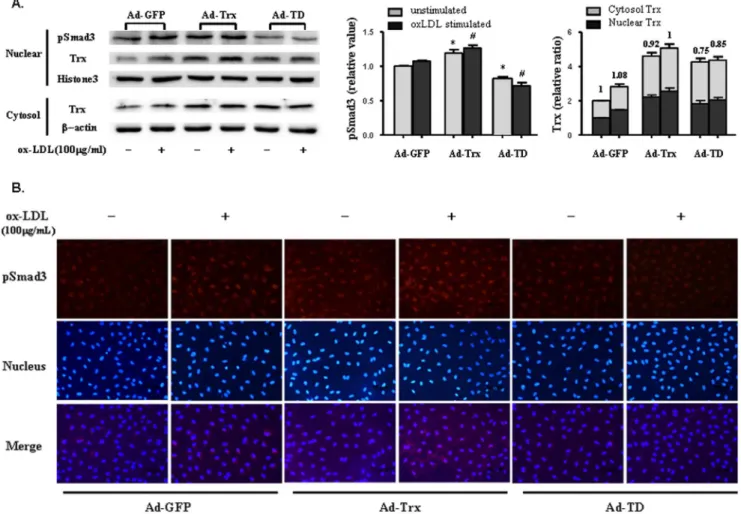
Trx and pSmad3 had high nuclear expression in Trx-overexpressing cells
Smad2, Smad3, and Smad4 are predominantly located in the cytoplasm. Upon TGF-b receptor activation, phosphorylated Smad2 and Smad3 translocate to the nucleus, coupled with the common mediator Smad4 [23]. Thus, the relative subcellular distribution of Smads may play an important role in TGF-b
signaling and regulate Smad signaling by localizing the proteins. Trx is a stress-induced protein and translocates to the nucleus under many stimulation conditions. The aforementioned results showed that Trx is a Smad3-interacting protein, and its subcellular distribution might regulate Smad3 signaling by localizing the protein. We extracted nuclear and cytosolic proteins to analyze Trx and pSmad3 expression with and without ox-LDL stimula-tion. As shown in Fig. 6A, Trx and pSmad3 expression was significantly enhanced in the nucleus of Ad-Trx cells compared with Ad-GFP cells, especially with ox-LDL stimulation. Nuclear pSmad3 expression was obviously decreased in Ad-TD cells in both basal and ox-LDL-stimulated conditions. After nuclear and cytosolic Trx expression was normalized to the unstimulated Ad-GFP group, the relative expression ratios of nuclear Trx to cytosolic Trx were obtained. The data indicated that dysfunctional Trx was more likely distributed to the cytoplasm. As shown in Fig. 6B, the immunofluorescent analysis yielded results that were similar to the Western blot results. Because Trx is a Smad3-interacting protein, these results indicate that functional Trx might promote the translocation of pSmad3 to the nucleus and contribute to further activation of the Smad3 signaling pathway.
Discussion
The present study showed that Trx inhibited the expression of the adhesion molecules VCAM-1 and ICAM-1 in HUVECs. We found that overexpression of functional Trx significantly enhanced Smad3 phosphorylation, whereas SIS3, a specific inhibitor of Smad3, reversed the Trx-induced inhibition of VCAM-1 and ICAM-1 expression after ox-LDL stimulation. These data indicate that Trx inhibited adhesion molecule expression via the Smad3 protein. Moreover, we found that Trx continued to interact with Smad3 and pSmad3, and this interaction might be responsible for the further nuclear translocation of pSmad3 in Trx-overexpressing HUVECs and activation of the Smad3 signaling pathway.
Ox-LDL is well-known to play a crucial role in the initiation and progression of atherosclerosis, which can be considered an inflammatory disease. Ox-LDL can induce proinflammatory actions in endothelial cells by increasing the expression of adhesion
molecules, induction of MCP-1 production, and direct chemoat-tractant effect or activation of AP-1 and its transcription factors [24]. Numerous studies have reported that TGF-b has an antiatherogenic effect. TGF-b was shown to prevent the ox-LDL-induced expression of adhesion molecules [4] and contribute to plaque stabilization [5]. In endothelial cells, HDL induced TGF-b2 and activated Smad2/3 to exert its antiatherosclerotic effect [25]. Recently, Guo et al. reported that ox-LDL upregulated TGF-b1 protein production and Smad3 phosphorylation through the Ras/ERK/PLTP pathway in human alveolar type II epithelial cells [26]. However, to date, the effect of ox-LDL in the TGF-b/ Smad signaling pathway in endothelial cells has not been reported. The present study found that ox-LDL reduced Smad3 expression but enhanced its phosphorylation and nuclear translocation in HUVECs.
The Trx system, including Trx, Trx reductase, and NADPH, is a ubiquitous thiol oxidoreductase system that regulates cellular reduction/oxidation (redox) status [27]. Trx reduces oxidized cysteine groups on proteins through an interaction with the redox-active center of Trx (Cys-Gly-Pro-Cys) to form a disulfide bond, which in turn can be reduced by TrxR and NADPH [9]. In the present study, wildtype Trx and redox-inactive dominant-negative mutant Trx (TD) were used to construct an adenovirus expression vector and infect HUVECs. The C32S/C35S mutant was a strong competitive inhibitor of TrxR, in which TrxR recognized the mutant with nearly equivalent affinity to Trx [28]. In contrast to the overexpression of Trx, the present data showed that TD overexpression increased ROS generation and adhesion protein expression but suppressed the Smad3 pathway by inhibiting Smad3 phosphorylation and nuclear translocation. These data indicate that inflammation related to the Smad3 pathway was regulated by the Trx redox site. Interestingly, we found that both Trx and TD promoted Smad3 phosphorylation under basal conditions, suggesting that Trx might contribute to some other unknown regulatory mechanism of Smad3 phosphorylation in addition to redox regulation.
through the MAPK, PI3K-AKT, or PKC pathway. Based on the present results, we speculate that the interaction between Ad-Trx/ TD and Smad3 protein might change the structure of Smad3 and facilitate its phosphorylation. In our study, ox-LDL stimulation induced Smad3 phosphorylation in HUVECs, and the Trx redox site affected this pathway. Thus, Trx overexpression further enhanced Smad3 phosphorylation, whereas TD overexpression downregulated Smad3 phosphorylation. Because Smad3 expres-sion was reduced in ox-LDL-stimulated Ad-GFP and Ad-Trx cells but reversed in Ad-TD cells, ox-LDL might play a role in Smad3
degradation, which was also affected by the Trx redox site. Oxidize LDL stimulation enhanced the interaction between pSmad3 and Trx, which might promote the translocation of pSmad3 to the nucleus and contribute to further activation of the Smad3 signaling pathway. Therefore we conclude that Trx might regulate Smad3 pathway by interaction or through kinase pathway depending on its redox activity.
The present study focused on Smad3 and not Smad2. Although highly related to Smad3, Smad2 lacks the ability to bind DNA [36,40–42]. Additionally, the functional properties of these two Figure 5. Smad3 is a newly recognized interaction partner of Trx.Reciprocal immunoprecipitation was performed using an anti-Trx antibody. Smad3 and pSmad3 were co-immunoprecipitated with Trx. Rabbit IgG served as a negative control. (A, B) Immunoblot with anti-Trx, anti-Smad3, or anti-pSmad3 antibody in wildtype HUVECs under basal or ox-LDL-stimulated conditions. The data from three separate experiments are expressed as mean6SEM. *p,0.05, compared with unstimulated HUVECs. (C, D) Immunoblot with anti-Trx, anti-Smad3, or anti-pSmad3 antibody in Ad-GFP (1, 2, 5, 6), Ad-Trx (3, 7), and Ad-TD (4, 8) cells under basal or ox-LDL-stimulated conditions. The data from three separate experiments are expressed as mean6SEM. *p,0.05, compared with unstimulated Ad-GFP cells;#p
,0.05, compared with unstimulated Ad-Trx cells;$p,0.05, compared with unstimulated Ad-TD cells.
proteins may be somewhat different. The gene-targeting of Smad2 or Smad3 revealed that Smad3 cannot compensate for the defects in Smad2-null mice during early development in vivo and that Smad3 may play exclusive roles in immune system function [43– 45]. For this reason, we investigated only Smad3 in the present study.
In conclusion, the present results suggested that Trx inhibited adhesion molecule expression via Smad3 protein. Although further studies are required, Trx redox was found to regulate Smad3 phosphorylation in ox-LDL-stimulated HUVECs, and Trx interacted with Smad3/pSmad3. Our study provides a potentially new molecular site for atherosclerosis prevention and treatment at the level of endothelial cells.
Acknowledgments
We thank Dr. J. Yodoi (Institute for Virus Research, Kyoto University) for providing the pcDNA3-Trx1 and pcDNA3-TD expression vectors. We thank Mr. Hanbang Guo (Beijing Institute of Geriatrics, Beijing Hospital) for human LDL isolation.
Author Contributions
Conceived and designed the experiments: BC RQ. Performed the experiments: BC WW. Analyzed the data: BC WW TS. Contributed reagents/materials/analysis tools: BC RQ. Wrote the paper: BC TS RQ.
References
1. Ross R (1999) Atherosclerosis is an inflammatory disease. Am Heart J 138: S419–S420.
2. Kaplan ZS, Jackson SP (2011) The role of platelets in atherothrombosis. Hematology Am Soc Hematol Educ Program 2011: 51–61.
3. Roberts AB, Sporn MB (1993) Physiological actions and clinical applications of transforming growth factor-beta (TGF-beta). Growth Factors 8: 1–9. 4. Chen H, Li D, Saldeen T, Mehta JL (2001) Transforming growth factor-b1
modulates oxidatively modified LDL-induced expression of adhesion molecules: role of LOX-1. Circ Res 89: 1155–1160.
5. Sakamoto Y, Miyazaki A, Tamagawa H, Wang GP, Horiuchi S (2005) Specific interaction of oxidized low-density lipoprotein with thrombospondin-1 inhibits transforming growth factor-bfrom its activation. Atherosclerosis 183: 85–93. 6. Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, et al. (2003)
Disruption of TGF-bsignaling in T cells accelerates atherosclerosis. J Clin Invest 112: 1342–1350.
7. Chen CL, Liu IH, Fliesler SJ, Han X, Huang SS, et al. (2007) Cholesterol suppresses cellular TGF-bresponsiveness: implications in atherogenesis. J Cell Sci 120: 3509–3521.
Figure 6. Role of Trx in nuclear translocation of pSmad3.(A)Top, Nuclear pSmad3 and Trx proteins in Ad-GFP, Ad-Trx, and Ad-TD cells.
Bottom, Cytosolic Trx expression in Ad-GFP, Ad-Trx, and Ad-TD cells. The cells were treated with or without ox-LDL (100mg/ml) stimulation for 6 h as
indicated.Left column, Quantitative nuclear pSmad3 protein is shown.Right column, Nuclear and cytosolic Trx expression was normalized to that of the unstimulated Ad-GFP group. The numbers above the columns indicate the relative expression ratio of nuclear Trx to cytosolic Trx. The data from three separate experiments are expressed as mean6SEM. *p,0.05, compared with unstimulated Ad-GFP cells;#
8. Feinberg MW, Jain MK (2005) Role of transforming growth factor-beta1/ Smads in regulating vascular inflammation and atherogenesis. Panminerva Med 47: 169–186.
9. Yamawaki H, Haendeler J, Berk BC (2003) Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res 93: 1029–1033.
10. Powis G, Montfort WR (2001) Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct 30: 421–455.
11. Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, et al. (2004) Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res 94: 1483–1491.
12. Miranda-Vizuete A, Ljung J, Damdimopoulos AE, Gustafsson JA, Oko R, et al. (2001) Characterization of Sptrx, a novel member of the thioredoxin family specifically expressed in human spermatozoa. J Biol Chem 276: 31567–31574. 13. Arner ES, Holmgren A (2000) Physiological functions of thioredoxin and
thioredoxin reductase. Eur J Biochem 267: 6102–6109.
14. Powis G, Mustacich D, Coon A (2000) The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med 29: 312–322.
15. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, et al. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17: 2596–2606.
16. Masutani H, Ueda S, Yodoi J (2005) The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ 12 Suppl 1: 991–998.
17. Chen B, Guan D, Cui ZJ, Wang X, Shen X (2010) Thioredoxin 1 downregulates MCP-1 secretion and expression in human endothelial cells by suppressing nuclear translocation of activator protein 1 and redox factor-1. Am J Physiol Cell Physiol 298: C1170–C1179.
18. Watanabe R, Nakamura H, Masutani H, Yodoi J (2010) Anti-oxidative, anti-cancer and anti-inflammatory actions by thioredoxin 1 and thioredoxin-binding protein-2. Pharmacol Ther 127: 261–270.
19. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, et al. (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2: 222–228.
20. Junn E, Han SH, Im JY, Yang Y, Cho EW, et al. (2000) Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol 164: 6287–6295.
21. Chung BH, Wilkinson T, Geer JC, Segrest JP (1980) Preparative and quantitative isolation of plasma lipoproteins: rapid, single discontinuous density gradient ultracentrifugation in a vertical rotor. J Lipid Res 21: 284–291. 22. Jinnin M, Ihn H, Tamaki K (2006) Characterization of SIS3, a novel specific
inhibitor of Smad3, and its effect on transforming growth factor-b1-induced extracellular matrix expression. Mol Pharmacol 69: 597–607.
23. Miyazono K (2000) TGF-bsignaling by Smad proteins. Cytokine Growth Factor Rev 11: 15–22.
24. Fan J, Watanabe T (2003) Inflammatory reactions in the pathogenesis of atherosclerosis. J Atheroscler Thromb 10: 63–71.
25. Norata GD, Callegari E, Marchesi M, Chiesa G, Eriksson P, et al. (2005) High-density lipoproteins induce transforming growth factor-b2 expression in
endothelial cells. Circulation 111: 2805–2811.
26. Guo LL, Chen YJ, Wang T, An J, Wang CN, et al. (2012) Ox-LDL-induced TGF-b1 production in human alveolar epithelial cells: involvement of the Ras/ ERK/PLTP pathway. J Cell Physiol 227: 3185–3191.
27. World CJ, Yamawaki H, Berk BC (2006) Thioredoxin in the cardiovascular system. J Mol Med (Berl) 84: 997–1003.
28. Oblong JE, Berggren M, Gasdaska PY, Powis G (1994) Site-directed mutagenesis of active site cysteines in human thioredoxin produces competitive inhibitors of human thioredoxin reductase and elimination of mitogenic properties of thioredoxin. J Biol Chem 269: 11714–11720.
29. Javelaud D, Mauviel A (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene 24: 5742–5750.
30. Massague J, Chen YG (2000) Controlling TGF-bsignaling. Genes Dev 14: 627– 644.
31. Lutz M, Knaus P (2002) Integration of the TGF-bpathway into the cellular signalling network. Cell Signal 14: 977–988.
32. Derynck R, Zhang Y, Feng XH (1998) Smads: transcriptional activators of TGF-bresponses. Cell 95: 737–740.
33. Kretzschmar M, Doody J, Massague J (1997) Opposing BMP and EGF signalling pathways converge on the TGF-bfamily mediator Smad1. Nature 389: 618–622.
34. Yakymovych I, Ten Dijke P, Heldin CH, Souchelnytskyi S (2001) Regulation of Smad signaling by protein kinase C. FASEB J 15: 553–555.
35. Moustakas A, Souchelnytskyi S, Heldin CH (2001) Smad regulation in TGF-b signal transduction. J Cell Sci 114: 4359–4369.
36. Shi Y, Massague J (2003) Mechanisms of TGF-bsignaling from cell membrane to the nucleus. Cell 113: 685–700.
37. Hashimoto S, Matsumoto K, Gon Y, Furuichi S, Maruoka S, et al. (1999) Thioredoxin negatively regulates p38 MAP kinase activation and IL-6 production by tumor necrosis factor-a. Biochem Biophys Res Commun 258: 443–447.
38. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, et al. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277: 20336–20342.
39. Watson JA, Rumsby MG, Wolowacz RG (1999) Phage display identifies thioredoxin and superoxide dismutase as novel protein kinase C-interacting proteins: thioredoxin inhibits protein kinase C-mediated phosphorylation of histone. Biochem J 343 Pt 2: 301–305.
40. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, et al. (1998) Direct binding of Smad3 and Smad4 to critical TGFb-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17: 3091–3100. 41. Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W (1998) Identification and
functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-b, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273: 21145–21152. 42. Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, et al. (1998) Human
Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1: 611–617.
43. Nomura M, Li E (1998) Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393: 786–790.
44. Zhu Y, Richardson JA, Parada LF, Graff JM (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94: 703–714.